
Abstract
Maltooligosyl trehalose trehalohydrolase (MTHase, EC 3.2.1.141) catalyzes the release of trehalose, a novel food ingredient, by splitting the α-1,4-glucosidic linkage adjacent to the α-1,1-glucosidic linkage of maltooligosyl trehalose. However, the high-yield preparation of recombinant MTHase has not yet been reported. In this study, a codon-optimized synthetic gene encoding Sulfolobus acidocaldarius MTHase was expressed in Escherichia coli. In initial expression experiments conducted using pET-24a (+) and E. coli BL21 (DE3), the MTHase activity was 10.4 U/mL and a large amount of the expression product formed inclusion bodies. The familiar strategies, including addition of additives, co-expression with molecular chaperones, and expression with a fusion partner, failed to enhance soluble MTHase expression. Considering the intermolecular disulfide bond of MTHase, expression was investigated using a system comprising plasmid pET-32a (+) and host E. coli Origami (DE3), which is conducive to cytoplasmic disulfide bond formation. The MTHase activity increased to 55.0 U/mL, a 5.3-fold increase. Optimization of the induction conditions in a 3-L fermentor showed that when the lactose was fed at 0.2 g/L/h beginning at an OD600 of 40 and the induction temperature was maintained at 30 °C, the MTHase activity reached a maximum of 204.6 U/mL. This is the first report describing a systematic effort to obtain high-efficiency MTHase production. The high yield obtained using this process provides the basis for the industrial-scale production of trehalose. This report is also expected to be valuable in the production of other enzymes containing disulfide bonds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maltooligosyl trehalose trehalohydrolase (MTHase, EC 3.2.1.141) catalyzes the conversion of maltooligosyl trehalose into trehalose and a maltooligosaccharide with lower molar mass by splitting the α-1,4-glucosidic linkage adjacent to the α-1,1-glucosidic linkage. It is among the key enzymes involved in the production of trehalose from starch [1]. Trehalose is a non-reducing disaccharide composed of two glucose molecules linked by an α-1,1-glucosidic bond. Because it protects proteins and lipid membranes from desiccation, heat, frost, and osmotic changes, trehalose is used as a preservative or stabilizer for cells, medicines, food, and cosmetics. Thus, trehalose is used in a variety of applications in many different industries [2,3,4,5]. Because the number of industrial applications of trehalose is gradually increasing, highly efficient, low-cost production of the enzymes used in trehalose production is attracting more attention.
MTHases have been reported in a variety of microorganisms, including Sulfolobus acidocaldarius, Sulfolobus solfataricus, Sulfolobus shibatae, Arthrobacter sp., Arthrobacter ramosus, Rhizobium sp., and others [2, 6,7,8]. Some researchers developed MTHase production systems based upon the isolated wild-type microorganisms [9, 10]. However, the low activities obtained with these systems generally restricted their use for commercial MTHase preparation. Higher MTHase yield would be more easily obtained through genetically engineered microorganisms constructed using recombinant DNA technology [11, 12].
Escherichia coli has been the most commonly used host for MTHase expression, but only a few researchers reported the MTHase yield; no systematic strategies for enhancing MTHase production have been explored [7, 13, 14]. Donatella de Pascale’s group expressed the MTHase from Sulfolobus solfataricus MT4 in E. coli Rb791 using a pTrc expression vector. The yield was 3630 U/L of medium [13]. Min Chang introduced the gene encoding MTHase from Corynebacterium glutamicum into E. coli BL21 (DE3) pLysS. In this system, soluble recombinant MTHase accounted for about 40% of total cell protein, which was 0.27 mg/mL (five times concentrated). The remainder of the recombinant protein was expressed as inclusion bodies [15].
Many proteins, especially cytoplasmic proteins, readily form insoluble aggregates when expressed as recombinant proteins using E. coli as the host. Several techniques have been developed to resolve this problem, including reducing the protein synthesis rate, adding various additives to the culture medium, optimizing the host and plasmid, co-expressing the protein with molecular chaperones, and expressing the protein with fusion partners [16]. For example, adding ethanol to the culture media induces the expression of chaperones and enhances the solubility of recombinant proteins [17, 18]. Some carbohydrates also enhance the expression of recombinant proteins by inducing osmotic stress responses [19, 20]. Betaine supplementation favors the native folding of recombinant proteins [21]. Fusion partners commonly used to solve solubility and folding issues include small ubiquitin-like modifier (SUMO), thioredoxin A (TrxA) and glutathione S-transferase [22,23,24,25].
