
Abstract
Feruloyl esterase (FAE)-encoding genes AnfaeA and AnfaeB were isolated from Aspergillus niger 0913. For overexpression of the two genes in Trichoderma reesei, constitutive and inductive expression plasmids were constructed based on parental plasmid pAg1-H3. The constructed plasmids contained AnfaeA or AnfaeB gene under the control of glyceraldehyde-3-phosphate dehydrogenase A gene (gpdA) promoter (from A. nidulans) or cellobiohydrolases I (cbh I) gene promoter (from T. reesei), and cbh I terminator from T. reesei. The target plasmids were transferred into T. reesei D-86271 (Rut-C30) by Agrobacterium tumefaciens-mediated transformation (ATMT), respectively. A high level of feruloyl esterase was produced by the recombinant fungal strains under solid-state fermentation, and the cbh I promoter was more efficient than the gpdA promoter in the expression of AnfaeA. The optimum temperatures and pH values were 50 °C and 5.0 for AnFAEA, and 35 °C and 6.0 for AnFAEB. The maximum production levels were 20.69 U/gsd for AnFAEA and 15.08 U/gsd for AnFAEB. The recombinant fungal enzyme systems could release 62.9% (for AnFAEA) and 52.2% (for AnFAEB) of total ferulic acids from de-starched wheat bran, which was higher than the 46.3% releasing efficiency of A. niger 0913. The supplement of xylanase from T. longibrachiatum in the enzymatic hydrolysis led to a small increment of the ferulic acids release.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ferulic acid (4-hydroxy-3-methoxycinnamic acid, FA) is the most plentiful phenolic phytochemical existing in plant materials, where it mainly cross-links to hemicellulosic carbohydrate moieties and to lignin via ester or ether bonds [1]. FA displays excellent antioxidant ability due to the existence of para hydroxy groups on the phenolic acid moiety [2]. In many countries, FA is approved to be as a food additive, mainly used for food preservative, flavor precursor, thickening agent or as functional promoting agent [1]. Ferulic acid is also a potential therapeutic agent with demonstrated antioxidant, anti-inflammatory, antimicrobial, hepatoprotective and UV-protective activities [3, 4]. In the pharmacological field, FA is mainly applied to synthesize bioactive compounds for prevention or treatment of special human diseases, e.g. diabetes, cancer, Alzheimer and cardiovascular diseases [1]. Additionally, FA can also be transformed to 4-vinyl guaiacol and vanillin by enzymatic conversion [5]. These chemicals are important flavouring agents in food and drink industries [6].
Agricultural by-products especially cereal grains contain 0.5–3% (w/w) extractable amount of FA, mostly in the trans-isomeric form, and esterified with the specific polysaccharides [3]. Among bran substrates, corn bran contains the highest amount of esterified ferulic acid (30 mg/g), while de-starched wheat bran (DSWB) can have the content up to 6 mg/g [2]. Preparation of FA from bran substrates is important for high-value utilization of agricultural by-products. The greatest advantage of FA extraction by enzymatic hydrolysis lies in the environmental friendliness, as well as the enzyme treatment is more specific at releasing the target products (FA) without damaging other valuable components that are easily destroyed during chemical extraction [2, 7].
Feruloyl esterases (FAEs, EC 3.1.1.73), also known as ferulic/cinnamic acid esterases, can catalyze the hydrolysis of ester linkages between FA and polysaccharides [8, 9], and has been known as a key factor in the liberation of FA from plant cell walls [10]. To date, different FAEs have been found in fungi and bacteria. Many fungal species belonging to Aspergillus such as A. niger and A. flavipes are active producers of FAEs [9, 11]. Based on substrate utilization and amino acid sequence analysis, FAEs were classified into four different types: A to D [12]. Type A FAEs prefer substrates that contain methoxy substitutions at C-3 and/or C-5 as found in ferulic and sinapic acids, and are active towards methyl p-coumarate. Type B FAEs show preference to substrates containing hydroxyl substitutions, such as caffeic acid or p-coumaric acid. The FAEs belonging to types C or D possess broad substrate specificity against synthetic HCA esters (methyl caffeate, methyl sinapate and methyl p-coumarate). Types A and D FAEs can release low quantities of diferulic acid from plant materials [2, 11].
