
Abstract
Primary hyperoxalurias (PH) comprise a group of three distinct metabolic diseases caused by derangement of glyoxylate metabolism in the liver. Recent years have seen advances in several aspects of PH research. This paper reviews current knowledge of the genetic and biochemical basis of PH, the specific epidemiology and clinical presentation of each type, and therapeutic approaches in different disease stages. Potential future specific therapies are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The first report of multiple oxalate renal calculi and nephrocalcinosis was by Lepoutre in 1925 [1]. Archer et al. [2] first used the term primary hyperoxaluria to describe two cases of extensive kidney stones that began in early childhood. Since then, the definition of primary hyperoxaluria has been extended to encompass a group of recessive inborn errors of glyoxylate metabolism characterized by overproduction of oxalate mainly in the liver and massive excretion of oxalate in the urine. Calcium oxalate is insoluble in body fluids and tends to precipitate primarily in the kidney, forming urinary calculi or diffuse nephrocalcinosis. Calcium oxalate crystals can obstruct the renal tubules, disrupt cellular function, and damage nearby cells by promoting inflammation and inducing oxidative stress [3–5]. As glomerular filtration rate (GFR) declines, the oxalate load produced by the liver can no longer be cleared by the kidneys, plasma oxalate increases, and calcium oxalate crystals are deposited in virtually all tissues in a devastating process termed systemic oxalosis. Patients can present at any age, ranging from infantile oxalosis with early-onset end-stage renal disease (ESRD) to nephrolithiasis first presenting in the seventh decade of life.
The most common, and most severe, form is primary hyperoxaluria type 1 (PH1; OMIM #259900) caused by absence, deficiency, or mistargeting of liver-specific enzyme alanine:glyoxylate aminotransferase (AGT). Primary hyperoxaluria types 2 (PH2; OMIM #260000) and 3 (PH3; OMIM #613616) are rarer and are caused by dysfunction of glyoxylate reductase–hydroxypyruvate reductase (GRHPR) and 4-hydroxy-2-oxoglutarate aldolase (HOGA1), respectively. Secondary hyperoxaluria, due to intestinal overabsorption or poisoning, can also occur and should be excluded prior to establishing the diagnosis of PH but is beyond the scope of this review.
Clinical manifestations
The most common clinical feature of PH is nephrolithiasis (present in up to 90 % of patients at diagnosis), which can be identified by ultrasound (US) performed for evaluation of hematuria, abdominal pain, and/or recurrent urinary tract infections [6, 7]. Diffuse nephrocalcinosis is noted in about half of such patients. A significant proportion (20–59 % [7, 8]) present with end-stage renal disease (ESRD), and some are only diagnosed after renal allograft failure. About 10 % are identified before symptoms occur by screening family members of a symptomatic patient. In two registries, diagnosis was typically made early, with median age at symptom onset and diagnosis 5.5–6 and 7.3–10.0 years, respectively [6, 7]. Between 18 % and 38 % of PH patients are diagnosed as adults [6, 8]. Time from initial symptoms and definitive diagnosis of PH may reflect a lack of familiarity with this condition by the treating physicians.
Primary hyperoxaluria type 1
PH1 is the most prevalent and severe form of primary hyperoxaluria. More than 50 % of patients progress to ESRD [9], with 28 % requiring renal replacement therapy (RRT) before the age of 15 [10]. Children with PH1 starting RRT are significantly younger than non-PH patients: median age 5.3 vs. 11.4 years [11]. Pyridoxine-responsive patients have a better prognosis. Infantile oxalosis is reported to account for 40 % of patients with PH requiring RRT [11], with inferior survival rates compared with non-PH patients: 86 %, 79 %, and 76 % at 1, 3, and 5 years from RRT initiation compared with 97 %, 94 %, and 92 % in non-PH patients [11]. This can be partly ascribed to systemic oxalosis, a process in which the decline in GFR results in plasma oxalate concentrations above the supersaturation threshold level. This inevitably leads to calcium oxalate deposition throughout the body, with devastating consequences (Fig. 1). Bone disease is most pronounced in the areas of rapid growth. Initially, transverse translucent symmetrical bands with thick, sclerotic margins can be seen at the end of the long bones. Advanced disease is characterized by cystic rarefaction of bones, loss of normal trabecular structure, and metaphyseal widening, causing multiple pathological fractures [12]. Calcium oxalate deposition was also noted in the periodontium, with bone loss and external tooth resorption [13]. Other manifestations include refractory anemia secondary to bone marrow replacement, hypothyroidism due to damage to the thyroid gland, myocardial deposition with conduction defects or cardiomyopathy, retinopathy, vasculopathy, and cerebrovascular infarction [14–19]. Interestingly, the liver, which is responsible for oxalate overproduction in patients with PH, is spared. Kidney transplantation without removal of the native liver results in rapid graft loss due to deposition of oxalate in the transplanted kidney [20].

Organs affected in primary hyperoxaluria (PH). Liver-produced oxalate induces kidney failure, and systemic oxalosis follows, damaging heart, blood vessels, bone, bone marrow, retina, and thyroid gland
Variable phenotypes have been reported, even within the same family, ranging from asymptomatic urinary oxalate excretion and nephrolithiasis in adulthood to early-onset ESRD and systemic oxalosis [21, 22]. Frishberg [23] described the extreme of intrafamilial phenotypic variability among patients with PH1 bearing the same genotype: The first baby in that family was born >30 years ago with ESRD. A US performed shortly before birth showed massive nephrocalcinosis, and the neonate succumbed to the disease at the age of 3 days. Four additional members of this family were diagnosed with PH1 between 2 and 7 months of age, developed ESRD associated with severe oxalosis, and died before the age of 3 years. Interestingly, their 20-year-old brother was diagnosed with PH1 upon screening of the entire family, and he remained absolutely asymptomatic with no evidence of nephrocalcinosis or nephrolithiasis. Takayama [24] reported a milder form of PH1 in Japanese patients, possibly reflecting differences in genetic background or epigenetic factors.
Primary hyperoxaluria type 2
PH2, described by Williams et al. [25], is rarer than PH1 and seems to have better long-term outcome [26]. Progression to ESRD occurs later than in PH1 [27, 28]. Hyperoxaluria-related graft failure after isolated renal transplantation has been described [29]. There is considerable clinical overlap between PH1 and PH2, making it impossible to differentiate clinically between them.