The MTHase from S. acidocaldarius, which has excellent thermostable and acidophilic properties, displays its highest activity at pH 5.5–6.0 and 75 °C [10]. This MTHase can catalyze the production of trehalose from starch at high yield when coupled with the maltooligosyl trehalose synthase from the same strain. The high reaction temperature, which accelerates the rate of the transformation process and reduces the risk of bacterial contamination, is beneficial for its use in industrial applications. However, the high-yield preparation of recombinant MTHase has not yet been reported. In this study, the gene encoding S. acidocaldarius MTHase was cloned and expressed in E. coli. Different strategies were investigated to identify a system that produced MTHase in good yield. Then fermentation conditions were optimized to enhance the yield of this MTHase (Fig. 1).

The experimental scheme of MTHase expression
Materials and methods
Bacterial strains, vectors and materials
Escherichia coli strain JM109 and the pMD™18-T Vector Cloning Kit (Takara, Dalian, China) were used for gene cloning. E. coli strains BL21 (DE3), Origami (DE3) and the plasmids pET-24a (+), pET-32a (+) were used for protein expression. The plasmid pG-Tf2, the enzymes used for DNA manipulations, and agarose were purchased from Takara (Dalian, China). Plasmid mini preparation kit agarose, and gel DNA purification kit were purchased from TIANGEN Biotech Co., Ltd (Beijing, China). Primer synthesis was performed by Shanghai Generay Biotech Co., Ltd (Shanghai, China). DNA sequencing was performed by Shanghai Sangon Biological Engineering Technology and Services Co., Ltd (Shanghai, China). Other analytically pure reagents were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
Construction of engineered E. coli for MTHase expression
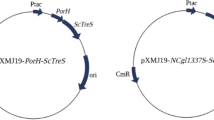
The gene treZ encoding the S. acidocaldarius MTHase (NCBI accession number WP_011278268) was analyzed, optimized for expression in E. coli, and synthesized by Shanghai Generay Biotech Co., Ltd (Fig. S1). Rarely used codons were replaced with codons preferred by E. coli while preserving the amino acid sequence, and the mRNA free energy and GC content of the gene were also optimized for protein expression. The synthetic gene, which was flanked by NdeI and HindIII sites (up- and downstream, respectively), was inserted into the plasmid pET-24a (+) to generate pET24a–treZ (Fig. S2). The genes encoding MTHase and the SUMO family protein SMT3 (NCBI accession number NP_010798.1) were amplified using pET24a–treZ and pET24a-sumo (laboratory stock) as template, respectively, then overlapping PCR was utilized to create a gene encoding an N-terminal fusion of sumo to treZ. The following primers were used to construct this gene. CCATATGTCGGACTCAGAAGTCAATCAAGAAGC, TTGCCGCCAAAGCTAAACATATACGTAGCACCACCAATC, GATTGGTGGTGCTACGTATATGTTTAGCTTTGGCGGCAA, and AAGCTTATTCCAGTTGATACACGC, where the underlined bases represented the NdeI and HindIII restriction enzyme sites. After ligation into the pMD18-T simple vector and verification of the resulting plasmid with DNA sequencing, sumo–treZ was liberated by NdeI and HindIII digestion and ligated into similarly digested pET-24a (+) to form plasmid pET24a–sumo–treZ (Fig. S3). Finally, the gene encoding MTHase, amplified from pET24a–treZ using primers CGGAATTCATGTTTAGCTTTGGCGGCAA and CCAAGCTTATTCCAGTTGATACACGC (EcoR I and HindIII restriction enzyme sites underlined), was ligated into pET-32a (+) using the approach described above, to form the expression plasmid pET32a–treZ. Chemically competent E. coli were transformed with pET24a–treZ, pET24a–sumo–treZ, or pET32a–treZ to produce candidate MTHase expression systems.