In plant cell walls, FA is linked to the lignin–carbohydrate complexes. Many studies have indicated that hemicelluloses-degrading enzymes, especially endoxylanase, were important for increasing the release of FA from biomaterials. Only 16.8% of the total alkali-extractable FA was released from DSWB by individual FAE (AnFaeA) from A. niger, and the extraction efficiency increased to 70% when the xylanases from the same fungus was added [13]. A high level of released FA from wheat bran depended on the synergistic action of the FAE from Fusarium oxysporum with the xylanase from Trichoderma longibrachiatum [14]. Enzymatic hydrolysis of agricultural wastes using single type C feruloyl esterase RuFae2 (the encoding gene from rumen microbial metagenome) only released a low level of FA. The addition of glycoside hydrolase (GH) 10 family endoxylanase (from Cellvibrio mixtus) led to increase in the release of FA, with the highest level of 6.7-fold for wheat bran [15]. The acquirement of feruloyl esterases and xylanase (maybe also other enzymes) at high levels is important for the industrial production of FA from biomass resources.
Trichoderma reesei, the anamorph of the ascomycete fungus Hypocrea jecorina, is one of the most important producers of cellulolytic and hemicellulolytic enzymes [16]. To date, all of the T. reesei strains used in the industry are derived from the wild-type strain QM6a, which is regarded as the T. reesei reference strain. Through three rounds of mutagenesis of the wild type, the cellulase hyperproducer strain RUT-C30 was isolated, and the extracellular protein yield was up to 20-fold to the parental strain [17, 18]. At least five xylanases (XYN1-5) could be produced by T. reesei, belonging to the GH families 10, 11 and 30, following the carbohydrate-active enzyme (CAZyme) classification [19]. At the same time, genome and secretome data proved that no feruloyl esterase-encoding gene existed in T. reesei [20]. In the present study, two FAE-encoding genes derived from A. niger were transferred into the genome of a cellulase hyper-producer T. reesei strain D-86271 (RUT-C30), and a high level of feruloyl esterase was produced by solid-state fermentation. The recombinant enzyme system could be used in the efficient release of FA from bran substrates.
Materials and methods
Strain, media and culture conditions
All microbial strains and plasmid used in this study are listed in Table 1. Aspergillus niger 0913 and Trichoderma reesei D-86271were used for cloning and heterologous expression of feruloyl esterase (fae) genes, respectively. Agrobacterium tumefaciens AGL-1 was used for fungal transformation. Escherichia coli strain DH5α was routinely used for plasmid propagation. Potato dextrose agar (PDA) medium was used for sporulation and preservation of fungal strains. For fungal fermentation, Mandel’s medium was prepared according to a documented method [21]. Escherichia coli cells were grown at 37 °C in Luria–Bertani (LB) medium supplemented with antibiotic when required. Solid-state fermentation (SSF) medium containing delignified wheat straw, wheat bran and 10× Mandel’s liquid was prepared as per previous description [22].
Cloning of AnfaeA and AnfaeB genes from A. niger
To clone AnfaeA and AnfaeB genes from A. niger 0913, the specific primers faeA_f1/faeA_r1 and faeB_f1/faeB_r1 were designed according to the corresponding genomic sequences of A. niger CBS 513.88 (accession numbers AM270190 and AM270291) with Primer Premier 6 software. All of the primers used in the study are listed in Table 2. Mycelia of A. niger 0913 were collected after 3 days culture of the strain in 50 ml PDA liquid medium at 28 °C, and its genomic DNA was isolated with a plant genomic DNA isolation kit (TransGen, Beijing, China). The DNA fragments containing the integrated AnfaeA or AnfaeB gene were amplified with the corresponding specific primer sets, and subcloned into the vector pEASY-Blunt. DNA sequencing was performed by the Invitrogen Biotechnologies Co. Ltd (Shanghai, China).
Plasmid construction and fungal transformation
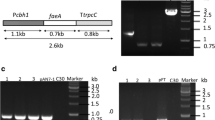
The vector pAg1-H3 was employed to construct fungal expression plasmids. To construct a constitutive expression plasmid, a 896 bp glyceraldehyde-3-phosphate dehydrogenase gene promoter (PgpdA, derived from Aspergillus nidulans) was isolated from plasmid pJL43RNAi by double digestion with restriction enzymes SacI and XbaI, and ligated into the same digested plasmid pBluescript II KS(+), yielding pBl-Pgpd. After digestion with ApaI and EcoRI, the 1.38 kb DNA fragment containing the AnfaeA gene was inserted into the corresponding sites of pBl-Pgpd, resulting in pBl-Pgpd–AnfaeA. pBl-Pgpd–AnfaeA was digested with ApaI and SacI, and one 2.3 kb DNA fragment was ligated into the terminated pAg1-H3 by the same restriction enzymes to give expression plasmid pPgpd–AnfaeA (Fig. 1).

