
Abstract
Exposure to organic dust (OD) in agriculture is known to cause respiratory symptoms including loss of lung function. OD exposure activates multiple signaling pathways since it contains a variety of microbial products and particulate matter. Previously, we have shown how OD exposure leads to the secretion of HMGB1 and HMGB1-RAGE signaling, and how this can be a possible therapeutic target to reduce inflammation. Cellular mitochondria are indispensable for homeostasis and are emerging targets to curtail inflammation. Recently, we have also observed that OD exposure induces mitochondrial dysfunction characterized by loss of structural integrity and deficits in bioenergetics. However, the role of HMGB1 in OD-induced mitochondrial dysfunction in human bronchial epithelial (NHBE) cells remains elusive. Therefore, we aimed to study whether decreased levels of intracellular HMGB1 or antibody-mediated neutralization of secreted HMGB1 would rescue mitochondrial dysfunction. Single and repeated ODE exposure showed an elongated mitochondrial network and cristolysis whereas HMGB1 neutralization or the lack thereof promotes mitochondrial biogenesis evidenced by increased mitochondrial fragmentation, increased DRP1 expression, decreased MFN2 expression, and increased PGC1α expression. Repeated 5-day ODE exposure significantly downregulated transcripts encoding mitochondrial respiration and metabolism (ATP synthase, NADUF, and UQCR) as well as glucose uptake. This was reversed by the antibody-mediated neutralization of HMGB1. Our results support our hypothesis that, in NHBE cells, neutralization of ODE-induced HMGB1 secretion rescues OD-induced mitochondrial dysfunction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Large-scale animal feeding operations generate many on-site contaminants that pose a threat to workers, animals, and the environment. Agriculture workers who are exposed to the airborne contaminants are at an increased risk of developing respiratory and other symptoms (Nordgren and Charavaryamath 2018). The on-site contaminants in animal feeding operations mainly include airborne organic dust (OD), gases, and other microbial components. The OD is a complex mixture of various microbial-associated molecular patterns (MAMPs) including endotoxin, peptidoglycans, Gram-positive bacterial components, (1 → 3)-β-D-glucans, and fungi (Poole and Romberger 2012; Nordgren and Charavaryamath 2018; Poole et al. 2019; Kelly and Poole 2019). Repeated exposure of workers to OD has been associated with respiratory disease development and severity. Exposed workers report a variety of respiratory symptoms and suffer from asthma, chronic bronchitis, and chronic obstructive pulmonary disease (COPD) (Charavaryamath and Singh 2006; May et al. 2012; Poole and Romberger 2012; Wunschel and Poole 2016; Warren et al. 2019). Due to the complex nature of the OD, several research studies have targeted various pattern recognition receptors (PRRs) and the respective downstream regulators. For example, a few groups have targeted various PRRs and signaling molecules including TLR2, TLR4, NOD2, protein kinase C, and MyD88 (Poole et al. 2007, 2010, 2011; Charavaryamath et al. 2008; Bailey et al. 2008; An et al. 2020). Recently, we examined various kinome (set of protein kinases) signaling pathways and showed a complex host response (Nath Neerukonda et al. 2018).
Airway epithelial cells are central to the first line of defense in the lungs (Whitsett and Alenghat 2015). Exposure of lung epithelial cells to OD has been shown to trigger oxidative stress, cytokine release, cellular dysfunction, and cell death (Charavaryamath and Singh 2006; Poole and Romberger 2012; Sethi et al. 2017; Bhat et al. 2019). Mitochondria (mt) are referred to as the powerhouse of the cells, and emerging data have identified a pivotal role of mitochondria in several inflammatory airway diseases (Prakash et al. 2017). Therefore, mitochondria represent an attractive therapeutic target in rescuing airway epithelial cells from the negative effects of chronic exposure to OD. Alterations in mitochondrial morphology and function have been documented in airway epithelial cells in COPD and asthma (Schumacker et al. 2014; Cloonan and Choi 2016; Piantadosi and Suliman 2017; Prakash et al. 2017; Fetterman et al. 2017; Aghapour et al. 2019). In addition to mitochondrial dysfunction, mitochondrial damage and release of mitochondrial components either into the cytosol or extracellularly induces inflammation (Cloonan and Choi 2012, 2016; Eisner et al. 2018; Aghapour et al. 2019; Cloonan et al. 2020). Mitochondrial damage-associated molecular patterns (mtDAMPs) perform as signaling molecules and activate various PRRs and induce a myriad of inflammatory cascades (Rubartelli and Lotze 2007; Zhang et al. 2010; Cloonan and Choi 2012). In our recent study, we showed increased mitochondrial fusion and mitophagy, along with the leakage of mtDAMPs such as mitochondrial DNA (mtDNA) and mitochondrial transcription factor A (TFAM) upon exposure of THP1 cells to OD extract (ODE) (Mahadev Bhat et al. 2021). Recent studies have shown that the release of TFAM along with mtDNA amplifies the release of TNFα and type 1 interferon, thus promoting a sterile inflammation via the cGAS-STING pathway (Cantaert et al. 2010; CHAUNG et al. 2012; Julian et al. 2013). Collectively, understanding the mechanism of how mitochondria promote inflammation can help in developing effective mitochondria-targeted therapies to curtail OD exposure-induced airway inflammation.
High mobility group box 1 (HMGB1) is known to be a key player in the induction of sterile inflammation and a possible therapeutic target (Andersson and Tracey 2011). HMGB1 is an endogenous nucleoprotein, evolutionarily conserved, ubiquitously present, and maintains nuclear homeostasis. This protein is secreted into the extracellular environment and acts as a prototypic DAMP and is important for oxidative stress response (Yang et al. 2015; Ugrinova and Pasheva 2017). Extracellular HMGB1 is a delayed mediator of inflammation when compared to other pro-inflammatory mediators and is shown to stimulate the release of multiple pro-inflammatory cytokines including tumor necrosis factor (TNF), interleukin (IL)-1, IL-6, IL-8, and macrophage inflammatory protein (MIP)-1 (Yang et al. 2015; Bhat et al. 2019; Massey et al. 2019, p. 1). The use of neutralizing monoclonal anti-HMGB1 antibodies in vivo has been shown to ameliorate tissue injury and reduce lethality (Zhou et al. 2009; Bhat et al. 2019). Contemporary publications have demonstrated that siRNA-mediated knockdown of HMGB1 in macrophages and dendritic cells suppressed the release of HMGB1 and reduced the cytokine storm (Qin et al. 2006; Ye et al. 2012). We have previously shown the pro-inflammatory effects of extracellularly secreted HMGB1 on bronchial epithelial cells on exposure to ODE and how its neutralization abrogates OD-induced inflammation (Nath Neerukonda et al. 2018; Bhat et al. 2019; Massey et al. 2019). Qi et al. directly correlated the alterations in mitochondrial function to the loss of HMGB1 and further demonstrated a significant decrease in basal OXPHOS and glycolysis along with decreased ATP output (Qi et al. 2015). Also, a lack of HMGB1 has been shown to promote mitochondrial fragmentation and loss of mitochondrial membrane potential (Tang et al. 2011). Therefore, we have raised pertinent questions as to what impact HMGB1 would have on mitochondrial morphology and function in bronchial epithelial cells on exposure to ODE.