Cultivation conditions for engineered E. coli
Shake-flask culture
A 10-µL sample of frozen glycerol stock was used to inoculate 10 mL of Luria–Bertani medium (g/L: yeast extract, 5.0; tryptone, 10.0; NaCl, 10.0). After cultivation at 37 °C in a rotary shaker (200 rpm) for 8 h, the seed culture (4% [v/v]) was used to inoculate 40 mL of Terrific Broth medium (g/L: yeast extract, 24.0; tryptone, 12.0; KH2PO4, 2.3; and K2HPO4, 16.4; glycerol, 5.0; and ampicillin 0.1). The resulting culture was shaken at 37 °C in a rotary shaker (200 rpm) until the optical density at 600 nm (OD600) reached 1.5; then, isopropyl β-d-thiogalactopyranoside (IPTG) was added to the culture at a final concentration of 0.1 mM to induce MTHase expression. At this point, the incubation temperature was reduced to 25 °C.
3-L fermentor
Escherichia coli Origami (DE3) cells harboring pET32a–treZ were cultivated using a fed-batch pattern in a 3.6-L fermentor (Labfors 5; Infors-HT Co., Ltd). The seed culture, prepared as described above, was used to inoculate a 1-L initial batch of semisynthetic medium (g/L: tryptone, 30.0; yeast extract, 20.0; K2HPO4, 14.6; MgSO4·7H2O, 2.0; (NH4)2-H-citrate, 1.0; glycerol, 8.0; ampicillin 0.1; and trace metal solution, 1.0 mL/L) at 37 °C. The trace metal solution contained (g/L) FeSO4·7H2O 10.0, ZnSO4·7H2O 5.3, CaCl2 2.0, CuSO4·5H2O 3.0, MnSO4·4H2O 0.5, Na2B4O7·10H2O 0.2, (NH4)6Mo7O24 0.1. During the entire fermentation process, the pH was maintained at 7.0 by automatic addition of 25% ammonia solution. The dissolved oxygen was maintained at about 30% of air saturation under a cascaded control of agitation and aeration rates, and the use of oxygen-enriched air. When the dissolved oxygen content and pH of the resulting culture suddenly spiked, which indicated the initial glycerol was completely consumed, feeding solution (g/L: tryptone, 50.0; yeast extract, 50.0; MgSO4·7H2O, 3.4; and glycerol, 500.0) was continuously added to the culture. When the OD600 of the culture reached a specified level (30, 40, or 50), lactose was fed at a constant rate (0.1, 0.2, 0.4, or 0.8 g/L/h) to induce MTHase expression. The temperature was kept at 37 °C during the growth phase, and at a specified value (25 °C, 30 °C, and 35 °C) during the induction phase.
Determination of biomass
A specified amount of the culture broth was collected, and the optical density at 600 nm (OD600) was measured using a spectrophotometer. If the OD600 value was higher than 0.8, the sample was diluted with 0.9% (w/v) sodium chloride solution. The dry cell weight (DCW) was measured as follows. Culture broth (10 mL) was collected and centrifuged at 13,200×g for 10 min. The supernatant was discarded, and the pellet was washed twice with 0.9% (w/v) sodium chloride solution. Finally, the washed pellet was dried to constant weight at 105 °C in a drying oven.
MTHase activity assay
Maltopentaose was dissolved to a concentration of 1% in 20 mM phosphate–citrate buffer (pH 6.0). S. acidocaldarius maltooligosyltrehalose synthase (MTSase) solution (10 µL), prepared in our previous study [26], was added to 0.48 mL of maltopentaose solution to produce maltotriosyl trehalose. After 2-h incubation at 60 °C, the reaction was stopped by heating for 10 min in boiling water. The product solution was incubated at 60 °C, 10 µL of appropriately diluted MTHase was added, and the mixture was allowed to react for exactly 10 min. Then reaction was terminated by heating for 10 min in boiling water. The amount of maltotriose released was measured using the 3,5-dinitrosalicylic acid reagent, as previously reported [27]. One unit (U) of MTHase activity was defined as the amount of MTHase needed to produce 1 µmol of trehalose per min under the assay conditions.