Primary hyperoxaluria type 3
PH3 is typically characterized by early-onset nephrolithiasis with normal renal function. First or recurrent stone formation during the second or third decade of life has also been observed. No patient so far has been reported with diffuse nephrocalcinosis or progression to ESRD [30]. Some patients achieve complete clinical remission despite persistent hyperoxaluria [31]. Furthermore, several siblings of PH3 patients, identified as having the same homozygous mutation based on family screening, remain asymptomatic into their third decade of life despite having persistent hyperoxaluria [30, 32]. The yet undefined pathogenetic mechanism of asymptomatic hyperoxaluria is intriguing, as it may pave the pathway to developing therapeutic strategies for patients with PH. One possible mechanism may be related to the fact that PH3 represents derangement in the catabolism of hydroxyproline, which is extremely high in the urine of healthy infants, reflecting high collagen turnover, but decreases gradually and stabilizes at approximately 10 years of age [33]. Similarly, 4-hydroxy-glutamate (4OHGlu) excretion, a metabolite of hydroxyproline, steadily declines with patient age, and there is a rough correlation between 4OHGlu and 4-hydroxyproline. A decline in 4OHGlu levels with age is also apparent in PH3 patients, presumably the result of a similar mechanism to that in controls [34].
Epidemiology
The exact prevalence of hyperoxaluria is unknown. Data is available only for PH1. European surveys suggest a prevalence of 0.7–2.9 per million population and a yearly incidence of 0.12–0.15 per million population or 1/120,000 live births [6, 9, 34, 35]. Higher prevalence is found in Mediterranean consanguineous populations [36]: PH1 accounts for 10 % of pediatric ESRD in Kuwait [37] and 13 % in Tunisia [38] compared with 0.5–2 % in Europe, North America, and Japan [11, 39, 40]. An adult dialysis unit from the Jenin District, Palestine, reported PH1 accounting for 10.7 % of prevalent patients [41].
PH2 and PH3 are presumably rarer, with a few dozen cases reported for each type [30, 31, 42–44]. PH3 became the second most prevalent form of PH [45]. Analysis of the US National Institutes of Health National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing, and the 1000 Genomes Project databases revealed higher-than-expected carrier frequency (CF) and incidence rate (IR), with PH1 and PH3 having similar CFs (∼1:174 vs ∼1:204) and IRs (∼1:119,714 vs ∼1:165,029). PH2 was ∼ 94 % less prevalent (CF ∼1:807, IR ∼1:2,602,898). The much higher prevalence of PH1 compared with PH3 in clinical registries may reflect underdiagnosis of the latter because of its much milder phenotype or lower penetrance [46]. The increasing availability of genetic testing may provide more accurate information in the future.
Genetics
PH1 is caused by mutations in the AGXT gene, a single-copy gene encoding the liver-specific enzyme AGT. AGXT is composed of 11 exons spanning 10 kbp and located on chromosome 2q36-q37 [47]. At least 178 pathogenic mutations have been described, involving all exons [48]. The majority are point mutations (missense, nonsense and splice-site mutations), and approximately a quarter are minor deletions or insertions [49]. The so-called minor allele haplotype is composed of a number of polymorphic variants (allelic frequency 5–10 %), all of which include a substitution of leucine to proline at codon 11 (c.32C > T, p.Pro11Leu). The minor allele has a reduced enzymatic capacity [50]. Furthermore, the P11L substitution introduces a weak mitochondrial targeting signal that in conjunction with a pathogenic variant, particularly c.508G > A (p.Gly170Arg), causes hyperoxaluria by peroxisome-to-mitochondrion mistargeting of AGT [51, 52]. Multiple mutations can be found even within a distinct ethnic group [53]. No clear genotype–phenotype correlation has been found [23, 54], except for pyridoxine responsiveness, which seems to be particularly beneficial in patients homozygous for the p.Gly170Arg or Phe152Ile mutations [55, 56].
In PH2, the gene encoding GRHPR is located in the centromeric region of chromosome 9 and contains nine exons spanning 9 kbp [57, 58]. At least 30 mutations in the GRHPR gene have been described [43, 59, 60]. A possible founder effect, of northern European origin, has been found for the two most common mutations [61].
In 2010, Belostotsky [30] identified mutations in the HOGA1 gene (formerly DHDPSL, chromosome 10q24.2) as causing PH3. About 20 pathogenic mutations in HOGA1 have been described, including a 3-bp deletion (c.944_946delAGG) prevalent among individuals of Ashkenazi Jewish descent. Beck et al. [31] described a potential European founder mutation (c.700 + 5G > T) as well as a triallelic PH mutation: homozygous HOGA1 and a heterozygous AGXT mutation. Heterozygous HOGA1 mutations were implicated as a putative risk factor for idiopathic calcium oxalate urolithiasis [62].
Biochemistry
Endogenous oxalate (HOOCCOOH) is a metabolic end product synthesized mainly in the liver and excreted almost exclusively in the urine. There is no evidence of oxalate being used or catabolized in mammalian tissue [63]. Most oxalate excreted in the urine of both normal individuals and PH patients is endogenous in origin, and only a small amount is derived from the diet. The main precursor of oxalate in humans is glyoxylate. Glyoxylate is produced either by peroxisomal oxidation of glycolate from vegetable matter or by catabolism of hydroxyproline derived from turnover of collagen, a major constituent of extracellular matrix and meat products, taking place in the mitochondria. Interestingly, there is a clear correlation between diet and intracellular compartmentalization of glyoxylate detoxification in different mammalian species: AGT in all herbivores studied is located either exclusively in the peroxisome, or in both the peroxisome and the mitochondria. Conversely, in carnivores, AGT is either mitochondrial or both mitochondrial and peroxisomal. Most omnivores have both mitochondrial and peroxisomal AGT. Humans are unique in having purely peroxisomal AGT despite having an omnivorous diet [64].
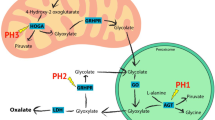
Glycolate is converted by glycolate oxidase (GO) to glyoxylate. Glyoxylate undergoes transamination to glycine by AGT, with alanine as the amino donor and pyridoxal 5’-phosphate (active vitamin B6 derivative) as a cofactor (Fig. 2). Hydroxyproline is catabolized to glyoxylate and pyruvate in the mitochondria. This glyoxylate is reduced to glycolate by glyoxylate reductase–hydroxypyruvate reductase (GRHPR). Glycolate then enters the peroxisome and is catabolized to glycine, as described above.

Cellular glyoxylate metabolism
AGT deficiency causes glyoxylate accumulation in PH1 [65]. Glyoxylate is mainly metabolized to oxalate by cytosolic lactate dehydrogenase (LDH) and partly reduced by cytosolic GRHPR to glycolate. This results in massive hyperoxaluria accompanied by glycolic aciduria, which is the hallmark of the disease. In about a third of cases, functional AGT is produced but is mistargeted to the mitochondria [66]. Lack of AGT in the peroxisome results in an identical PH1 phenotype.
GRHPR deficiency in PH2 [25] results in glyoxylate accumulation and hyperoxaluria. Hydroxypyruvate, which is normally reduced by GRHPR to D-glycerate, is converted by LDH to L-glycerate in its absence. The urine of PH2 patients is therefore characterized by hyperoxaluria combined with L-glyceric aciduria.