In the current manuscript, we tested a hypothesis that ODE-exposure of primary normal human bronchial epithelial (NHBE) cells induces mt-damage, dysfunction, and targeting HMGB1 is protective. This study examined the impact of HMGB1 on mitochondrial biogenesis and function on ODE exposure by using NHBE cells. We tested this hypothesis by using a single (acute) and repeated (chronic) ODE exposure model with siRNA-mediated knockdown of HMGB1 and by neutralization of secreted HMGB1, respectively. Here, we demonstrated that ODE exposure promotes the release of mtDAMPs and activation of cGAS-STING-mediated signaling while knockdown of HMGB1 promotes mitochondrial biogenesis on single exposure to ODE. In the repeated exposure model, neutralization of extracellular HMGB1 is protective and maintains mitochondrial morphology, as well as upregulates pathways involved in mitochondrial respiration and biogenesis. Using an air–liquid interface (ALI) model, we show that neutralization of extracellular HMGB1 rescues ODE exposure-induced damages to epithelial integrity via its effect on tight junction proteins.
Materials and methods
Chemicals and reagents
We purchased NHBE - growth medium (PneumaCult-Ex media; BEGM) and differentiation media (PneumaCult-ALI media) from Stemcell Technologies. MTT, CM-H2DCFDA, 2-NBDG, MitoTracker green, and MitoSOX Red dyes were purchased from Invitrogen (Thermo Fisher Scientific), and Rhod-2AM dye was obtained from Abcam. Griess’ reagent was purchased from Sigma–Aldrich. Antibodies used and their details have been listed in Supplementary Tables 4 and 5, respectively. Primers used and their sequences are listed in Supplementary Table 2 and were synthesized at the Iowa State University’s DNA facilities. TaqMan® Gene Expression Assay was purchased from Invitrogen (Thermo Fisher Scientific). HMGB1 siRNA kit was purchased from Integrated DNA Technologies (IDT), and anti-HMGB1 neutralizing antibody sample (Batch # Q2g7-47C, stock concentration 1.8 mg/mL) was a generous gift from Dr. Kevin Tracey’s laboratory (Feinstein Institutes for Medical Research, Northwell Health, NY).
Organic dust extract preparation
Organic dust (OD) was collected and an aqueous extract was prepared as previously described (Romberger et al. 2002; Bhat et al. 2019). Settled surface dust samples from swine housing facilities were collected, and 1 g of the dust was placed into sterile Hank’s balanced salt solution (10 mL; Gibco). The solution was incubated for one hour at room temperature and centrifuged for 20 min at 1365 × g to remove the larger coarse particles and microbes. The final supernatant was filter-sterilized (0.22 μm), a process that also removes coarse particles. Stock (100%) ODE aliquots were frozen at − 20 °C until use in experiments. The filter-sterilized organic dust extract (ODE) samples were considered 100% and diluted to 1–5% (v/v) before use in our experiments.
Endotoxin estimation
The concentration of endotoxin in the ODE samples was measured using the Pyrochrome® chromogenic endotoxin assay kit (Associates of Cape Cod, Inc., East Falmouth, MA) (Table 1). First, the samples were diluted in a ratio of 1:10 in endotoxin-free water. The samples along with reconstituted pyrochrome lysate were added to an endotoxin-free 96-well plate in a sample to lysate ratio of 1:4. Based on the manufacturer’s recommendation, the standard was reconstituted and added to the plate in a sample to lysate ratio of 1:4. The microplate was incubated at 37 °C with shaking and the absorbance was read at 405 nm (every 10 min, three readings over a total of 30 min) using the Gen 5™ software in the BioTek® ELx808™ spectrophotometer.
Cell culture and treatments
De-identified human primary airway epithelial cells were obtained from Mayo Clinic, Rochester, MN. These cells were collected at Mayo Clinic under an approved Institutional Review Board (IRB) protocol. The lung tissues from patients undergoing thoracic surgeries for focal, non-infectious causes were collected. A trained pathologist identified the normal lung areas, and airway epithelial cells were isolated from the third to sixth generation human bronchi. Iowa State University’s Institutional Review Board considered these de-identified cells as exempt from approval. De-identified NHBE cells were also received through Dr. Kristina L. Bailey (University of Nebraska Medical Center, Omaha, NE). The NHBE cells were isolated from the human donors through the Live on Nebraska, an organ, and tissue donation program. All the donors were deceased, and an informed consent from the relatives was obtained by the Live on Nebraska. These de-identified NHBE cells were exempt from the Iowa State University’s Institutional Review Board (IRB) approval. All work was performed under an approved protocol from Iowa State University’s Institutional Biosafety Committee (IBC) approved protocol (IBC 19–004). Cells used were derived from a total 3 donors (n = 3) to perform all the experiments in this manuscript. For each of the experiments, all three donors were used with technical replicates for certain assays. All data represented here has been normalized to their respective donor controls which were time matched with the treatments to keep the data consistent and decrease variability due to heterogeneity between donors.
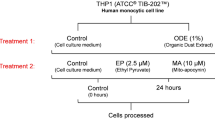
NHBE cells were seeded onto type-I bovine collagen (30 μg/mL; Stemcell Technologies)-coated T-75 flasks. Cells were grown submerged in BEGM (PneumaCult-EX) supplemented with 50X growth supplement (Stemcell Technologies), 100 IU/mL penicillin, 100 μg/mL streptomycin (Gibco), and 1 µg/mL of Amphotericin B (Sigma–Aldrich). The cells were incubated in a humidified chamber with 5% CO2 at 37 ˚C until approximately around 60–70% confluence was achieved. All groups with treatment details are outlined in Fig. 1.