Results and discussion
Expression of S. acidocaldarius MTHase in E. coli BL21 (DE3)
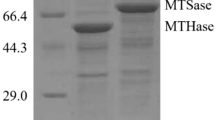
An E. coli codon-optimized synthetic gene (treZ) encoding S. acidocaldarius MTHase was prepared by Shanghai Generay Biotech Co., Ltd. To ensure optimal expression, rare codons in the coding sequence were replaced with preferred codons and the free energy of mRNA was decreased 13.5 kcal/mol. Engineered E. coli BL21 (DE3) harboring the plasmid pET24a–treZ, which expresses treZ with a T7 promoter, was cultivated in a shake flask. MTHase expression was induced through the addition of 0.05 mmol/L IPTG and further cultivation for 24 h. SDS-PAGE analysis showed a band around 59 kDa, which is consistent with the calculated molar mass of MTHase, in both the soluble and insoluble intracellular fractions (Fig. 2). The MTHase activity was determined to be 10.4 U/mL (228.8 U/g wet cell) in the soluble intracellular fraction, which was much higher than the expression level (3.4 U/g wet cell) previously reported [12].

SDS-PAGE analysis of recombinant MTHase in shake flasks. M, molecular mass standard proteins; 1, intracellular soluble fraction of E. coli BL21 (DE3)/pET24a–treZ; 2, intracellular soluble fraction of E. coli Origami (DE3)/pET32a–treZ; 3, intracellular insoluble fraction of E. coli BL21 (DE3)/pET24–treZ; 4, intracellular insoluble fraction of E. coli Origami (DE3)/pET32a–treZ
Although MTHase was successfully expressed in E. coli BL21 (DE3), the MTHase in the soluble fraction was unacceptably low; much of the target protein was located in insoluble inclusion bodies. Further experiments were conducted to attempt to address this issue. A series of additives including ethyl alcohol (2%, v/v), glycine betaine (20 mmol/L), sorbitol (1%, w/v), saccharose (0.4 mol/L), and dithiothreitol (DTT, 1 mmol/L), were added to the culture medium. Only the addition of sucrose increased both cell growth (by 5.2%) and MTHase activity (by 5%). Other additives had no effect on MTHase production (Fig. S4). The plasmid pG-Tf2 harboring the gene sequence groES–groEL–tig was inserted into E. coli/pET24a–treZ for co-expression of MTHase with the chaperones GroES, GroEL and trigger factor. Unfortunately, the MTHase activity was 10.3 U/mL, almost the same as that obtained without chaperone co-expression. Finally, an N-terminal fusion of SUMO with MTHase was prepared. Once again, expression of MTHase activity was not increased. Thus, it seemed that the typical strategies used to improve recombinant protein expression in E. coli failed to improve MTHase production in E. coli BL21 (DE3).
Expression of S. acidocaldarius MTHase in E. coli Origami (DE3)
The inability to produce soluble MTHase in high yield in E. coli BL21 (DE3) and the failure of typical solubilization strategies suggested that MTHase may be not suitable for expression in E. coli BL21 (DE3), and that this was related to some property of MTHase. The native form of MTHase is a dimer covalently linked by a single intermolecular disulfide bond between the cysteine-297s of the two molecules. Because the reducing environment of the E. coli cytoplasm is not conducive to disulfide bond formation [28], it seemed reasonable that the inability to form the proper disulfide bond caused the formation of inclusion bodies.
To address disulfide bond formation, a commercial plasmid bearing the TrxA tag, pET-32a (+), was used in this study. TrxA is a low molecular weight protein (109 aa) that catalyzes the formation of disulfide bonds and improves protein folding [28]. Use of this plasmid was paired with use of the E. coli Origami strain. This strain contains mutant thioredoxin reductase (trxB) and glutathione reductase (gor) genes, which enable recombinant proteins to form disulfide bonds more efficiently in the cytoplasm [28, 29]. The codon-optimized synthetic gene MTHase encoding was inserted into pET-32a (+) to form a gene encoding an N-terminal TrxA fusion. This plasmid was inserted into E. coli Origami (DE3). Initial expression experiments were conducted in shake flasks. SDS-PAGE analysis of the soluble fraction of the engineered E. coli displayed a single major band at approximately 71 kDa, which is consistent with the calculated molar mass of the Trx–MTHase fusion. This band was thicker than that obtained from E. coli BL21 (DE3), while the inclusion body content obviously decreased (Fig. 2). Correspondingly, the MTHase activity was 42.0 U/mL, fourfold higher than that obtained from the engineered E. coli BL21 (DE3). When the N-terminal TrxA was removed using enterokinase, the MTHase activity increased to 55.0 U/mL, 5.3-fold greater than that obtained using E. coli BL21 (DE3). The combined use of pET-32a (+) and E. coli Origami (DE3), which is more beneficial for the formation of cytoplasmic disulfide bonds than the previous expression system, efficiently improved the production of active MTHase, suggesting that formation of the disulfide bond plays an important role in the correct folding of MTHase.