Deficiency in HOGA1, the final enzyme in mitochondrial hydroxyproline catabolism, is responsible for PH3 [30, 67]. The mechanism by which HOGA1 deficiency causes hyperoxaluria has not been elucidated. A putative explanation is that under normal conditions, glyoxylate is converted to glycolate in the mitochondria, rather than in the cytosol where it can be catabolized to oxalate (Fig. 2). Glycolate exits the mitochondria and enters the peroxisome where it is metabolized to glycine. HOGA1 deficiency results in accumulation of mitochondrial 4-hydroxy-2-oxoglutarate (HOG), which leaks to the cytosol, where it can be converted to glyoxylate by a yet unknown pyruvate aldolase, and oxidized by cytosolic LDH to oxalate [32]. Alternatively, it has been suggested that HOG builds up and down-regulates mitochondrial GRHPR activity, resulting in a phenotype reminiscent of PH2 [68].
Diagnosis
PH are rare diseases and can present at any age from infancy to late adulthood. A high index of suspicion is essential to avoid delay in diagnosis and provide timely treatment. All children with nephrolithiasis or nephrocalcinosis should be evaluated for an underlying metabolic disorder, as these are prevalent in the pediatric population. This includes measuring urinary excretion of sodium, calcium, phosphorus, magnesium, uric acid, oxalate, and citrate, as well as checking spot urine pH and amino acids and measuring urine volume. Adults with recurrent calcium oxalate kidney stones should also be evaluated for PH. Our suggestion for a simple diagnostic flowchart is depicted in Fig. 3. Evidence-based guidelines for diagnostic workup have been published [69, 70].

Diagnostic schema for suspected hyperoxaluria. Ca-Ox calcium oxalate, Ca calcium, P phosphorus, Mg magnesium, UA uric acid, PTH parathyroid hormone, vit D vitamin D, AGT alanine:glyoxylate aminotransferase, HOGA 4-hydroxy-2-oxoglutarate aldolase, GRHPR glyoxylate reductase–hydroxypyruvate reductase, 4-OH-Glu 4-hydroxyglutamate, PH1/PH2/PH3 primary hyperoxaluria types 1/2/3
Passed stones are composed of virtually pure calcium oxalate monohydrate (COM, also known as whewellite) in patients with PH1 [71]. PH stones have a light surface color and are composed of an aggregation of different-sized crystals, as opposed to idiopathic calcium oxalate calculi, which are strongly pigmented. Stones from PH3 patients (and one treatment-naïve PH1 patient) have exhibited both COM and calcium oxalate dehydrate (COD, weddellite) crystals [72]. Abundant oxalate crystals in a kidney biopsy, especially if associated with interstitial inflammatory reaction and granuloma formation, may also be a clue for the diagnosis of PH.
Urinary oxalate should be measured in a 24-h urine collection, if possible. PH patients generally have oxalate excretion that exceeds twice the upper limit of normal (<0.45 mmol/1.73 m2/24 h). Levels >0.5 mmol/1.73 m2/24 h should prompt further investigations, including ruling out secondary/enteric hyperoxaluria [69, 70]. If urine collection is not feasible (e.g., infants), a spot urinary oxalate/creatinine ratio can be performed but must be interpreted according to age, as this ratio is highest in premature newborns and decreases rapidly in the first year of life. It should be noted that children with markedly reduced GFR (<30 ml/min/1.73 m2) may have normal urinary oxalate excretion. Measurements of oxalate levels in plasma are helpful but not easily available [70]. Elevated urinary glycolic acid is characteristic of PH1 but has also been found in PH3 patients [30, 72]. Glyceric aciduria is suggestive of PH2 although not invariably found. Elevated urinary level of 4OHGlu effectively identifies patients with PH3 [32, 34]. As 4OHGlu levels were found to be markedly elevated in stored newborn screening dried-blood sample, this metabolite may be used to screen for PH3 [34].
Although there is overlap in urinary oxalate excretion between the three forms of PH, it has been suggested that patients with PH3 tend to have lower oxalate levels but higher urinary calcium and uric acid concentrations compared with those with PH1 or PH2 [62].
As mutation analysis technology advances, most patients suspected of having PH are diagnosed by genetic testing. Since PH1 accounts for >80 % of PH patients, it seems logical to test AGXT first, and if no mutation is found, GRHPR and HOGA1 should be screened for rarer mutations. Liver biopsy with enzymatic analysis is reserved for the minority of patients with progressive kidney disease who have a phenotype suggestive of PH without pathogenic mutations (unclassified PH). Normal AGT and GRHPR activity would exclude PH1 and PH2, respectively, in these cases, and enable safer decision making about dialysis and transplantation modalities. Molecular diagnosis can provide families with the option of premarital, prenatal, or pregestational diagnosis to ensure the birth of an unaffected child. Mutation screening should also be performed in asymptomatic siblings of affected individuals in order to consider early treatment. Genetic counseling is of utmost importance in low-resource populations with a high rate of consanguinity where RRT is not readily available and many young children may die of PH1.
Treatment
Supportive measures
In the early stages of the disease, conservative measures are essential to preserve renal function and prevent nephrolithiasis. Adequate hydration and oral citrate supplements are important to increase calcium oxalate solubility and prevent crystallization [73]. High fluid intake has been shown to be an effective interventional measure in stone formers [74]. The recommended daily fluid intake in patients with PH is 2–3 l/m2 body surface area. In infants and toddlers, nasogastric tube or gastrostomy feeding around the clock is often required to ensure adequate water intake and consequently dilute urine. Special attention should be paid during situations of increased fluid losses (diarrhea, vomiting, fever) or limited fluid intake. Fluid administration IV is indicated in these circumstances to prevent calcium oxalate deposits. Administration of potassium citrate at 0.1–0.15 mg/kg body weight leads to alkalinization of the urine, which increases calcium oxalate solubility but also inhibits crystallization by forming complexes with calcium. With declining kidney function and the risk of hyperkalemia, sodium citrate may be given instead.
Pyridoxine (vitamin B6), a cofactor of AGT, was reported to be beneficial in about a third of patients with PH1, specifically those with p.Gly170Arg or Phe152Ile mutations [55]. Fargue [75] showed that pyridoxine increases net expression, catalytic activity, and peroxisomal import of the most common mistargeted mutant form of AGT (i.e., Gly170Arg in the background of the polymorphic minor allele) in Chinese hamster ovary (CHO) cell line. Partial effect of pyridoxine has also been observed for two other common AGT mutations in the minor allele background. The first prospective study of vitamin B6 treatment in patients with PH1 with preserved kidney function was recently published by Hoyer-Kuhn et al. [76]. The starting dose was 5 mg/kg body weight administered in increments of 5 mg/kg every 6 weeks up to a final dose of 20 mg/kg daily. Six of 12 patients showed >30 % reduction in urinary oxalate levels, but none achieved complete biochemical remission. Whether moderate reduction in urinary oxalate excretion has bearing on the rate of kidney disease progression remains to be proven. Higher doses of vitamin B6 should be avoided, as sensory neuropathy syndromes have been described in adults taking >200 mg/kg daily [77].