ODE exposure of NHBE cells and air–liquid interface culture. Three experimental designs were used to study the impact of HMGB1 on mitochondria during ODE exposure. For acute exposure, primary NHBE cells were transfected with HMGB1 targeted siRNA to induce knockdown followed by exposure to either media (control) or ODE (1%) for 24 h (a). For chronic exposure, NHBE cells were treated with media alone (control) or ODE (1%) followed by media (control) or HMGB1 neutralization antibody (10 µg/mL) for 8 h per day for 5 days (b). Air–liquid interface (ALI) culture model was developed by seeing NHBE cells onto a semi-permeable membrane to develop, and the differentiated cells were exposed to ODE, with or without HMGB1 neutralization for 1 h per day for 5 days (c)
The stock and working concentrations of the treatments are outlined in Table 2. Our in vitro models of single and repeated (five days) ODE exposure and treatments are represented in Fig. 1a and b, respectively. Cells were treated with either medium (control) or ODE (1% v/v) after HMGB1-specific siRNA-mediated knock-down or co-treatment with anti-HMGB1 antibody for 24 h (acute exposure) or 8 h a day for 5 days (chronic exposure), respectively, with corresponding time-matched controls. Following treatments, samples were processed for various assays.
Air–liquid interface culture and treatment
Following subculture, NHBE cells (from passage 2 to 3) were seeded onto 10.5-mm trans-well inserts (polyester membrane inserts with 0.4-μm sized pores; Corning) coated with type-I bovine collagen (30 μg/mL; Stemcell Technologies, Vancouver, Canada) in 12-well plates. Cells were seeded at a density of ~ 5 × 103/mm2 in PneumaCult-EX expansion media (Stemcell Technologies) onto the apical side of the insert, with the media in the basal chamber as well. The cells were maintained in submerged culture conditions for 2–3 days in a humidified chamber with 5% CO2 at 37 ˚C until a confluent monolayer was developed. Fresh medium (0.5 mL in the apical chamber and 1.0 mL in the basal chamber) was added every 48 h to replace the spent media. Following confluence and development of monolayer, the expansion media in the apical and basal side was carefully removed, and an ALI was developed by addition of the PneumaCult-ALI differentiation media (Stemcell Technologies) in the basal chamber alone and incubated in a humidified chamber with 5% CO2 at 37 ˚C. Fresh medium (1.0 mL in the basal chamber) was replenished every 48 h, and the apical surface of the monolayer was washed with DPBS twice every 48 h to prevent mucus buildup. Post airlift the cells were maintained for 20 days and differentiation was assessed by observing cilia development using a brightfield inverted microscope (40 × objective, EVOSM5000 Imaging System, Thermo Fisher Scientific) and measuring FoxJ1 and Muc5B mRNA and protein expression (data not shown).
Post differentiation, the apical surface of the cells was treated with either medium (control) or ODE (1% v/v) followed by a co-treatment with either anti-HMGB1 neutralization antibody (10 μg/mL) or isotype-matched IgG controls (corresponding time-matched control) for 1 h per day for 5 days. Following the treatments, samples were processed after 5 days for various assays. The exposure and treatments are outlined in Fig. 1c.
siRNA-mediated knockdown of HMGB1
Three (R1, R2, and R3) custom-designed double-stranded anti-HMGB1 siRNAs (DsiRNA), scrambled RNA (negative control, NC), HPRT1 DsiRNA (positive control), and a fluorescent transfection efficiency control (TYE563) were purchased (Integrated DNA Technologies Inc., Coralville, IA) to maximize the probability of achieving successful HMGB1 knockdown. Lipofectamine 2000 (Thermo Fisher) was used to transfect DsiRNA into the NHBE cells. We have listed the sequences of siRNAs and NC siRNA in Supplementary Table 1.
To carry out transfection with various siRNAs, NHBE cells were first cultured in the BEGM without antibiotics. For each transfection, 10 nM of (DsiRNA) for HMGB1, scrambled siRNA (negative control), HPRT1 DsiRNA (positive control), and a fluorescent transfection control (TYE563) were diluted in Opti-MEM media and gently mixed with lipofectamine 2000 according to the manufacturer’s instructions and protocols as per a published manuscript (Chang et al. 2012). Following incubation for 20 min at room temperature, the transfection mixture was added to the cells and the cells were further incubated at 37 °C for 24 h. Knockdown was authenticated by qRT-PCR and western blot analysis of the target gene and protein, respectively (Supplementary Fig. 1). Transfection was verified by fluorescent microscopy for TYE563 (fluorescent transfection control), and images were acquired using Nikon Eclipse TE2000-U inverted fluorescence microscope (Nikon, Tokyo, Japan). Images were processed using HCImage live 4 software (Hamamatsu Corporation, Sewickley, PA).
Cell viability assay and MTT assay
Before executing each experiment, cell viability was quantified. Live/dead cell count was determined by 4% trypan blue dye (EMD Millipore) exclusion, and percentage viability was calculated. The population of cells with more than 95% viability was used for the experiments.
The MTT assay has been commonly used in the estimation of LC50 and cell viability by measuring the formazan produced when mitochondrial dehydrogenase enzymes cleave the tetrazolium ring (Latchoumycandane et al. 2005). We used the MTT assay to determine the LC50 of the treatment groups in NHBE cells listed in Fig. 1a and b. Cells were seeded (20,000 cells/well) in a 96-well culture plate and treated for 24 h and 8 h/day for 5 days. After the treatment, the cells were washed with PBS and incubated with 200 µl of 0.25% (w/v) MTT in BEGM for 3 h at 37 °C. The supernatant was removed, and MTT crystals were solubilized in 200 µl of dimethyl sulfoxide. Mitochondrial activity was measured by quantifying fluorescence using SpectraMax spectrophotometer (Molecular Devices Corporation) at 570 nm with the reference wavelength at 630 nm.