To clarify whether the improvement in soluble MTHase expression was caused by the thioredoxin fusion or oxidative cytoplasmic expression in E. coli Origami (DE3), E. coli BL21 (DE3) harboring pET-32a (+), and E. coli Origami (DE3) harboring pET-24a (+) were constructed. The MTHase activities obtained from the two strains were 17.9 U/mL and 38.8 U/mL, respectively, indicating that the improvement in soluble MTHase expression depended on a synergistic effect of the two mechanisms and that oxidative cytoplasmic expression in E. coli Origami (DE3) played a larger role in the process.
Enhancing MTHase production in E. coli Origami (DE3) by optimizing induction in a 3-L fermentor
MTHase production in E. coli Origami (DE3) was scaled up in a 3-L fermentor. The fermentation was performed using a fed-batch cultivation process used in a previous study to achieve high cell density [30]. As with all proteins produced using an inducible expression system, the method of induction was considered the primary factor determining the yield of the target protein [31]. Many reports have shown that optimal induction, which is closely related to the balance between cell growth and protein expression, occurs under different conditions for different proteins [31,32,33]. In this study, the point at which induction was initiated, the lactose concentration used, and the induction temperature were investigated in an effort to optimize MTHase production.
Effect of induction point on cell growth and MTHase production
Protein expression generally places a metabolic burden on cells. This burden decreases biomass production and protein yield, especially when the induction is performed too early. Late induction also limits induction efficiency and protein production. Therefore, induction is usually initiated when the biomass in the fermentor reaches a certain level. The optimized induction point is generally at the early- or mid-log phase [30, 33]. In addition, according to the previous study [21, 30, 31], high cell density (an OD600 of more than 100) could usually be achieved using the semisynthetic medium and fed-batch cultivation. To identify the optimal induction point for MTHase production, the inducer lactose was fed into the culture when the OD600 reached 30, 40, and 50, respectively. As shown in Fig. 3, cell growth was similar during the growth phase, but differences in the biomass gradually appeared when induction was initiated at different biomass levels. Earlier induction gave rise to lower biomass, as expected. The highest DCW (70.8 g/L) was obtained when induction was begun at an OD600 of 50. This DCW was 43.0% greater than that observed when induction was initiated at an OD600 of 30 and 6.9% higher than that observed when induction was initiated at an OD600 of 40. The lowest MTHase activity (143.4 U/mL) was also observed at the earliest induction point. When induction was delayed until the biomass reached OD600 40 or 50, the MTHase production improved. A slightly higher value (184.0 U/mL) was seen at an OD600 of 40 than at an OD600 of 50, but the difference was only 5.1%. The specific MTHase activities were also close, which were 16.4 U/mg and 15.8 U/mg, respectively. This range of induction points available for high-yield MTHase production is beneficial for controlling the fermentation process under practical production conditions.