Schematic chart of the expression plasmids for feruloyl esterase genes. The plasmids pPgpd–AnfaeA, pPcbhI–AnfaeA and pPcbhI–AnfaeB were constructed using plasmid pAg1-H3 as parental plasmid. LB left arm; RB right arm. hph, hygromycin resistance gene; PgpdA, the glyceraldehyde-3-phosphate dehydrogenase A gene promoter from A. nidulans; PcbhI or TcbhI, the cellobiohydrolases I gene promotor or terminator from T. reesei D-86271
The inductive expression plasmids in which target genes were controlled by the cellobiohydrolase I (cbh I) promoter from T. reesei were also constructed. A 1088 bp cbh1 terminator (Tcbh1) was amplified from T. reesei D-86271 with primers Tcbh1_f and Tcbh1_r. Then, the obtained fragment was inserted into the XhoI-ApaI sites of pAg1-H3 after digestion with the same enzymes, yielding pAg-Tcbh1. The 1554 bp cbh1 promoter fragment (containing the signal peptide and the first two amino acid codons of the mature cbh1gene) was isolated from T. reesei D-86271 strain by primers Pcbh1_fs and Pcbh1_rs, and ligated into KpnI-XhoI sites of pAg-Tcbh1, resulting in pAg-PTcbh1. The AnfaeA (837 bp) or AnfaeB (1590 bp) gene fragments (deleted native signal sequences) were amplified by primer sets faeA_f3/faeA_r3 or faeB_f3/faeB_r3 (containing a His tag), respectively. After gel purifications, the gene fragment was ligated into plasmid pAg-PTcbh1 (terminated with XhoI) by Hieff Clone™ One Step Pcr Cloning Kit (Yeasen, Shanghai), and yielding pPcbh1–AnfaeA or pPcbh1–AnfaeB (Fig. 1).
The plasmids were introduced into the T. reesei strain D-86271 by A. tumefaciens-mediated transformation (ATMT) method as described previously [25, 26]. After co-culture of fungal spores and Agrobacterium cells at 25 °C for 48 h, fungal transformants were selected on PDA plates supplemented with 100 µg/ml hygromycin and 400 µg/ml cefotaxime sodium. The fungal transformants were confirmed by PCR with gene-specific primer sets.
Preparation of enzymes by fungal solid-state fermentation
Approximately, 1 × 108 fungal conidia were inoculated into a 250-ml Erlenmeyer flask containing 50 ml Mandel’s medium (10 g/L glucose as carbon source) and incubated at 28 °C and 180 rpm for 48 h. Two milliliters of the fungal cultures were transferred into a 250-ml Erlenmeyer flask containing 3 g SSF substrates. The fermentation was conducted in a Plant Growth Chamber MLR-352H-PC (Panasonic, Ehime Prefecture, Japan) at 30 °C and 70% humidity for 13 days. After the fermentation, the culture of SSF from each flask was mixed fully with 30 mL distilled water containing Tween-80 at the final concentration of 0.1% (v/v). Then, the mixture was shaken at 25 °C and 120 rpm for 2 h, and centrifuged at 7000 rpm for 10 min. The supernatant was stored at 4 °C for subsequent enzyme determination.
Determination of enzyme activities and protein concentration
Feruloyl esterase activities, cellulose activity on filter paper (FPase) and xylanase were assayed in 50 mM sodium citrate buffer (pH 6.0 or 4.8) using methyl ferulate (MFA), Whatman No. 1 filter paper or xylan-beechwood as the substrates, respectively. The reaction conditions (except temperature) were the same as previously described [22]. The released reducing sugar was quantified by the 3,5-dinitrosalicylic acid (DNS) method using glucose or xylose standard curves [27]. The released free FA from substrates was analyzed by an Agilent 1260 Infinity high-performance liquid chromatography (HPLC) system (Agilent Technologies, CA, USA) equipped with a ZORBAX Eclipse Plus C18 reversed-phase column. One unit (U) of enzyme activity was defined as the amount of enzyme that produces 1 µmol of glucose, xylose or free FA, from the appropriate substrates, per minute under the detection conditions.