The main source of oxalate is endogenous production by the liver. Strict restriction of oxalate-containing food is therefore unnecessary, especially as intestinal oxalate absorption is reduced in patients with PH [78]. Theoretically, diet restricted in meat and gelatin, sources of hydroxyproline, may benefit patients with PH3, but this approach has not been studied systematically to date.
Extracorporeal shock-wave lithotripsy (ESWL) in patients with PH is unadvisable. ESWL demonstrated suboptimal results in children with PH, as calcium oxalate stones are difficult to fragment [79]. Moreover, repeated treatments may cause parenchymal damage and shockwave-induced renal damage has been shown to promote calcium oxalate deposition in a rat model of hyperoxaluria [80].
Oxalobacter formigenes is an anaerobic oxalate-degrading bacterium. Uncontrolled observations have reported a reduction in urinary and plasma oxalate levels in PH patients treated orally with O. formigenes [81, 82]. A randomized controlled trial of orally administered O. formigenes in PH1 patients with preserved renal function failed to show a significant reduction in urinary oxalate excretion [83].
Dialysis
Dialysis in children with PH and ESRD serves a dual purpose: clearance of excess fluids and uremic toxins, and removal of the oxalate load produced by the liver that can no longer be cleared via the kidneys. As oxalate clearance in peritoneal dialysis is inefficient, the modality of choice is intensified daily hemodialysis [84, 85]. A combination of daily nocturnal continuous-cycling peritoneal dialysis (CCPD) with hemodialysis has also been suggested. However, no dialysis strategy can offset oxalate generation by the liver [85]. Ultimately, PH1 patients on chronic dialysis will develop systemic oxalate deposition, with severe clinical consequences. Hemodialysis should therefore be limited to patients awaiting liver or combined liver and kidney transplantation (LKT) and/or those who are too small to be transplanted. Hemodialysis or hemofiltration is also used after transplantation to help remove heavy oxalate loads.
Transplantation
Historically, kidney transplantation outcome in patients with PH has been poor. The 1990 report of the European Dialysis and Transplant Registry [20] reports 3-year graft survival of 17–23 % in PH patients. Graft loss was mostly due to oxalate deposition in the transplanted kidney. LKT prevents disease recurrence in the renal allograft [86, 87], as liver transplantation with removal of the native liver corrects the metabolic defect [88]. PH patients receiving combined or sequential LKT have a better death-censored graft survival than those receiving kidney transplantation alone (KTA) [89, 90]. The 5-year survival of children with PH undergoing KTA or LKT is 14 % and 76 %, respectively [10]. These results are inferior to those reported in adults with PH who undergo kidney transplantation alone and may reflect the impact on young children of the most severe form of PH. Patients with ESRD due to PH accumulate a massive amount of oxalate in various body tissues, as described above (systemic oxalosis). Recovery of renal function hence results in immediate and significant oxaluria, even after removal of the native liver and liver transplantation. In order to prevent oxalate-induced damage to the renal allograft, Kidney Disease Improving Global Outcomes (KDIGO) guidelines [91] recommend urinary dilution with aggressive hydration and frequent dialysis for patients with delayed graft function in the immediate posttransplantation period. Harps [92] reported excellent results in nine pediatric patients who received continuous venovenous hemofiltration (CVVH) treatments immediately after combined LKT for PH1. Adequate hydration (3 l/m2/day) and anti-crystallization therapy with orally administered potassium/sodium citrate should be continued until urinary oxalate excretion returns to normal levels, a process that may take several years [93]. Orthophosphate and magnesium oxide may also help prevent calcium oxalate deposition in the transplanted kidney [91]. A recent study reported successful long-term results of KTA in four of five patients with the p.Gly170Arg mutation maintained with pharmacological doses of pyridoxine [94]. After clearing oxalate stores, urine oxalate levels were normal in most samples. These results must be interpreted cautiously because of the small number of patients and the fact that all of them were >30 years, suggesting a milder disease phenotype.
Preemptive liver transplantation has been shown to preserve renal function [95, 96]. Taking into account the fact that disease progression is highly variable, even within families, and that liver transplantation carries significant risks, it is advisable to delay the procedure until GFR declines substantially. It should be emphasized that the native liver, which is completely normal aside from the enzymatic defect, has to be removed, as opposed to auxiliary liver transplantation implemented in some patients with acute hepatic failure. Sequential transplantation with the liver first may benefit patients who do not have a suitable kidney donor. Liver transplantation, combined with intensive dialysis prior to renal transplantation, allows for a decrease in total body oxalate load. Both living and deceased donors have been used. Furthermore, sequential liver and kidney transplantation from a single living donor has been reported [97].
There is scarce data on transplantation outcome in PH2 patients [98]. Kidney transplantation alone has been performed with varying success, and graft failure due to oxalate deposition has been reported [27]. At this time, there are no reports of PH3 patients requiring RRT.
Future strategies for treatment
Several experimental therapies for PH are under investigation. Neonatal liver-cell transplantation transiently reduced plasma oxalate levels, without populating the native liver, in an infantile oxalosis patient [99]. This procedure could serve as a bridge to liver transplantation. Its effectiveness is limited due to persistent oxalate production by native hepatocytes, which is the underlying difficulty in any potential therapeutic modality for patients with PH that does not provide adequate repopulation of the native liver tissue. The main problem remains the accumulation of substrates, mainly glyoxylate—which is oxidized to oxalate—rather than enzymatic deficiency per se.
AGXT gene transfer using adenoassociated virus vector in a mouse model of PH1 reduced oxaluria to normal levels and restored AGT activity in whole liver extracts [100]. Human studies have not been performed as yet.
Some AGT mutations have been shown to cause protein homeostasis defects, such as destabilization, misfolding, and mistargeting. This suggests that kinetic stabilizers and protein homeostasis modulators may be suitable pharmacological therapies in PH1 [101, 102], similar to the effect of pyridoxine, which acts not only as a coenzyme increasing catalytic activity but also as a molecular chaperone stabilizing the mutated monomers and improving the productive folding and dimerization of the enzyme [103].
Enzyme replacement therapy, which has been successful in a number of metabolic derangements, mainly lysosomal storage diseases, would be challenging in PH, as the enzyme would have to be directed into the peroxisomes of the hepatocytes and in an efficient system that would correct the vast majority of native cells. Another potential strategy for treating patients with PH is substrate reduction therapy (SRT), which is an attempt to decrease oxalate production by inhibiting synthesis of its precursor, glyoxylate. The most appealing target for treating PH1 would be to develop specific inhibitors of HAO1, encoding GO, which would block oxidation of glycolate to glyoxylate. Preliminary results show that Hao1-deficient mice are asymptomatic and that introducing this knockout gene into PH1 mice results in a marked reduction in urinary oxalate excretion [104]. This observation concurs with the report of loss-of-function mutations in the Hao1 gene, resulting in a child with asymptomatic glycolic aciduria [105].