Transmission electron microscopy
For transmission electron microscopy (TEM), cells were seeded (1 × 106 cells/well) in 12-well plates with poly D-lysine hydrobromide (Sigma–Aldrich, St. Louis, MO)-coated coverslips and chronically exposed to the treatments as outlined in Fig. 1b. Post-treatment NHBE cells were washed twice with DPBS and fixed with 3% glutaraldehyde (w/v) and 1% paraformaldehyde (w/v) in 0.1 M cacodylate buffer, pH 7.4 for 48 h at 4 °C. Coverslips were rinsed 3 times in 0.1 M cacodylate buffer and then post-fixed in 1% osmium tetroxide in 0.1 M cacodylate buffer for 1 h (room temp.). The samples were rinsed in deionized distilled water and en bloc stained with 2% aqueous uranyl acetate for 30 min, dehydrated in a graded ethanol series, cleared with ultra-pure acetone, and infiltrated and embedded using EmBed (EPON) formula epoxy resin (Electron Microscopy Sciences, Ft. Washington, PA) by inverting onto BEEM® capsules. Resin blocks were polymerized for 48 h at 65 °C. Coverslips were separated from the resin by using liquid nitrogen, trapping the cell monolayer in the resin. Thick and ultrathin sections were prepared using a Leica UC6 ultramicrotome (North Central Instruments, Minneapolis, MN). Ultrathin sections were collected onto the copper grids and images were captured using a JEOL 2100 scanning and transmission electron microscope (Japan Electron Optic Laboratories, Peabody, MA) with a Gatan OneView 4 K camera (Gatan Inc., Pleasanton, CA).
Immunofluorescence microscopy
NHBE cells were seeded (1 × 106 cells/well) in 12-well plates with Poly D-lysine hydrobromide (Sigma–Aldrich, St. Louis, MO)-coated coverslips and exposed to the treatments as outlined in Fig. 1a and b. At the respective time points, the media was removed, and cells were washed with 1 × PBS. Cells were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and washed with PBS. Cells were blocked for an hour using a blocking solution containing 10% normal donkey serum (EMD Millipore, Burlington, MA), 0.2% Triton X 100, and PBS. Coverslips with cells were incubated with anti-HMGB1 (1:1000 dilution, rabbit polyclonal) antibody in antibody diluent solution (2.5% normal donkey serum, 0.25% sodium azide, 0.2% Triton X 100, PBS) (Abcam, Cambridge, MA) with overnight incubation at 4 °C. Next, coverslips were incubated with donkey anti-rabbit biotin-conjugated secondary antibody (1:400, diluted in antibody diluent, HMGB1) (Jackson ImmunoResearch, West Grove, PA) for an hour at room temperature. Next, 1:300 dilution of streptavidin Cy3 in PBS (HMGB1) was added. Coverslips were mounted onto slides using VECTASHIELD antifade mounting medium with 4',6-diamidino-2-phenylindole, dihydrochloride (DAPI, Vector Labs, Burlingame, CA), and images were captured using Nikon Eclipse TE2000-U inverted fluorescence microscope (Nikon, Tokyo, Japan). Images were processed using HCImage live 4 software (Hamamatsu Corporation, Sewickley, PA).
Subcellular fractionation
Whole-cell and subcellular protein lysate extractions (cytosol and MT) were conducted at 4 °C using cold reagents. For whole-cell protein lysates, cell pellets were subjected to lysis using RIPA buffer with HALT protease and phosphatase inhibitor (Thermo Fisher Scientific). Subcellular fractionation of cell pellets for isolation of mitochondria was carried out using the Mitochondria Isolation Kit for Cultured Cells (Thermo Fisher Scientific) as per the manufacturer’s instructions. The cytosolic fraction and isolated mitochondria were lysed with RIPA buffer (with protease and phosphatase inhibitors) for 30 min at 4 °C and periodic sonication on ice, followed by centrifugation to collect lysate. Protein concentrations of the fractions were quantified by Bradford assay (Bio-Rad) and stored at − 80 °C until use.
mtDNA isolation and long-range PCR
To assess mitochondrial DNA (mtDNA) leakage into cellular cytosol, mtDNA was isolated from mitochondria-free cytosolic fraction of the cells using the Genomic DNA Purification kit (Thermo Fisher Scientific) as per the manufacturer’s instructions. The purity and concentration of the isolated DNA were quantified using NanoVue Plus spectrophotometer (GE Healthcare, UK). Due to low concentrations, the mtDNA was first amplified using long-range PCR, and the primers used are listed in Supplementary Table 2. PCR reactions were performed at 94 °C for 1 min followed by 30 cycles at 98 °C for 10 s, 60 °C for 40 s, and 68 °C for 16 min and a final elongation for 10 min (Liu et al. 2015). The presence of mtDNA was confirmed by separating the PCR product by electrophoresis on a 0.8% agarose gel stained with ethidium bromide. We ensured that there were equal amounts of template mtDNA in each sample (for qPCR reaction) by adjusting the concentration of amplified mtDNA that was obtained.
Real-time — qPCR
Total RNA was isolated using TRIzol extraction methods (Seo et al. 2014) and RNA concentration was measured using NanoVue Plus spectrophotometer (GE Healthcare, UK). One microgram of RNA was used to synthesize cDNA using the high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific) following the manufacturer’s instructions. qRT-PCR was performed using 500 ng of cDNA in a 10 μL reaction volume for each target in triplicates. The housekeeping genes 16S rRNA (for mtDNA fold change) and 18S rRNA (Thermo Fisher Scientific) were used. No-template controls and dissociation curves were run for all reactions to exclude cross-contamination. The primers for genes of interest listed in Supplementary Table 2 were synthesized at Iowa State University’s DNA Facility. The qRT-PCR reactions were run in a QuantStudio-3™ (Thermo Fisher Scientific) detection system, and the data was analyzed using 2−ΔΔCT method (Livak and Schmittgen).
Targeted gene array
qRT-PCR using TaqMan® Gene Expression Assays (Life Technologies, Carlsbad, CA) was performed for 96 targets specific for human mitochondrial energy metabolism listed in Supplementary Table 3. Five hundred nanograms of cDNA in a 10 μL reaction volume was prepared for each target in duplicates. The reaction conditions were as follows: 50 °C for 2 min, 95 °C for 2 min, followed by 40 cycles at 95 °C for 1 s, and 60 °C for 20 s. Data analysis was performed using the relative quantification app on Thermo Fisher cloud and analyzed using 2−ΔΔCT method (Livak and Schmittgen 2001). The genes with a fold change of ≥ 10 and ≤ 10 were selected and represented on a heat map. Using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, upregulated and downregulated pathways with significantly altered mRNA transcripts with FDR corrected p-value cutoff < 0.05 were selected and represented by gene counts.