The recombinant protein (for AnfaeB gene) was purified from crude extracts by an Ni–NTA purification system (Qiagen, Valencia, CA, USA) and detected by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis. The optimum pH values or temperatures for the recombinant enzymes were detected in universal pH buffer (pH 3–10) [28], or at different temperatures from 30 to 70 °C. Extracellular proteins were quantified by the BCA assay kit (Thermo Tech, USA).
Release of FA from wheat bran by enzymatic hydrolysis
The de-starched wheat bran (DSWB) was prepared with the previous method [22]. Five hundred milligrams of bran was suspended in 10 ml of 0.1 M sodium citrate buffer (pH 6.0) with 0.02% (w/v) sodium azide to prevent bacterial contamination. Approximately, 10 mg crude enzyme from different T. reesei transformants or the parental strain with or without endoxylanase from T. longibrachiatum (Sigma, USA) was added into the reaction buffer, and the mixture incubated at 35 °C or 45 °C and 120 rpm on a shaker. After 24 h, the reaction was stopped by treatment at 99 °C for 30 min. The samples were subjected to analysis of FA content after centrifugation at 10,000g for 15 min by the HPLC method. The amount of alkali-extractable FA in the DSWB was considered as 100% for the enzymatic hydrolysis [22, 29].
Results and discussion
Cloning of AnfaeA and AnfaeB from A. niger
The 1.38 and 2.08 kb DNA fragments containing AnfaeA or AnfaeB genes were amplified from A. niger 0913, and the gene sequences were deposited in GenBank under the accession numbers KY412895 and KY412896, respectively. The newly isolated genes have high similarities with the reported AnfaeA or AnfaeB from other A. niger strains. The deduced 281 amino acid residues of AnfaeA gene is the same as the reported feruloyl esterase A (GenBank: XP_001393337) from A. niger CBS 513.88, except that 183 Arg is substituted by His. The 521 amino acid residues encoded by the AnfaeB gene showed 100% identity with the homologous protein (GenBank: XP_001396085) from A. niger CBS 513.88. At the same time, the protein sequence changes for AnFAEB of A. niger 0913 are 21 Pro to Ser and 326 Ser to Tyr compared to FAEB (GenBank: AJ309807) from A. niger CMICC 298302 [30]. Indeed, feruloyl esterase B from A. niger should be divided into the type C sub-class according to the phylogenetic analysis [12].
Overexpression of AnfaeA and AnfaeB in T. reesei
The three expression plasmids, pPgpd–AnfaeA, pPcbh1–AnfaeA and pPcbh1–AnfaeB, were constructed and introduced into T. reesei D-86271 by the ATMT method, respectively. For each plasmid, at least 20 fungal transformants appeared on the selected plates. The randomly selected transformants (five to eight for each plasmid) were confirmed by PCR amplification with gene-specific primers (see supplementary Figure s1). To detect the expression of the target gene, solid-state fermentation of recombinant T. reesei strains was conducted using delignified wheat straw and wheat bran as substrates. FAE activities were detected in the crude extracts from most fungal transformants using MFA as the detection substrate. Meanwhile, the parental strain D-86271 could not produce feruloyl esterase. Under the same fermentation conditions, T. reesei transformants Pcbh1–AnfaeA produced higher FAE than the transformants Pgpd–AnfaeA (Table 3). The highest production levels were 19.55 U/gds (gram dry substrate) and 16.63 U/gds for fungal transformants Pcbh1–AnfaeA and Pgpd–AnfaeA, respectively. The data indicated that the cbhI promotor combined with the signal of the cbhI gene was more efficient than the gpdA promotor combined with the native signal in the expression of the AnfeaA gene. The FAE activities produced by transformants Pcbh1–AnFaeB were lower than those by transformants Pcbh1–AnfaeA under the same fermentation and detection conditions. The production level of FAE by these recombinant T. reesei strains was significantly higher (about 4 to 5-fold) than wild-type A. niger strain 0913 (4.09 U/gds) (Table 3).