Conclusion
The three types of PH demonstrate a wide clinical spectrum, ranging from relatively benign nephrolithiasis to early-onset ESRD and severe systemic oxalosis. PH should be suspected in every child with kidney stone disease or nephrocalcinosis. Early diagnosis is essential for optimal treatment. Genetic testing can offer families prenatal or pregestational diagnosis. Patient advocacy groups may assist families to cope with the disease by obtaining information and joining support groups, e.g., the Oxalosis and Hyperoxaluria Foundation (OHF, http://www.ohf.org/); European Hyperoxaluria Consortium (Oxaleurope, http://www.oxaleurope.org/); and the Rare Kidney Stone Consortium (RKSC, https://rarediseasesnetwork.epi.usf.edu/RKSC/). Hopefully, future advances will provide better therapeutic modalities for children with PH.
Summary points
-
1.
PH is characterized by hepatic oxalate overproduction, leading to renal deposition of calcium oxalate crystals.
-
2.
Three distinct PH entities have been described: mutations in AGXT cause PH1, the most severe form, often leading to ESRD. GRHPR and HOGA1 mutations cause PH2 and PH3, respectively. PH2 is rare and clinically similar to PH1. PH3 has a milder phenotype and may be underdiagnosed.
-
3.
PH treatment is challenging. Patients with ESRD need frequent intense hemodialysis to prevent systemic oxalosis.
-
4.
Liver transplantation prevents recurrence in transplanted kidney.
-
5.
Genetic diagnosis and counseling can ensure the birth of unaffected children.
Multiple-choice questions (answers are provided following the reference list)
-
1.
Which of the following is an indication for metabolic screening of lithogenic risk factors, including hyperoxaluria?
-
a.
A single renal stone in a 3-year-old boy
-
b.
Four episodes of renal colic in a 25-year-old woman
-
c.
Renal failure with severely hyperechoic kidneys in a 4-month-old baby
-
d.
All of the above
-
a.
-
2.
What is the recommended approach to treating early-stage hyperoxaluria?
-
a.
Sodium restriction and urine alkalinizetion
-
b.
Extracorporal shockwave lithotripsy
-
c.
Hyperhydration and citrate supplements
-
d.
Restriction of oxalate-rich food
-
a.
-
3.
A 7 year-old boy is referred after having passed two kidney stones. Two older sisters suffered a few episodes of renal colic at a young age, but repeated US scans are negative for stones or nephrocalcinosis. All three siblings have elevated urinary oxalate levels. Family history of ESRD is negative. What is the most likely diagnosis?
-
a.
PH1
-
b.
PH2
-
c.
PH3
-
d.
Secondary hyperoxaluria
-
a.
-
4.
Which of the following is not recommended for treating a child with advanced renal failure due to PH?
-
a.
Early start of peritoneal dialysis
-
b.
Preemptive liver transplantation
-
c.
Combined liver and kidney transplantation
-
d.
Daily hemodialysis
-
a.
References
Lepoutre C (1925) Calculs multiples chez un enfent: Infiltration du parenchyme renal par des depots crystallins. J Urol 20:424
Archer HE, Dormer AE, Scowen EF, Watts RW (1957) Primary hyperoxaluria. Lancet 273:320–322
Mulay SR, Kulkarni OP, Rupanagudi KV, Migliorini A, Darisipudi MN, Vilaysane A, Muruve D, Shi Y, Munro F, Liapis H, Anders HJ (2013) Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest 123:236–246
Schepers MS, Van Ballegooijen ES, Bangma CH, Verkoelen CF (2005) Crystals cause acute necrotic cell death in renal proximal tubule cells, but not in collecting tubule cells. Kidney Int 68:1543–1553
Schepers MS, Van Ballegooijen ES, Bangma CH, Verkoelen CF (2005) Oxalate is toxic to renal tubular cells only at supraphysiologic concentrations. Kidney Int 68:1660–1669
Van Woerden CS, Groothoff JW, Wanders RJ, Davin JC, Wijburg FA (2003) Primary hyperoxaluria type 1 in The Netherlands: prevalence and outcome. Nephrol Dial Transplant 18:273–279
Lieske JC, Monico CG, Holmes WS, Bergstralh EJ, Slezak JM, Rohlinger AL, Olson JB, Milliner DS (2005) International registry for primary hyperoxaluria. Am J Nephrol 25:290–296
Van der Hoeven SM, van Woerden CS, Groothoff JW (2012) Primary hyperoxaluria type 1, a too often missed diagnosis and potentially treatable cause of end-stage renal disease in adults: results of the Dutch cohort. Nephrol Dial Transplant 27:3855–3862
Kopp N, Leumann E (1995) Changing pattern of primary hyperoxaluria in Switzerland. Nephrol Dial Transplant 10:2224–2227
Cochat P, Deloraine A, Rotily M, Olive F, Liponski I, Deries N (1995) Epidemiology of primary hyperoxaluria type 1. Nephrol Dial Transplant 10:3–7
Harambat J, Van Stralen KJ, Espinosa L, Groothoff JW, Hulton SA, Cerkauskiene R, Schaefer F, Verrina E, Jager KJ, Cochat P, (ESPN/ERA-EDTA) (2012) Characteristics and outcomes of children with primary oxalosis requiring renal replacement therapy. Clin J Am Soc Nephrol 7:458–465
Fisher D, Hiller N, Drukker A (1995) Oxalosis of bone: report of four cases and a new radiological staging. Pediatr Radiol 25:293–295
Guerra EN, Vianna L, Sobreira MN, de Araújo FN, de Melo NSJ (2012) Oral manifestations of hyperoxaluria. Craniofac Surg 22:2191–2192
Frishberg Y, Feinstein S, Rinat C, Drukker A (2000) Hypothyroidism in primary hyperoxaluria type 1. J Pediatr 136:255–257
Jorge P, García González MJ, Rebollo SG, García S, Bosa F, Laynez I, De la Rosa A (2013) Myocardial infiltration by oxalate: a rare case of cardiomyopathy by accumulation of oxalate in a 53-year-old woman. J Am Coll Cardiol 62:e525
Chou LY, Donohue WL (1952) Oxalosis; possible inborn error of metabolism with nephrolithiasis and nephrocalcinosis due to calcium oxalate as the predominating features. Pediatrics 10:660–666
Brancaccio D, Poggi A, Ciccarelli C, Bellini F, Galmozzi C, Poletti I, Maggiore Q (1981) Bone changes in end-stage oxalosis. Am J Roentgenol 136:935–939
Meredith TA, Wright JD, Gammon JA, Fellner SK, Warshaw BL, Maio M (1984) Ocular involvement in primary hyperoxaluria. Arch Ophthalmol 102:584–587