Effect of induction point (OD600) on cell growth and MTHase production. a Cell growth, b MTHase activity, c specific MTHase activity. Filled squares: 30, filled circles: 40, filled triangles: 50
Effect of induction temperature on cell growth and MTHase production
Induction temperature is another important factor for both cell growth and protein production [21]. In this study, three induction temperatures, 25, 30, and 35 °C, were investigated. As shown in Fig. 4, the cell growth rate and biomass increased with increasing induction temperature. Cell growth was obviously inhibited when the induction temperature was 25 °C; the peak DCW was only 42.6 g/L. The highest DCW (73.2 g/L), obtained when the induction temperature was 35 °C, was 1.7- and 1.1-fold of that obtained at 25 °C and 30 °C, respectively. However, high biomass did not cause high MTHase production. Among the three induction temperatures, the highest MTHase activity (184.0 U/mL, 16.4 U/mg) was observed at 30 °C. At 25 °C, the MTHase activity (142.5 U/mL, 19.8 U/mg) equated to a production of 3.3 × 103 U per g DCW, 17.8% higher than that observed at 30 °C. Because lower induction temperatures decrease the protein synthesis rate and help appropriate protein folding, these temperatures help to generate the soluble form of recombinant proteins [16, 21]. This is probably why the lower temperature led to higher MTHase production for per g DCW. However, the low induction temperature also resulted in a lower final biomass, which hampered the final yield of the target protein. Thus, induction at 25 °C resulted in the lowest MTHase yield. The final biomass value was highest when protein expression was induced at 35 °C. However, the final MTHase activity was only 94.6% of that seen when expression was induced at 30 °C. Higher induction temperatures are widely considered to make the folding of newly synthesized polypeptides too fast. Rapid folding leads to folding errors that cause the formation of insoluble inclusion bodies and limit the production of active enzyme [21]. In this study, induction of MTHase at 35 °C had only a slightly negative effect on MTHase production, compared with induction at 30 °C. Considering the cost of cooling required to maintain the lower temperature, an induction temperature between 30 and 35 °C should be acceptable under practical production conditions.

Effect of induction temperature on cell growth and MTHase production. a Cell growth, b MTHase activity, c specific MTHase activity. Filled squares: 25 °C, filled circles: 30 °C, filled triangles: 35 °C
Effect of the inducer lactose feeding rate on cell growth and MTHase production
Lactose is a low-cost, non-toxic inducer used with T7 promoter-based expression systems. Expression from the T7 promoter can normally be fully induced using high concentrations of lactose. However, cell growth and protein should be balanced to obtain the maximal level of protein production when expression is induced with lactose [34, 35]. In this study, lactose feeding was performed at four different feeding rates, 0.1, 0.2, 0.4 and 0.8 g/L/h, to determine the optimal feeding rate. As shown in Fig. 5, the biomass reached its highest value (69.1 g/L) when the lactose feeding rate was the lowest (0.1 g/L/h). Increasing the lactose feeding rate reduced the biomass to some extent; the lowest DCW (52.4 g/L) was seen at 0.8 g/L/h. The highest MTHase activity, 204.6 U/mL (19.5 U/mg), was obtained at a lactose feeding rate of 0.2 g/L/h. This activity was 1.6-fold of that seen at a lactose feeding rate of 0.1 g/L/h. Higher lactose feeding rates (> 0.2 g/L/h) decreased the final MTHase activity, especially 0.8 g/L/h. This phenomenon may have been caused by the metabolic burden or the presence of misfolded proteins caused by excessive induction [31, 36].

Effect of inducer lactose feeding rate on cell growth and MTHase production. a Cell growth, b MTHase activity, c specific MTHase activity. Filled squares: 0.1 g/L/h, open circles: 0.2 g/L/h, filled upright triangles: 0.4 g/L/h, unfilled downward triangles: 0.8 g/L/h
Conclusion
Sulfolobus acidocaldarius MTHase was expressed in E. coli, and the MTHase activity obtained using the pET-32a/E. coli Origami (DE3) system was 5.3-fold greater than that obtained using the pET-24a/E. coli BL21 (DE3) system. This supports that disulfide bond formation plays an important role in the proper folding of overexpressed cytoplasmic MTHase. When the fermentation conditions were optimized, the MTHase activity reached 204.6 U/mL (Table 1). To the best of our knowledge, this is the first report describing a systematic effort to obtain high-efficiency MTHase production. This report is also expected to be valuable in the production of other enzymes containing disulfide bonds.