The recombinant AnFAEB was purified from the crude extracts by the Ni–NTA system. SDS-PAGE analysis indicated that the molecular weight (Mw) of purified AnFAEB was similar to its theoretical value of 56.42 kDa (including the His tag) (Fig. 2). Approximately, 2 mg target proteins was purified from 1 ml of crude extracts. Feruloyl esterases are widely used in food, biofuel, farming and biosynthesis industries [9, 31]. FAE genes derived from A. niger were successfully expressed in different host cells [8, 13, 32]. By analysis of the literature (see supplementary Table S1), it was demonstrated that host cells (species or strain) and fermentation conditions were important for the production of FAEs. Pichia pastoris is a high efficiency host for the heterologous expression of FAEs [13]. The utilization of agricultural wastes as main substrates in the fermentation of T. reesei makes this fungus a competitive host in the industrial production of feruloyl esterases.

SDS–PAGE analysis of purified recombinant AnFaeB. M, protein marker. 1, crude enzyme. 2, pure enzyme. In each lane, 80 µg of crude enzyme or 15 µg of purified enzyme was loaded for electrophoresis on 10% SDS–PAGE gel
Characterization of the recombinant FAEs
The optimum pH for the activity of AnFAEA was 5.0, and the relative activity was above 90% in the pH range of 4.5–6.0. Lower or higher pH values significantly decreased the activity of AnFAEA (Fig. 3a). AnFAEA displayed the maximum activity at 50 °C and retained the most activity at the temperature range from 30 to 45 °C. At 55 °C, the enzyme lost about 40% of its activity (Fig. 3b). The optimum pH and temperature of the recombinant AnFAEA were consistent with the FAEA derived from other A. niger strains [11, 32]. The optimum pH of the recombinant AnFAEB was 6.0 (Fig. 3a), which was in agreement with published data [33]. The recombinant AnFAEB displayed the highest activity at 35 °C (Fig. 3b), which was significantly lower than the previously reported value of 50 °C [33, 34]. The molecular mass of native or homologously expressed FAEB is 74 kDa, which is higher than the theoretical value of 55.6 kDa largely due to the N-glycosylation [30, 33]. It was speculated that the thermal sensitivity of recombinant AnFAEB was due to the lack of glycosylation in the heterologous expression by T. reesei D-86271 (Fig. 2). At the same time, different substrates were used in the detection of FAE activities.

Effect of pH (a) and temperature (b) on the enzyme activities of AnFAEA and AnFAEB. a Optimum pH values of the recombinant AnFAEA and AnFAEB. These enzyme activities were assayed at 40 °C in universal pH buffer (pH 3–10). b Optimum temperatures of the recombinant enzymes AnFAEA and AnFAEB. The activities were determined in 50 mM sodium citrate buffer (pH 6.0) at different temperatures. Purified AnFAEB was used in the detection. The relative activities were calculated using the maximum enzyme activity as 100%. Error bars represent standard deviations from three independent assays
Release of FA from wheat bran by the recombinant enzyme systems
The fungal transformants pPcbh1–AnfaeA4 and pPcbh1–AnfaeB5 were picked out to prepare enzymes and release FA from wheat bran. After 7–9 days of fermentation, the highest FAE activities were detected in the crude extracts of the fungal transformants (Fig. 4). The maximum activity levels were 20.69 U/gds for AnFAEA and 15.08 U/gds for AnFAEB. The levels of FPase, xylanase and total extracellular proteins produced by the recombinant T. reesei strains were close to those of their parental strain (Fig. 5), suggesting that the expression of exogenous FAE genes did not significantly affect the normal enzyme production of T. reesei D-86271.