Rao NM, Yallapragada A, Winden KD, Saver J, Liebeskind DS (2013) Stroke in primary hyperoxaluria type I. J Neuroimaging 24:411–413
Broyer M, Brunner FP, Brynger H, Dykes SR, Ehrich JH, Fassbinder W, Geerlings W, Rizzoni G, Selwood NH, Tufveson G (1990) Kidney transplantation in primary oxalosis: data from the EDTA Registry. Nephrol Dial Transplant 5:332–336
Godwin JT, Fowler MF, Dempsey EF, Henneman PH (1958) Primary hyperoxaluria and oxalosis; report of a case and review of the literature. N Engl J Med 259:1099–1103
Hall EG, Scowen EF, Watts RW (1960) Clinical manifestations of primary hyperoxaluria. Arch Dis Child 35:108–112
Frishberg Y, Rinat C, Shalata A, Khatib I, Feinstein S, Becker-Cohen R, Weismann I, Wanders RJ, Rumsby G, Roels F, Mandel H (2005) Intra-familial clinical heterogeneity: absence of genotype-phenotype correlation in primary hyperoxaluria type 1 in Israel. Am J Nephrol 25:269–275
Takayama T, Nagata M, Ichiyama A, Ozono S (2005) Primary hyperoxaluria type 1 in Japan. Am J Nephrol 25:297–302
Williams HE, Smith LH Jr (1968) L-glyceric aciduria. A new genetic variant of primary hyperoxaluria. N Engl J Med 278:233–238
Chlebeck PT, Milliner DS, Smith LH (1994) Long-term prognosis in primary hyperoxaluria type II (L-glyceric aciduria). Am J Kidney Dis 23:255–259
Hicks NR, Cranston DW, Charlton CA (1983) Fifteen-year follow-up of hyperoxaluria type II. N Engl J Med 309:796
Marangella M, Petrarulo M, Cosseddu D (1994) End-stage renal failure in primary hyperoxaluria type 2. N Engl J Med 330:1690
Naderi G, Latif A, Tabassomi F, Esfahani ST (2014) Failure of isolated kidney transplantation in a pediatric patient with primary hyperoxaluria type 2. Pediatr Transplant 18:E69–73
Belostotsky R, Seboun E, Idelson GH, Milliner DS, Becker-Cohen R, Rinat C, Monico CG, Feinstein S, Ben-Shalom E, Magen D, Weissman I, Charon C, Frishberg Y (2010) Mutations in DHDPSL are responsible for primary hyperoxaluria type III. Am J Hum Genet 87:392–399
Beck BB, Baasner A, Buescher A, Habbig S, Reintjes N, Kemper MJ, Sikora P, Mache C, Pohl M, Stahl M, Toenshoff B, Pape L, Fehrenbach H, Jacob DE, Grohe B, Wolf MT, Nürnberg G, Yigit G, Salido EC, Hoppe B (2013) Novel findings in patients with primary hyperoxaluria type III and implications for advanced molecular testing strategies. Eur J Hum Genet 21:162–172
Belostotsky R, Pitt JJ, Frishberg Y (2012) Primary hyperoxaluria type III-a model for studying perturbations in glyoxylate metabolism. J Mol Med 90:1497–1504
Shih VE (2003) Amino acid analysis. In: Blau N, Duran M, Blaskovics ME, Gibson KM (eds) Physician’s guide to the laboratory diagnosis of metabolic diseases, 2nd edn. Springer, Berlin, pp 11–26
Pitt JJ, Willis F, Tzanakos N, Belostotsky R, Frishberg Y (2014) 4-hydroxyglutamate is a biomarker for primary hyperoxaluria type 3. JIMD Rep. doi:10.1007/8904_2013_291
Hoppe B, Latta K, von Schnakenburg C, Kemper MJ (2005) Primary hyperoxaluria-the German experience. Am J Nephrol 25:276–281
Cochat P, Exanthus J, Basmaison O (1999) Fifth workshop on primary hyperoxaluria. Nephrol Dial Transplant 14:2784–2789
Al-Eisa AA, Samhan M, Naseef M (2004) End-stage renal disease in Kuwaiti children: an 8-year experience. Transplant Proc 36:1788–1791
Kamoun A, Lakhoua R (1996) End-stage renal disease of the Tunisian child: epidemiology, etiologies, and outcome. Pediatr Nephrol 10:479–482
Hattori S, Yosioka K, Honda M, Ito H (2002) The 1998 report of the Japanese national registry data on pediatric end-stage renal disease patients. Pediatr Nephrol 17:456–461
North American Pediatric Renal Trials and Collaborative Studies (2011) NAPRTCS 2011 Annual Dialysis Report. https://web.emmes.com/study/ped/annlrept/annualrept2011.pdf
Abumwais JQ (2012) Etiology of chronic renal failure in Jenin district, Palestine. Saudi J Kidney Dis Transpl 23:158–160
Milliner DS, Wilson DM, Smith LH (2001) Phenotypic expression of primary hyperoxaluria: comparative features of types I and II. Kidney Int 59:31–36
Johnson SA, Rumsby G, Cregeen D, Hulton SA (2002) Primary hyperoxaluria type 2 in children. Pediatr Nephrol 17:597–601
Takayama T, Nagata M, Ozono S, Nonomura K, Cramer SD (2007) A novel mutation in the GRHPR gene in a Japanese patient with primary hyperoxaluria type 2. Nephrol Dial Transplant 22:2371–2374
Rare Kidney Stone Consortium [Online] (2013) Rare kidney stone consortium registry. [Cited: 05 11, 2014.] http://www.rarekidneystones.org/hyperoxaluria/physicians.html
Hopp K, Cogal AG, Hakonarson H, Milliner DS, Harris PC (2013) Estimated incidence of primary hyperoxaluria using population allele frequencies of disease variants [Abstract]. J Am Soc Nephrol 24:529A
Purdue PE, Lumb MJ, Fox M, Griffo G, Hamon-Benais C, Povev S, Danpure CJ (1991) Characterization and chromosomal mapping of a genomic clone encoding human alanine:glyoxylate aminotransferase. Genomics 10:34–42
Rumsby, G(2013) Alanine:glyoxylate aminotransferase (agxt) gene (primary hyperoxaluria type 1) mutation database [Online]. [Cited: April 22, 2014.] http://www.uclh.nhs.uk/OurServices/ServiceA-Z/PATH/PATHBIOMED/CBIO/Documents/AGXT%20mutation%20database.pdf
Williams E, Acquaviva C, Amoroso A, Chevalier F, Coulter-Mackie M, Monico CG, Giachino D, Owen T, Robbiano A, Salido E, Waterham H, Rumsby G (2009) Primary hyperoxaluria type 1: update and additional mutation analysis of the agxt gene. Hum Mutat 30:910–917
Lumb MJ, Danpure CJ (2000) Functional synergism between the most common polymorphism in human alanine:glyoxylate aminotransferase and four of the most common disease-causing mutations. J Biol Chem 275:36415–36422
Purdue PE, Takada Y, Danpure CJ (1990) Identification of mutations associated with peroxisome-to-mitochondrion mistargeting of alanine/glyoxylate aminotransferase in primary hyperoxaluria type 1. J Cell Biol 111:2341–2351