Western blot analysis
Lysates (whole cell, cytosol, and mt containing equal amounts of protein (20 μg/sample), along with a molecular weight marker (Bio-Rad), were run on 10–15% sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE) as previously described (Bhat et al. 2019). Proteins were transferred to a nitrocellulose membrane and nonspecific binding sites were blocked with Licor Odyssey blocking buffer. The membranes were then incubated with different primary antibodies and dilutions listed in Supplementary Table 4. Next, membranes were incubated with one of the following secondary antibodies: Alexa Fluor 680 goat anti-mouse, Alexa Fluor 680 donkey anti-rabbit, or IRDye 800CW donkey anti-rabbit (1:10,000; LI-COR) listed in Supplementary Table 5. To confirm equal protein loading, blots were probed with relevant housekeeping proteins listed in Supplementary Table 4. Western blot images were captured using Odyssey® CLx IR imaging system (LI-COR Biotechnology), and analysis was performed using ImageJ program (National Institutes of Health).
Intracellular reactive oxygen species
Intracellular reactive oxygen species (ROS) production was measured using chloromethyl derivative of dichlorodihydrofluorescein diacetate (CM-H2DCFDA) (Thermo Fisher Scientific, USA). A working solution of 10 µM of DCFDA in DPBS was used. NHBE cells (50,000 cells/well) were seeded in a 96 well plate and incubated in a 5% CO2 incubator to reach confluence. The cells were incubated with CM-H2DCFDA working solution at 37 °C for 30 min, following treatments as outlined in Fig. 1a. The cells were then washed, and fluorescence intensity of the oxidized form of H2DCFDA was measured at excitation/emission wavelengths of 488/535 nm using SpectraMax spectrophotometer (Molecular Devices Corporation). The results were expressed as mean fluorescence intensity relative to control.
Mitochondrial morphology and mitochondrial superoxide
NHBE cells were seeded (50,000 cells/well) in a 96-well culture plate and treated as outlined in Fig. 1a and b. Following treatment, the media was removed and MitoTracker green (200 nM), and MitoSOX red (5 μM) dye diluted in BEGM was added into each well and incubated at 37 °C for 15 min. Following incubation, the cells were washed with DPBS and fluorescence intensity was measured by spectrophotometer reading taken at excitation/emission wavelengths of 485/520 nm and 510/580 nm, respectively, SpectraMax spectrophotometer (Molecular Devices Corporation). The results were expressed as percentage fluorescence relative to control.
NHBE cells were also seeded (1 × 106 cells/well) in 12-well plates with poly D-lysine hydrobromide (Sigma–Aldrich, St. Louis, MO)-coated coverslips and exposed to the treatments as outlined in Fig. 1a and b. At the respective time points, the media was removed, and cells were washed with DPBS. These were stained with MitoTracker Red CMXRos (200 nM) and incubated at 37 °C for 15 min. The cells were washed with DPBS and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and washed with PBS again. Coverslips were mounted onto slides using VECTASHIELD antifade mounting medium with 4',6-diamidino-2-phenylindole, dihydrochloride (DAPI, Vector Labs, Burlingame, CA), and imaged using Nikon Eclipse TE2000-U inverted fluorescence microscope (Nikon, Tokyo, Japan). Images were processed using HCImage live 4 software (Hamamatsu Corporation, Sewickley, PA).
Mitochondrial glucose uptake
Cellular glucose uptake by NHBE cells was measured using fluorescent d-glucose analog 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy-D-glucose (2-NBDG) (Zou et al. 2005). Cells were seeded (50,000 cells/well) in a 96-well culture plate and treated as outlined in Fig. 1a and b. After treatment, the media was removed, and the cells were glucose starved by adding Krebs–Ringer Buffer Solution (KRB) solution for 15 min. Cells were washed with DPBS, and 100 μL of 50 μg/mL 2-FITC-labeled NBDG in BEGM was added into each well and incubated at 37 °C for 20 min. Following incubation, the cells were washed with DPBS, and fluorescence intensity was measured by spectrophotometer reading taken at an excitation/emission wavelengths of 466/550 nm using SpectraMax spectrophotometer (Molecular Devices Corporation). The results were expressed as percentage fluorescence relative to control.
Mitochondrial calcium influx
Mitochondrial calcium influx ([Ca2+]mito) in NHBE cells was measured using the Rhod-2AM dye. After treatment, the mitochondria were isolated using the Mitochondria Isolation Kit for Cultured Cells (Thermo Fisher Scientific). Using Bradford assay, we measured the protein concentration so as to maintain consistency in the number of mitochondria loaded into the wells of a 96-well plate. A protein concentration of 100 µg was loaded into each well and Rhod-2AM (Abcam, 10 µM) dye was added and incubated at 37 °C for 30 min to stain the mitochondria. The plate was later read at an excitation/emission wavelengths of 552/581 nm using SpectraMax spectrophotometer (Molecular Devices Corporation).
NHBE cells were also seeded (1 × 106 cells/well) in 12-well plates with poly D-lysine hydrobromide (Sigma–Aldrich, St. Louis, MO)-coated coverslips and exposed to the treatments as outlined in Fig. 1a and b. At the respective time points, the media was removed, and cells were washed with DPBS. These were stained with Rhod-2AM (Abcam, 10 µM) and incubated at 37 °C for 30 min. The cells were washed with DPBS and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and washed with PBS again. Coverslips were mounted onto slides using VECTASHIELD antifade mounting medium with 4',6-diamidino-2-phenylindole, dihydrochloride (DAPI, Vector Labs, Burlingame, CA), and imaged using EVOS M5000 imaging system (Thermo Fisher Scientific).
Griess assay
Griess assay was performed as described (Gordon et al. 2011) to indirectly measure nitric oxide (NO) as direct measurement of NO using electron paramagnetic resonance spectroscopy is expensive, cumbersome, and semi-quantitative (Csonka et al. 2015). Concentration of secreted nitric oxide was measured (representing reactive nitrogen species (RNS)) as nitrite levels in cell culture media using Griess reagent (Sigma–Aldrich) and sodium nitrite standard curve, prepared using a stock solution of 200 µM. The assay was performed in a 96 well-plate, and absorbance was measured at 550 nm using SpectraMax spectrophotometer (Molecular Devices Corporation). The results were expressed as µM concentration of nitrite secreted.