Production of feruloyl esterase by the recombinant T. reesei strains in solid-state fermentation. T. reesei transformants PcbhI–AnfaeA4 and PcbhI–AnfaeB5 were cultured in SSF medium at 30 °C for 2 weeks. The activities of FAEs in crude extract were assayed under optimum temperature and pH value, using MFA as substrate. Error bars represent standard deviations from three independent assays

Comparison of FAE, FPase, xylanase and extracellular proteins produced by T. reesei parental strain and the recombinant strains. The T. reesei strains (D-86271, pPcbhI–AnfaeA4 and pPcbhI–AnfaeB5) were cultured in SSF medium at 30 °C for 11 days. The activities of FAE, FPase and xylanase in crude extract were assayed as described in Materials and methods, and extracellular protein concentrations were determined with the BCA method. Error bars represent standard deviations from three independent assays
The optimum temperature of recombinant AnFAEB was lower than the reported value as described above. Release of FA from DSWB by the crude enzymes of transformant pPcbh1–AnfaeB5 was compared under different temperatures for 24 h. Under 35 °C, the highest level of FA was released from DSWB. The releasing efficiency had a slight decrement at lower or higher hydrolysis temperatures. By incubation at 50 °C, the liberated FA from DSWB was decreased by 7.51% (see supplementary Figure s2). Further, the enzymatic hydrolysis of DSWB was conducted by the crude extracts of different fungal strains at 45 or 35 °C (only for AnFaeB). The result of HPLC detection indicated that 62.9 and 52.2% of total FA were released by the crude enzymes of transformants pPcbh1–AnfaeA4 and pPcbh1–AnfaeB5, respectively (Fig. 6). The crude enzymes of A. niger 0913 released 46.3% FA under the same conditions. No detectable FA was released by the enzymes from T. reesei parental strain. Obviously, AnFAEA was more efficient than AnFAEB in releasing FA, which was in agreement with a previous report [30]. Different types of FAEs show preferences to synthesized or natural substrates. FAEA was more active than FAEB against wheat arabinoxylan, and FAEB was more active than FAEA against sugar beet pectin [30]. The constructed recombinant T. reesei strains could be used in preparation of various hydroxycinnamic acids from different plant cell walls.

Release of FA from de-starched wheat bran by crude enzymes from different T. reesei strains. In a 10 ml reaction buffer (pH 6.0), 20 mg crude enzymes with or without 30 U xylanase (TlXylanase, from T. longibrachiatum) per gram dry substrate (gds) was added to release FA from DSWB at 45 °C or 35 °C (only for AnFAEB) for 24 h. FA content was determined by HPLC analysis, and the release efficiency was calculated using alkali-extractable FA as 100%. Error bars represent standard deviations from three independent experiments
Endoxylanase plays a very important role in synergy with FAEs to enhance the liberation of FA from plant materials [14, 35]. In this study, the exogenous T. longibrachiatum xylanases enhanced the releasing efficiencies by 9.62, 5.50 and 6.88% for A. niger 0913, transformant pPcbh1–AnfaeA4 and transformants pPcbh1–AnfaeB5, respectively (Fig. 6). The different increments suggested that the xylanases (or enzyme system) of T. reesei were more efficient than those of A. niger in releasing FA from DSWB. Bartolome et al. found that various xylanases showed strong effect on the liberation of FA from DSWB by FAE-III, and the A. niger xylanases displayed a lower effect compared to other xylanases [36, 37]. At the same time, at least two kinds of FAEs exist in the crude extract of A. niger 0913 according to the expression characteristic of AnfaeA and AnfaeB genes in A. niger [30, 38]. The co-enzymatic hydrolysis with FAEA and FAEB decreased the release of FA from pre-treated WIP (water-insoluble pentosan of wheat arabinoxylan) by an unknown mechanism [30]. These reasons could explain why the recombinant T. reesei strains were more efficient than A. niger 0913 in releasing FA from wheat bran.
In conclusion, FAE-encoding genes AnfaeA and AnfaeB derived from A. niger 0913 were successfully expressed in T. reesei D-86271 under the control of the gpdA promoter and/or cbh I promoter. The cbh I promoter was more efficient than the gpdA promoter in the expression of the AnfaeA gene. Under solid-state fermentation, the maximum FAE productions were 20.69 U/gsd for AnFAEA and 15.08 U/gsd for AnFAEB by the corresponding recombinant strains. The optimum pH values and temperatures were 5.0 and 50 °C for the recombinant AnFAEA, and 6.0 and 35 °C for the recombinant AnFAEB. The recombinant fungal strains (especially containing the AnfaeA gene) could be used to efficiently release FA from de-starched wheat bran.