References
Fang TY, Tseng WC, Shih TY, Wang MY (2008) Identification of the essential catalytic residues and selectivity-related residues of maltooligosyltrehalose trehalohydrolase from the thermophilic archaeon Sulfolobus solfataricus ATCC 35092. J Agric Food Chem 56:5628–5633
Kato M (1999) Trehalose production with a new enzymatic system from Sulfolobus solfataricus KM1. J Mol Catal B Enzym 6:223–233
Richards AB, Krakowka S, Dexter LB, Schmid H, Wolterbeek APM, Waalkens-Berendsen DH, Shigoyuki A, Kurimoto M (2002) Trehalose: a review of properties, history of use and human tolerance, and results of multiple safety studies. Food Chem Toxicol 40:871–898
Ohtake S, Wang YJ (2011) Trehalose: current use and future applications. J Pharm Sci US 100:2020–2053
Sebaaly C, Greige-Gerges H, Stainmesse S, Fessi H, Charcosset C (2016) Effect of composition, hydrogenation of phospholipids and lyophilization on the characteristics of eugenol-loaded liposomes prepared by ethanol injection method. Food Biosci 15:1–10
Mukai K, Tabuchi A, Nakada T, Shibuya T, Chaen H, Fukuda S, Kurimoto M, Tsujisaka Y (1997) Production of trehalose from starch by thermostable enzymes from Sulfolobus acidocaldarius. Starch Starke 49:26–30
Kim YH, Kwon TK, Park S, Seo HS, Cheong JJ, Kim CH, Kim JK, Lee JS, Choi YD (2000) Trehalose synthesis by sequential reactions of recombinant maltooligosyltrehalose synthase and maltooligosyltrehalose trehalohydrolase from Brevibacterium helvolum. Appl Environ Microbiol 66:4620–4624
Yamamoto T, Maruta K, Watanabe H, Yamashita H, Kubota M, Fukuda S, Kurimoto M (2001) Trehalose-producing operon treYZ from Arthrobacter ramosus S34. Biosci Biotechnol Biochem 65:1419–1423
Nakada T, Maruta K, Mitsuzumi H, Kubota M, Chaen H, Sugimoto T, Kurimoto M, Tsujisaka Y (1995) Purification and characterization of a novel enzyme, maltooligosyl trehalose trehalohydrolase, from Arthrobacter sp Q36. Biosci Biotechnol Biochem 59:2215–2218
Nakada T, Ikegami S, Chaen H, Kubota M, Fukuda S, Sugimoto T, Kurimoto M, Tsujisaka Y (1996) Purification and characterization of thermostable maltooligosyl trehalose trehalohydrolase from the thermoacidophilic archaebacterium Sulfolobus acidocaldarius. Biosci Biotechnol Biochem 60:267–270
Demain AL, Vaishnav P (2009) Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv 27:297–306
Schiraldi C, Di Lernia I, De Rosa M (2002) Trehalose production: exploiting novel approaches. Trends Biotechnol 20:420–425
de Pascale D, Sasso MP, Di Lernia I, Di Lazzaro A, Furia A, Farina MC, Rossi M, De Rosa M (2001) Recombinant thermophilic enzymes for trehalose and trehalosyl dextrins production. J Mol Catal B Enzym 11:777–786
Seo JS, An JH, Baik MY, Park CS, Cheong JJ, Moon TW, Park KH, Choi YD, Kim CH (2007) Molecular cloning and characterization of trehalose biosynthesis genes from hyperthermophilic archaebacterium Metallosphaera hakonesis. J Microbiol Biotechnol 17:123–129
Chang M, Qiao Y, Ding HB (2011) Cloning and expression of maltooligosyltrehalose trehalohydrolase gene from Corynebacterium glutamicum. J Agri Sci Technol 13:47–52
Sorensen HP, Mortensen KK (2005) Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb Cell Fact 4:1
Barroso JF, Elholm M, Flatmark T (2003) Tight binding of deoxyribonucleotide triphosphates to human thymidine kinase 2 expressed in Escherichia coli. Purification and partial characterization of its dimeric and tetrameric forms. Biochemistry 42:15158–15169
Okamoto-Kainuma A, Yan W, Fukaya M, Tukamoto Y, Ishikawa M, Koizumi Y (2004) Cloning and characterization of the dnaKJ operon in Acetobacter aceti. J Biosci Bioeng 97:339–342
Blackwell JR, Horgan R (1991) A novel strategy for production of a highly expressed recombinant protein in an active form. FEBS Lett 295:10–12
Kagawa N, Cao QW (2001) Osmotic stress induced by carbohydrates enhances expression of foreign proteins in Escherichia coli. Arch Biochem Biophys 393:290–296