TEER
TEER is the potential difference across the epithelium, which is a measure of the tightness of the cell–cell contacts within the epithelium, and TEER is measured using a pair of electrodes (Srinivasan et al. 2015). NHBE cells were plated on 10.5-mm trans-well inserts in a 12-well plate and allowed to grow. Cell confluence was monitored by TEER measurements with the epithelial Voltohmmeter 2 (EVOM2, WPI, Sarasota, FL). The value obtained is an indication of the integrity of the epithelial cell monolayers. Once TEER value reached a steady-state, treatments outlined in Fig. 1c were added to the apical surface of the cells, and TEER measurements were performed every 12 h for 5 days. The mean of four measurements per insert was calculated. The electrical resistance of inserts without cells was subtracted from all samples, and the resistance values obtained were multiplied with the surface area of the inserts for measuring the TEER values (Ω cm2).
Statistical analysis
Data analysis was performed using GraphPad Prism 8.0 software (La Jolla, CA, USA). Raw data were analyzed with either Student’s t-test or using one-way ANOVA, and Tukey’s post hoc test was performed to compare all the treatment groups. A p-value of < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, and * indicates different from control. Samples for all the assays were derived from the same experiment and were processed in parallel.
Results
Endotoxin levels in ODE samples
Endotoxin activity of the ODE samples is listed in Table 1. From the seven samples, we found that endotoxin values ranged from 0.8067 ± 0.0008 to 1.433 ± 0.02 EU/mL (Table 1). Sample # 7 (endotoxin level 1.263 ± 0.7 EU/mL) was used for all experiments listed in this manuscript.
Suppression of HMGB1 expression in ODE-exposed NHBE cells promotes mitochondrial fission
To assess the impact of HMGB1 on mitochondria, primary NHBE cells were transfected with HMGB1 targeted siRNA to induce knockdown. Successful transfection was first confirmed by using a fluorescently tagged DsiRNA (DsiRNA TYE 563; 10 nmol) that produced positive immunofluorescence in the cytoplasm of NHBE cells at 24 h post-transfection (Supplementary Fig. 1a-a’’). After establishing an optimal transfection protocol, NHBE cells were transfected with three HMGB1 targeting siRNA sequences (R1, R2, and R3; 10 nmol each) and a negative control siRNA (scrambled DsiRNA; 10 nmol) and incubated for 24 h. The knockdown was confirmed by western blot and qPCR of samples from all the groups. Quantified data showed a decrease in HMGB1 levels for all three sequences and no change in the negative control (Supplementary Fig. 1b–d). The siRNA sequence that induced the highest percentage of knockdown (siRNA.R3) was selected to perform all the experiments.
NHBE cells with and without HMGB1 knockdown were exposed to either media (control) or ODE for 24 h (Fig. 1a). mRNA fold change of HMGB1 was measured and was found to increase on ODE exposure but remained low in cells with HMGB1 knockdown (Supplementary Fig. 1e). The impact of lack of HMGB1 on mitochondrial morphology was investigated. On ODE exposure, the expression of mitochondrial fusion proteins mitofusin 1 (MFN1), mitofusin 2 (MFN2), and optic atrophy 1 (OPA1) were measured. The expression of MFN1 and MFN2 decreased in NHBE cells with and without HMGB1 exposed to ODE, while OPA1 levels were significantly upregulated in NHBE cells treated with ODE alone (Fig. 2a–f). Mitochondrial fission protein, dynamin-related protein 1 (DRP1), level was significantly upregulated in the ODE-exposed cells lacking in HMGB1 (Fig. 2g, h). To further corroborate whether the mitochondrial morphological changes observed are associated with the number of active mitochondria, change in mitochondrial mass was measured by quantifying MitoTracker green fluorescence. There was a significant decrease in the mitochondrial mass on exposure to ODE in cells with and without HMGB1 (Fig. 2i). ODE-exposed cells stained with MitoTracker red dye were visualized to accurately identify the morphological changes. In NHBE cells treated with ODE alone, the mitochondrial network appeared filamentous and branched with few fragmented or globular mitochondria (Fig. 2j’). By contrast, mitochondria appeared more fragmented or globular in ODE-exposed NHBE cells lacking HMGB1 indicating mitochondrial fission (Fig. 2j’’’).

HMGB1 knockdown promotes mitochondrial fission on OD exposure. Changes in mitochondrial morphology in NHBE cells with or without siRNA-mediated HMGB1 knockdown and treated with ODE (1%) for 24 h were assessed. Immunoblotting was performed to measure the mitochondrial fusion proteins MFN1 (a, b), MFN2 (c, d), and OPA1 (e, f) and fission protein DRP1 (g, h) and compared. Mitochondrial mass was measured (i), and the morphology was visualized by staining with Mito-tracker dye (j-j’’’; scale bar = 100 μm). All the protein bands were normalized over β-actin (37 kD), and percentage intensity relative to control was analyzed. Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 3–6/treatment (* indicates significant difference from control)
ODE exposure increases BNIP3 expression in NHBE cells
We examined whether the lack of HMGB1 during ODE exposure impacts mitochondria-targeted autophagy, referred to as mitophagy. The expression of the two important mediators of mitophagy, PTEN-induced kinase 1 (PINK1) and the E3 ubiquitin protein ligase Parkin, were measured. NHBE cells treated with ODE for 24 h showed no change in PINK1 but significantly decreased Parkin expression (Fig. 3a–d). ODE-exposed NHBE cells lacking HMGB1 showed a significant decrease in both PINK1 and Parkin on exposure to ODE. The expression of BNIP3, a mitochondrial Bcl-2 homology 3 (BH3)-only protein, known to activate mitochondrial permeability transition (MPT), was also measured (Ney 2015). BNIP3 levels were significantly increased on exposure to ODE alone, while cells treated with anti-HMGB1 siRNA showed a decrease in the expression of BNIP3 (Fig. 3e, f).