References
Perez-Rodriguez N, Moreira CD, Agrasar AT, Dominguez JM (2016) Feruloyl esterase production by Aspergillus terreus CECT 2808 and subsequent application to enzymatic hydrolysis. Enzyme Microb Tech 91:52–58
Gopalan N, Rodriguez-Duran LV, Saucedo-Castaneda G, Nampoothiri KM (2015) Review on technological and scientific aspects of feruloyl esterases: a versatile enzyme for biorefining of biomass. Bioresource Technol 193:534–544
Kumar N, Pruthi V (2014) Potential applications of ferulic acid from natural sources. Biotechnol Rep 4:86–93
Shirai A, Watanabe T, Matsuki H (2016) Inactivation of foodborne pathogenic and spoilage micro-organisms using ultraviolet—a light in combination with ferulic acid. Lett Appl Microbiol 64:96–102
Mathew S, Abraham TE (2006) Bioconversions of ferulic acid, an hydroxycinnamic acid. Crit Rev Microbiol 32:115–125
Abokitse K, Wu MQ, Bergeron H, Grosse S, Lau PCK (2010) Thermostable feruloyl esterase for the bioproduction of ferulic acid from triticale bran. Appl Microbiol Biot 87:195–203
Mathew S, Abraham TE (2004) Ferulic acid: an antioxidant found naturally in plant cell walls and feruloyl esterases involved in its release and their applications. Crit Rev Biotechnol 24:59–83
Gong YY, Yin X, Zhang HM, Wu MC, Tang CD, Wang JQ, Pang QF (2013) Cloning, expression of a feruloyl esterase from Aspergillus usamii E001 and its applicability in generating ferulic acid from wheat bran. J Ind Microbiol Biot 40:1433–1441
Topakas E, Vafiadi C, Christakopoulos P (2007) Microbial production, characterization and applications of feruloyl esterases. Process Biochem 42:497–509
Uraji M, Arima J, Inoue Y, Harazono K, Hatanaka T (2014) Application of two newly identified and characterized feruloyl esterases from Streptomyces sp. in the enzymatic production of ferulic acid from agricultural biomass. Plos One 9:1–7
Dilokpimol A, Makela MR, Aguilar-Pontes MV, Benoit-Gelber I, Hilden KS, de Vries RP (2016) Diversity of fungal feruloyl esterases: updated phylogenetic classification, properties, and industrial applications. Biotechnol Biofuels 9:231
Crepin VF, Faulds CB, Connerton IF (2004) Functional classification of the microbial feruloyl esterases. Appl Microbiol Biot 63:647–652
Wu H, Li H, Xue Y, Luo G, Gan L, Liu J, Mao L, Long M (2017) High efficiency co-production of ferulic acid and xylooligosaccharides from wheat bran by recombinant xylanase and feruloyl esterase. Biochem Eng J 120:41–48
Moukouli M, Topakas E, Christakopoulos P (2008) Cloning, characterization and functional expression of an alkalitolerant type C feruloyl esterase from Fusarium oxysporum. Appl Microbiol Biot 79:245–254
Wong DWS, Chan VJ, Liao H, Zidwick MJ (2013) Cloning of a novel feruloyl esterase gene from rumen microbial metagenome and enzyme characterization in synergism with endoxylanases. J Ind Microbiol Biot 40:287–295
Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, Chapman J, Chertkov O, Coutinho PM, Cullen D et al (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26:1193–1193
Bischof RH, Ramoni J, Seiboth B (2016) Cellulases and beyond: the first 70 years of the enzyme producer Trichoderma reesei. Microb Cell Fact 15:106
Peterson R, Nevalainen H (2012) Trichoderma reesei RUT-C30–thirty years of strain improvement. Microbiology 158:58–68
Ramoni J, Marchetti-Deschmann M, Seidl-Seiboth V, Seiboth B (2017) Trichoderma reesei xylanase 5 is defective in the reference strain QM6a but functional alleles are present in other wild-type strains. Appl Microbiol Biotechnol 101:4139–4149
Gong WL, Zhang HQ, Liu SJ, Zhang LL, Gao PJ, Chen GJ, Wang LS (2015) Comparative secretome analysis of Aspergillus niger, Trichoderma reesei, and Penicillium oxalicum during solid-state fermentation. Appl Biochem Biotech 177:1252–1271
Mandels M, Andreotti RE (1978) Problems and challenges in the cellulose to cellulase fermentation. Process Biochem 13:6–13
Long L, Ding D, Han Z, Zhao H, Lin Q, Ding S (2016) Thermotolerant hemicellulolytic and cellulolytic enzymes from Eupenicillium parvum 4–14 display high efficiency upon release of ferulic acid from wheat bran. J Appl Microbiol 121:422–434