Duan X, Chen J, Wu J (2013) Optimization of pullulanase production in Escherichia coli by regulation of process conditions and supplement with natural osmolytes. Bioresour Technol 146:379–385
Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR (2004) SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genom 5:75–86
Peroutka Iii RJ, Orcutt SJ, Strickler JE, Butt TR (2011) SUMO fusion technology for enhanced protein expression and purification in prokaryotes and eukaryotes. Methods Mol Biol 705:15–30
Smith DB, Johnson KS (1988) Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67:31–40
Lavallie ER, Diblasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM (1993) A thioredoxin gene fusion expression system that circumvents inclusion body formation in the Escherichia coli cytoplasm. Biotechnology 11:187–193
Han C, Su LQ, Hong RY, Wu SX, Wu J (2017) A comparative study of maltooligosyltrehalose synthase from Sulfolobus acidocaldarius expressed in Pichia pastoris and Escherichia coli. Process Biochem 60:35–41
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal chem 31:426–428
Bessette PH, Aslund F, Beckwith J, Georgiou G (1999) Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc Natl Acad Sci USA 96:13703–13708
de Marco A (2009) Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb Cell Fact 8:26
Cheng J, Wu D, Chen S, Chen J, Wu J (2011) High-level extracellular production of alpha-cyclodextrin glycosyltransferase with recombinant Escherichia coli BL21 (DE3). J Agric Food Chem 59:3797–3802
Su LQ, Hong RY, Wu J (2015) Enhanced extracellular expression of gene-optimized Thermobifida fusca cutinase in Escherichia coli by optimization of induction strategy. Process Biochem 50:1039–1046
Su LQ, Huang Y, Wu J (2015) Enhanced production of recombinant Escherichia coli glutamate decarboxylase through optimization of induction strategy and addition of pyridoxine. Bioresource Technol 198:63–69
Donovan RS, Robinson CW, Glick BR (1996) Review: optimizing inducer and culture conditions for expression of foreign proteins under the control of the lac promoter. J Ind Microbiol 16:145–154
Su LQ, Ma Y, Wu J (2015) Extracellular expression of natural cytosolic arginine deiminase from Pseudomonas putida and its application in the production of L-citrulline. Bioresour Technol 196:176–183
Kilikian BV, Suarez ID, Liria CW, Gombert AK (2000) Process strategies to improve heterologous protein production in Escherichia coli under lactose or IPTG induction. Process Biochem 35:1019–1025
Lebendiker M, Danieli T (2014) Production of prone-to-aggregate proteins. FEBS Lett 588:236–246
Acknowledgements
This work received financial support from the National Natural Science Foundation of China (31771916, 31501419), the Natural Science Foundation of Jiangsu Province (BK20180082), the National Science Fund for Distinguished Young Scholars (31425020), the National First-class Discipline Program of Light Industry Technology and Engineering (LITE2018-03), and the 111 Project (No. 111-2-06).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
The codon-optimized synthetic gene (treZ) sequence (TIFF 4432 KB)
Fig. S2
The construction diagram of recombinant plasmid pET24a-treZ (TIFF 3362 KB)
Fig. S3
The construction diagram of recombinant plasmid pET24a-sumo-treZ (TIFF 4512 KB)
Fig. S4
Effects of different additives on cell growth and MTHase production. ■ OD600, □ MTHase activity (TIFF 35 KB)
Fig. S5
SDS-PAGE analysis of recombinant MTHase in the 3-L fermentor. M, molecular mass standard proteins; 1–8, the MTHase samples corresponding with that in Table 1 (TIF 6446 KB)
Rights and permissions
About this article

Cite this article
Su, L., Wu, S., Feng, J. et al. High-efficiency expression of Sulfolobus acidocaldarius maltooligosyl trehalose trehalohydrolase in Escherichia coli through host strain and induction strategy optimization. Bioprocess Biosyst Eng 42, 345–354 (2019). https://doi.org/10.1007/s00449-018-2039-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-018-2039-4