OD exposure promotes apoptosis in NHBE cells. Immunoblotting of NHBE cells, with or without siRNA-mediated HMGB1 knockdown and treated with ODE (1%) for 24 h, was performed to measure expression of mitophagy markers, PINK1 (a, b) and Parkin (c, d), and BNIP3 (e, f). All the protein bands were normalized over β-actin (37 kD), and percentage intensity relative to control was analyzed. Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 3/treatment (* indicates significant difference from control)
ODE exposure induces mtDNA release into extracellular fluid
Using qPCR, we measured the levels of mtDNA released into the mt-free cytosol and cell culture supernatant (extracellular spaces) samples. NHBE cells treated with ODE for 24 h showed a significant increase in mtDNA levels both in the mt-free cytosol and supernatant samples, while ODE-exposed cells lacking HMGB1 had decreased levels in both cytosol and supernatant samples (Fig. 4a, b). In addition, ODE-exposed cells showed increased expression of TLR9, a receptor that mediates cellular response to unmethylated CpG dinucleotides found in bacterial DNA and eukaryotic mtDNA to mount an innate immune response (Fig. 4c, d). Next, we examined whether the mtDNA released from the mitochondrial matrix is activating the cGAS-STING pathway. Although no significant change in cyclic GMP-AMP synthase (cGAS) and interferon regulatory factor 3 (IRF3) expression were observed, a significant increase in interferon gamma inducible protein 16 (IFI16) protein expression in cells treated with ODE alone was documented (Fig. 4e–j). In ODE-exposed cells lacking HMGB1, the levels of these proteins were comparable to controls or significantly decreased in the case of IRF3 (Fig. 4i, j). mRNA fold change of type 1 interferons, ifna1, ifna4, and ifnb, which are downstream targets of IFI16 and IRF3, was measured. Cells exposed to ODE for 24 h showed a significant increase in the mRNA levels of ifna1, ifna4, and ifnb, while the levels remained comparable to controls in cells lacking HMGB1 (Fig. 4k–m).

Acute OD exposure-mediated release of mtDNA stimulates IFN production. Mitochondrial DNA (mtDNA) leakage into the cytosol (a) and extracellularly (b) in NHBE cells, with or without siRNA-mediated HMGB1 knockdown and treated with ODE (1%) for 24 h was analyzed via qPCR. Immunoblotting was performed to measure expression of TLR9 (c, d), cGAS (e, f), IFI16 (g, h), and IRF3 (i, j). mRNA fold change of gene targets of mtDNA release, ifna1 (k), ifna4 (l), and ifnb (m), was measured by qPCR. All the protein bands were normalized over β-actin (37 kD), and percentage intensity relative to control was analyzed. Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 3/treatment (* indicates significant difference from control)
To investigate whether mtDNA is released during repeated exposure of ODE, NHBE cells were treated with media alone (control) or ODE (1%) followed by media (control) or HMGB1 neutralization antibody (10 µg/mL) for 8 h per day for 5 days (Fig. 1b). Following a 5-day exposure, there was an increase in cytosolic mtDNA content in cells treated with HMGB1 neutralization antibody and an increase in extracellular mtDNA in cells treated with ODE alone (Supplementary Fig. 2a, b). There was also an increase in ifna4 and a decrease in ifna and ifnb mRNA levels in cells treated with HMGB1 neutralization antibody (Supplementary Fig. 2c–e).
Mitochondrial biogenesis is promoted in ODE-exposed NHBE cells lacking HMGB1
Mitochondrial biogenesis in NHBE cells was assessed by measuring peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) and nuclear factor erythroid 2-related factor 2 (NRF2) protein expression. NHBE cells with and without HMGB1 exposed to ODE for 24 h showed a significant increase in PGC1α and NRF2 expression compared to controls (Fig. 5a–d). Expression of TFAM, a downstream target of PGC1α and NRF2, in both the mitochondria and mitochondria-free cytosol were measured. In ODE-exposed NHBE cells lacking HMGB1, we noted a small decrease in TFAM levels in the mitochondria (~ 10%) and an increase in the cytosolic levels (~ 10%) (Fig. 5e–g). We observed that the cytosolic levels of TFAM significantly decreased in cells treated with ODE alone (Fig. 5e, g). mRNA fold change of downstream targets of NRF2, glutathione S-transferase P (gstp1), heme oxygenase 1 (hmox1), and NAD(P)H quinone dehydrogenase 1 (nqo1), was measured. In ODE-exposed NHBE cells lacking HMGB1, we documented a significant decrease in gstp1, while we noted an increase in mRNA levels of hmox1 and nqo1. In ODE-treated cells, while no significant change was observed in the expression of gstp1, a significant increase was observed in hmox1 and nqo1, when compared to control and cells lacking HMGB1 (Fig. 5h–j).

Lack of HMGB1 promotes mitochondrial biogenesis in OD exposed NHBE cells. Markers of mitochondrial biogenesis in NHBE cells with or without siRNA-mediated HMGB1 knockdown and treated with ODE (1%) for 24 h were measured. Immunoblotting of whole cell lysates of NHBE cells was performed to measure PGC1α (a, b) and NRF2 expression (c, d). TFAM expression was compared in the mitochondrial (e, f) and cytosolic (e, g) fractions of the NHBE cells. mRNA fold change of gene targets downstream of NRF2, gstp1 (h), hmox1 (i), and nqo1 (j), was measured by qPCR. All the protein bands were normalized over β-actin (37 kD), and percentage intensity relative to control was analyzed. Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 3/treatment (* indicates significant difference from control)
On the other hand, in the repeated long-term exposure model, we observed that ODE induced a significant decrease in mRNA fold change of pgc1a and nrf2, and HMGB1 neutralization significantly increased pgc1a and nrf2 levels. However, HMGB1 neutralization did not increase the levels to that of controls (Supplementary Fig. 3a, b). mRNA fold change of tfam is significantly reduced as well (Supplementary Fig. 3c). mRNA levels of nqo1 were significantly reduced, while that of hmox1 increased in both treatments (Supplementary Fig. 3d–e).
Single ODE exposure leads to release of cytochrome c
To assess the impact of the increased mtDNA leakage, we measured the release of other mitochondrial DAMPs (mtDAMPs) and reactive species generation. In addition to TFAM and mtDNA, cytochrome c is another prominent mtDAMP which is a key player in apoptosis (Cai et al. 1998; Ott et al. 2002). We measured the expression of cytochrome c in mitochondrial and mitochondria-free cytosolic fractions of NHBE cells. ODE exposure of NHBE cells increased the cytosolic cytochrome c levels but not the mitochondrial cytochrome c levels (Fig. 6a–c). ODE-exposed cells lacking HMGB1 showed a significant decrease in both mitochondrial and cytosolic cytochrome c protein levels. Griess assay results showed an increase in RNS levels on exposure to ODE, with levels comparable to control in cells lacking HMGB1 (Fig. 6d). To corroborate this, we measured the mRNA fold change in nos2, the gene coding for inducible nitric oxide synthase (iNOS), and observed an increased expression in cells treated with ODE alone while nos2 mRNA fold change decreased in cells lacking HMGB1 (Fig. 6e). In addition, we documented an increase in intracellular reactive oxygen species (ROS) levels in ODE-treated cells, which was significantly decreased in cells lacking HMGB1 (Fig. 6f). On the other hand, the mitochondrial superoxide levels were significantly decreased in both cases compared to control (Fig. 6g). We observed a decrease in mitochondrial calcium levels ([Ca2+]mito) in cells exposed to ODE alone whereas [Ca2+]mito levels increased in cells lacking HMGB1 (Fig. 6h) (Fig. 6i’, i’’’). In the repeated ODE exposure model, ODE exposure alone decreased the ([Ca2+]mito) whereas neutralization of HMGB1 increased the ([Ca2+]mito) (Supplementary Fig. 4a-a’’’) (Supplementary Fig. 4b). There was no change in the levels of mitochondrial superoxide (Supplementary Fig. 4c).