Khang CH, Park SY, Rho HS, Lee YH, Kang S (2006) Filamentous fungi (Magnaporthe grisea and Fusarium oxysporum). Methods Mol Biol 344:403–420. Agrobacterium protocols, 2/e, volume 2. Edited by Wang K, Humana, Totowa
Ullán RV, Godio RP, Teijeira F, Vaca I, García-Estrada C, Feltrer R, Kosalkova K, Martín JF (2008) RNA-silencing in Penicillium chrysogenum and Acremonium chrysogenum: validation studies using beta-lactam genes expression. J Microbiol Meth 75:209–218
Mullins ED, Chen X, Romaine P, Raina R, Geiser DM, Kang S (2001) Agrobacterium-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathology 91:173–180
Long LK, Yang J, An Y, Liu G (2012) Disruption of a glutathione reductase encoding gene in Acremonium chrysogenum leads to reduction of its growth, cephalosporin production and antioxidative ability which is recovered by exogenous methionine. Fungal Genet Biol 49:114–122
de Cassia Pereira J, Marques NP, Rodrigues A, de Oliveira TB, Boscolo M, da Silva R, Gomes E, Martins DAB (2015) Thermophilic fungi as new sources for production of cellulases and xylanases with potential use in sugarcane bagasse saccharification. J Appl Microbiol 118:928–939
Turner BL (2010) Variation in pH optima of hydrolytic enzyme activities in tropical rain forest soils. Appl Environ Microbiol 76:6485–6493
Levasseur A, Navarro D, Punt PJ, Belaich JP, Asther M, Record E (2005) Construction of engineered bifunctional enzymes and their overproduction in Aspergillus niger for improved enzymatic tools to degrade agricultural by-products. Appl Environ Microb 71:8132–8140
de Vries RP, VanKuyk PA, Kester HCM, Visser J (2002) The Aspergillus niger faeB gene encodes a second feruloyl esterase involved in pectin and xylan degradation and is specifically induced in the presence of aromatic compounds. Biochem J 363:377–386
Faulds CB (2010) What can feruloyl esterases do for us? Phytochem Rev 9:121–132
Levasseur A, Saloheimo M, Navarro D, Andberg M, Monot F, Nakari-Setala T, Asther M, Record E (2006) Production of a chimeric enzyme tool associating the Trichoderma reesei swollenin with the Aspergillus niger feruloyl esterase A for release of ferulic acid. Appl Microbiol Biot 73:872–880
Levasseur A, Benoit I, Asther M, Asther M, Record E (2004) Homologous expression of the feruloyl esterase B gene from Aspergillus niger and characterization of the recombinant enzyme. Protein Expres Purif 37:126–133
Kroon PA, Faulds CB, Williamson G (1996) Purification and characterization of a novel esterase induced by growth of Aspergillus niger on sugar-beet pulp. Biotechnol Appl Biochem 23:255–262
Faulds CB, Williamson G (1995) Release of ferulic acid from wheat bran by a ferulic acid esterase (FAE-III) from Aspergillus niger. Appl Microbiol Biotechnol 43:1082–1087
de Vries RP, Michelsen B, Poulsen CH, Kroon PA, van den Heuvel RH, Faulds CB, Williamson G, van den Hombergh JP, Visser J (1997) The faeA genes from Aspergillus niger and Aspergillus tubingensis encode ferulic acid esterases involved in degradation of complex cell wall polysaccharides. Appl Environ Microbiol 63:4638–4644
Bartolomé B, Faulds CB, Tuohy M, Hazlewood GP, Gilbert HJ, Williamson G (1995) Influence of different xylanases on the activity of ferulic acid esterase on wheat bran. Biotechnol Appl Biochem 22:65–73
de Vries RP, Visser J (1999) Regulation of the feruloyl esterase (faeA) gene from Aspergillus niger. Appl Environ Microbiol 65:5500–5503
Acknowledgements
We are grateful to Prof. Gang Liu (Institute of Microbiology, CAS) for providing the plasmid pAg1-H3 and Agrobacterium tumefaciens AGL-1. This work was supported by grants from a 948 Research Project (No. 2013-4-16) from the State Forestry Administration of China, the Natural Science Fund for Provincial Colleges and University of Jiangsu Province, China (15KJB220003), the Research Fund for the Advanced Talents, Nanjing Forestry University (GXL201311) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Long, L., Zhao, H., Ding, D. et al. Heterologous expression of two Aspergillus niger feruloyl esterases in Trichoderma reesei for the production of ferulic acid from wheat bran. Bioprocess Biosyst Eng 41, 593–601 (2018). https://doi.org/10.1007/s00449-018-1894-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-018-1894-3