Purdue PE, Allsop J, Isaya G, Rosenberg LE, Danpure CJ (1991) Mistargeting of peroxisomal L-alanine:glyoxylate aminotransferase to mitochondria in primary hyperoxaluria patients depends upon activation of a cryptic mitochondrial targeting sequence by a point mutation. Proc Natl Acad Sci U S A 88:10900–10904
Rinat C, Wanders RJ, Drukker A, Halle D, Frishberg Y (1999) Primary hyperoxaluria type I: a model for multiple mutations in a monogenic disease within a distinct ethnic group. J Am Soc Nephrol 10:2352–2358
Lorenzo V, Alvarez A, Torres A, Torregrosa V, Hernández D, Salido E (2006) Presentation and role of transplantation in adult patients with type 1 primary hyperoxaluria and the I244T AGXT mutation: single-center experience. Kidney Int 70:1115–1119
Monico CG, Rossetti S, Olson JB, Milliner DS (2005) Pyridoxine effect in type I primary hyperoxaluria is associated with the most common mutant allele. Kidney Int 67:1704–1709
van Woerden CS, Groothoff JW, Wijburg FA, Annink C, Wanders RJ, Waterham HR (2004) Clinical implications of mutation analysis in primary hyperoxaluria type 1. Kidney Int 66:746–752
Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP (1999) The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Hum Mol Genet 8:2063–2069
Rumsby G, Cregeen DP (1999) Identification and expression of a cDNA for human hydroxypyruvate/glyoxylate reductase. Biochim Biophys Acta 1446:383–388
Cregeen DP, Williams EL, Hulton S, Rumsby G (2003) Molecular analysis of the glyoxylate reductase (GRHPR) gene and description of mutations underlying primary hyperoxaluria type 2. Hum Mutat 22:947
Rumsby, G (2012) Glyoxylatereductase/hydroxypyruvate reductase (GRHPR) gene (Primary hyperoxaluria type 2). [Online] [Cited: April 22, 2014.] http://www.uclh.nhs.uk/OurServices/ServiceA-Z/PATH/PATHBIOMED/CBIO/Documents/GRHPR%20mutation%20database.pdf
Webster KE, Ferree PM, Holmes RP, Cramer SD (2000) Identification of missense, nonsense, and deletion mutations in the GRHPR gene in patients with primary hyperoxaluria type II (PH2). Hum Genet 107:176–185
Monico CG, Rossetti S, Belostotsky R, Cogal AG, Herges RM, Seide BM, Olson JB, Bergstrahl EJ, Williams HJ, Haley WE, Frishberg Y, Milliner DS (2011) Primary hyperoxaluria type III gene HOGA1 (formerly DHDPSL) as a possible risk factor for idiopathic calcium oxalate urolithiasis. Clin J Am Soc Nephrol 6:2289–2295
Abratt VR, Reid SJ (2010) Oxalate-degrading bacteria of the human gut as probiotics in the management of kidney stone disease. Adv Appl Microbiol 72:63–87
Birdsey GM, Lewin B, Holbrook JD, Simpson VR, Cunningham AA, Danpure CJ (2005) A comparative analysis of the evolutionary relationship between diet and enzyme targeting in bats, marsupials and other mammals. Proc Biol Sci 272:833–840
Danpure CJ, Jennings PR (1986) Peroxisomal alanine:glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett 201:20–24
Danpure CJ, Cooper PJ, Wise PJ, Jennings PR (1989) An enzyme trafficking defect in two patients with primary hyperoxaluria type 1: peroxisomal alanine/glyoxylate aminotransferase rerouted to mitochondria. J Cell Biol 108:1345–1352
Williams EL, Bockenhauer D, van't Hoff WG, Johri N, Laing C, Sinha MD, Unwin R, Viljoen A, Rumsby G (2012) The enzyme 4-hydroxy-2-oxoglutarate aldolase is deficient in primary hyperoxaluria type 3. Nephrol Dial Transplant 27:3191–3195
Riedel TJ, Knight J, Murray MS, Milliner DS, Holmes RP, Lowther WT (2012) 4-Hydroxy-2-oxoglutarate aldolase inactivity in primary hyperoxaluria type 3 and glyoxylate reductase inhibition. Biochim Biophys Acta 1822:1544–1552
Cochat P, Hulton SA, Acquaviva C, Danpure CJ, Daudon M, De Marchi M, Fargue S, Groothoff J, Harambat J, Hoppe B, Jamieson NV, Kemper MJ, Mandrile G, Marangella M, Picca S, Rumsby G, Salido E, Straub M, van Woerden CS (2012) Primary hyperoxaluria type 1: indications for screening and guidance for diagnosis and treatment. Nephrol Dial Transplant 27:1729–1736
Milliner DS (2005) The primary hyperoxalurias: an algorithm for diagnosis. Am J Nephrol 25:154–160
Daudon M, Jungers P, Bazin D (2008) Peculiar morphology of stones in primary hyperoxaluria. N Engl J Med 359:100–102
Jacob DE, Grohe B, Geßner M, Beck BB, Hoppe B (2013) Kidney stones in primary hyperoxaluria: new lessons learnt. PLoS One 8:e70617
Leumann E, Hoppe B, Neuhaus T, Blau N (1995) Efficacy of oral citrate administration in primary hyperoxaluria. Nephrol Dial Transplant 10(Suppl 8):14–16
Borghi L, Meschi T, Amato F, Briganti A, Novarini A, Giannini A (1996) Urinary volume, water and recurrences in idiopathic calcium nephrolithiasis: a 5-year randomized prospective study. J Urol 155:839–843
Fargue S, Rumsby G, Danpure CJ (2013) Multiple mechanisms of action of pyridoxine in primary hyperoxaluria type 1. Biochim Biophys Acta 1832:1776–1783
Hoyer-Kuhn H, Kohbrok S, Volland R, Franklin J, Hero B, Beck BB, Hoppe B (2014) Vitamin B6 in primary hyperoxaluria I: first prospective trial after 40 years of practice. Clin J Am Soc Nephrol 9:468–477
Schaumburg H, Kaplan J, Windebank A, Vick N, Rasmus S, Pleasure D, Brown MJ (1983) Sensory neuropathy from pyridoxine abuse. A new megavitamin syndrome. N Engl J Med 309:445–448
Sikora P, von Unruh GE, Beck B, Feldkötter M, Zajaczkowska M, Hesse A, Hoppe B (2008) [13C2]oxalate absorption in children with idiopathic calcium oxalate urolithiasis or primary hyperoxaluria. Kidney Int 73:1181–1186
Al-Abadi E, Hulton SA (2013) Extracorporal shock wave lithotripsy in the management of stones in children with oxalosis-still the first choice? Pediatr Nephrol 28:1085–1089
Xue YQ, He DL, Chen XF, Li X, Zeng J, Wang XY (2009) Shock wave induced kidney injury promotes calcium oxalate deposition. J Urol 182:762–765
Hoppe B, Beck B, Gatter N, von Unruh G, Tischer A, Hesse A, Laube N, Kaul P, Sidhu H (2006) Oxalobacter formigenes: a potential tool for the treatment of primary hyperoxaluria type 1. Kidney Int 70:1305–1311
Hoppe B, Dittlich K, Fehrenbach H, Plum G, Beck BB (2011) Reduction of plasma oxalate levels by oral application of Oxalobacter formigenes in 2 patients with infantile oxalosis. Am J Kidney Dis 58:453–455
Hoppe B, Groothoff JW, Hulton SA, Cochat P, Niaudet P, Kemper MJ, Deschênes G, Unwin R, Milliner D (2011) Efficacy and safety of Oxalobacter formigenes to reduce urinary oxalate in primary hyperoxaluria. Nephrol Dial Transplant 26:3609–3615
Hoppe B, Graf D, Offner G, Latta K, Byrd DJ, Michalk D, Brodehl J (1996) Oxalate elimination via hemodialysis or peritoneal dialysis in children with chronic renal failure. Pediatr Nephrol 10:488–492
Illies F, Bonzel KE, Wingen AM, Latta K, Hoyer PF (2006) Clearance and removal of oxalate in children on intensified dialysis for primary hyperoxaluria type 1. Kidney Int 70:1642–1648
Millan MT, Berquist WE, So SK, Sarwal MM, Wayman KI, Cox KL, Filler G, Salvatierra O Jr, Esquivel CO (2003) One hundred percent patient and kidney allograft survival with simultaneous liver and kidney transplantation in infants with primary hyperoxaluria: a single-center experience. Transplantation 76:1458–1463
Jamieson NV, European PHI Transplantation Study Group (2005) A 20-year experience of combined liver/kidney transplantation for primary hyperoxaluria (PH1): the European PH1 transplant registry experience 1984–2004. Am J Nephrol 39:282–289
Watts RW, Calne RY, Williams R, Mansell MA, Veall N, Purkiss P, Rolles K (1985) Primary hyperoxaluria (type I): attempted treatment by combined hepatic and renal transplantation. Q J Med 57:697–703
Cibrik DM, Kaplan B, Arndorfer JA, Meier-Kriesche HU (2002) Renal allograft survival in patients with oxalosis. Transplantation 74:707–710
Bergstralh EJ, Monico CG, Lieske JC, Herges RM, Langman CB, Hoppe B, Milliner DS (2010) Transplantation outcomes in primary hyperoxaluria. Am J Transplant 10:2493–2501
Kasiske BL, Zeier MG, Craig JC, Ekberg H, Garvey CA, Green MD, Jha V, Josephson MA, Kiberd BA, Kreis HA, McDonald RA, Newmann JM, Obrador GT, Chapman JR, Vincenti FG, Balk EM, Wagner M, Raman G, Earley A, Abariga S, Kidney Disease: Improving Global Outcomes (KDIGO) Transplant Work Group (2009) KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant 9(Suppl 3):S1–155
Harps E, Brinkert F, Ganschow R, Briem-Richter A, van Husen M, Schmidtke S, Herden U, Nashan B, Fischer L, Kemper MJ (2011) Immediate postoperative intensive care treatment of pediatric combined liver-kidney transplantation: outcome and prognostic factors. Transplantation 91:1127–1131
Ruder H, Otto G, Schutgens RB, Querfeld U, Wanders RJ, Herzog KH, Wölfel P, Pomer S, Schärer K, Rose GA (1990) Excessive urinary oxalate excretion after combined renal and hepatic transplantation for correction of hyperoxaluria type 1. Eur J Pediatr 150:56–58
Lorenz EC, Lieske JC, Seide BM, Meek AM, Olson JB, Bergstralh EJ, Milliner DS (2014) Sustained pyridoxine response in primary hyperoxaluria type 1 recipients of kidney alone transplant. Am J Transplant 14:1433–1438
Nolkemper D, Kemper MJ, Burdelski M, Vaismann I, Rogiers X, Broelsch CE, Ganschow R, Müller-Wiefel DE (2000) Long-term results of pre-emptive liver transplantation in primary hyperoxaluria type 1. Pediatr Transplant 4:177–181
Shapiro R, Weismann I, Mandel H, Eisenstein B, Ben-Ari Z, Bar-Nathan N, Zehavi I, Dinari G, Mor E (2001) Primary hyperoxaluria type 1: improved outcome with timely liver transplantation: a single-center report of 36 children. Transplantation 72:428–432
Mor E, Nesher E, Ben-Ari Z, Weissman I, Shaharabani E, Eizner S, Solomonov E, Rahamimov R, Braun M (2013) Sequential liver and kidney transplantation from a single living donor in two young adults with primary hyperoxaluria type 1. Liver Transpl 19:646–648
Lam CW, Yuen YP, Lai CK, Tong SF, Lau LK, Tong KL, Chan YW (2001) Novel mutation in the GRHPR gene in a Chinese patient with primary hyperoxaluria type 2 requiring renal transplantation from a living related donor. Am J Kidney Dis 38:1307–1310
Beck BB, Habbig S, Dittrich K, Stippel D, Kaul I, Koerber F, Goebel H, Salido EC, Kemper M, Meyburg J, Hoppe B (2012) Liver cell transplantation in severe infantile oxalosis-a potential bridging procedure to orthotopic liver transplantation? Nephrol Dial Transplant 27:2984–2989
Salido E, Rodriguez-Pena M, Santana A, Beattie SG, Petry H, Torres A (2011) Phenotypic correction of a mouse model for primary hyperoxaluria with adeno-associated virus gene transfer. Mol Ther 19:870–875
Santana A, Salido E, Torres A, Shapiro LJ (2003) Primary hyperoxaluria type 1 in the Canary islands: a conformational disease due to I244T mutation in the P11L-containing alanine:glyoxylate aminotransferase. Proc Natl Acad Sci 100:7277–7282
Mesa-Torres N, Fabelo-Rosa I, Riverol D, Yunta C, Albert A, Salido E, Pey AL (2013) The role of protein denaturation energetics and molecular chaperones in the aggregation and mistargeting of mutants causing primary hyperoxaluria type I. PLoS One 8:e71963
Cellini B, Montioli R, Oppici E, Astegno A, Voltattorni CB (2014) The chaperone role of the pyridoxal 5′-phosphate and its implications for rare diseases involving B6-dependent enzymes. Clin Biochem 47:158–165
Salido E, Pey AL, Rodriguez R, Lorenzo V (2012) Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta 1822:1453–1464
Frishberg Y, Zeharia A, Lyakhovetsky R, Bargal R, Belostotsky R (2014) Mutations in HAO1 encoding glycolate oxidase cause isolated glycolic aciduria. J Med Genet 51:526–529
Acknowledgments
The authors are indebted to Dr. Ruth Belostotsky for designing Fig. 2.
Author information
Authors and Affiliations
Corresponding author
Additional information
Answers
1. d
2. c
3. c
4. a
Rights and permissions
About this article

Cite this article
Ben-Shalom, E., Frishberg, Y. Primary hyperoxalurias: diagnosis and treatment. Pediatr Nephrol 30, 1781–1791 (2015). https://doi.org/10.1007/s00467-014-3030-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-014-3030-1