Acute OD exposure causes cytochrome c release into the cytosol. Immunoblotting of mitochondrial and mitochondria-free cytosolic fractions of NHBE cells with or without siRNA-mediated HMGB1 knockdown and treated with ODE (1%) for 24 h, to detect the presence of cytochrome C. Cytochrome C expression was compared between the mitochondrial (a, b) and cytosolic (a, c) fractions of the cells. Concentration of secreted nitrites was measured using Griess assay (d), and mRNA levels of nos2 (e) was measured by qPCR. Using CM-H2DCFDA and Mito-SOX dyes, the levels of superoxide anions (SOX) intracellularly (f) and within the mitochondria (g) were measured, respectively. Intramitochondrial calcium levels in mitochondria isolated from the treated cells were quantified (h) and visualized (i-i’’’; scale bar = 100 μm) by Rhod-2AM staining. For western blot, all the protein bands were normalized over β-actin (37 kD) and percentage intensity relative to control analyzed. Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 3–6/treatment (* indicates significant difference from control)
Repeated ODE exposure promotes mitochondrial fusion
Using transmission electron microscopy (TEM), we assessed the ultrastructural changes in NHBE cells following exposure to ODE with or without HMGB1 neutralization for 8 h/day for 5 days (Fig. 1b). Compared to controls, ODE-treated NHBE cells showed change in mitochondrial morphology. Mitochondria appeared to be more elongated with cristolysis, and in some cases, there was mitochondrial hypertrophy (Fig. 7a, a’). In contrast, neutralization of HMGB1 decreased the ODE-induced cristolysis and mitochondrial shape resembled that of controls (Fig. 7a, a’’). We stained the cells with MitoTracker dye to assess changes in the number of active mitochondria. Mitotracker stained cells revealed no change in mitochondrial mass among any of the treatments (Fig. 7c). However, we did observe noticeable ODE-induced changes in mitochondrial morphology. Compared to controls, ODE-exposed NHBE cells showed filamentous mitochondria and mitochondria appeared to be more clustered close to the nucleus (Fig. 7b, b’). With HMGB1 neutralization, the mitochondria were more fragmented and globular and were less clustered near the nuclei when compared to ODE-exposed cells (Fig. 7b’’). Using MTT assay, we measured the cell viability, and HMGB1 neutralization rescued the ODE-induced decrease in cell viability (Fig. 7d).

Antibody-mediated neutralization of HMGB1 maintains mitochondrial morphology and cell viability. Transmission electron microscopy (TEM) of NHBE cells treated with medium or ODE (1%) followed by medium or HMGB1 neutralization antibody (10 µg/mL) for 8 h per day for 5 days show changes in the mitochondrial morphology. A number of mitochondria displayed fused and elongated mitochondrial morphology (a-a’’; scale bar, 0.5–1 µm) on treatment with ODE. Cells treated with ODE followed by HMGB1 neutralization showed noticeably healthier mitochondria with a few morphological changes. Mitochondrial mass was measured (c), and the morphology was visualized by staining with MitoTracker dye (b-b’’; scale bar = 100 μm). MTT assay was performed to measure cell viability on treatment (d). Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 6/treatment (* indicates significant difference from control)
HMGB1 neutralization rescues mitochondrial respiration in repeated ODE exposure models
A targeted analysis of expression of genes associated with mitochondrial respiration was performed. A pre-designed gene array with 96 genes (including housekeeping genes) specific to various components involved in mitochondrial respiration was used, and fold changes were measured by qPCR (Supplementary Table 3). The change in gene expression between ODE-exposed NHBE cells, with or without HMGB1 neutralization for 8 h/day for 5 days, was calculated relative to control by the 2−ΔΔCT method, and genes with a fold change ≤ 10 or ≥ 10 were represented on a heat map (Livak and Schmittgen 2001). Neutralization of HMGB1 increased the expression of genes of protein subunits involved in mitochondrial respiration such as various subunits of ATP synthase (atp), cytochrome c oxidase (cox), ubiquinol-cytochrome c reductase (uqcr), and NADH:ubiquinone oxidoreductase (naduf) when compared to ODE-treated cells (Fig. 8a). Using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, upregulated and downregulated pathways associated with mitochondria bioenergetics were analyzed (Fig. 8b–e). Pathways related to the metabolism, respiratory electron transport, and mitochondria biogenesis which were downregulated with ODE exposure were significantly upregulated on HMGB1 neutralization (Fig. 8c, d).

HMGB1 neutralization upregulates genes associated with mitochondrial respiration. mRNA expression profiles of genes specific to mitochondrial respiration in NHBE cells treated with medium or ODE (1%) followed by medium or HMGB1 neutralization antibody (10 µg/mL) for 8 h per day for 5 days were measured using a targeted gene array. Heatmap of mRNA transcripts of genes that had a tenfold change in expression was selected (a). Selected upregulated (b, d) and downregulated (c, e) pathways from reactome pathway analysis of significantly altered mRNA transcripts with FDR corrected p-value cutoff < 0.05 were selected and represented by gene counts. Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 2/treatment (* indicates significant difference from control)
Single ODE exposure induces hypoxia response in NHBE cells lacking HMGB1
Oxygen sensing and adaptations in NHBE cells (with or without anti-HMGB1 siRNA treatment) exposed to ODE for 24 h were measured. An increase in hypoxia-inducible factor 1-alpha (HIF1α) protein expression was observed in cells lacking HMGB1 while the expression of HIF1α was significantly reduced with ODE exposure alone (Fig. 9a, b). mRNA fold change of ubiquitin ligase Von Hippel–Lindau tumor suppressor (vhl) was significantly increased in both ODE-exposed NHBE cells with or without HMGB1 (Fig. 9c). mRNA fold change of genes involved in glucose uptake, solute carrier family 2 member 6 (slc2a6), and glycolysis, enolase 1 (eno1), was measured. A significant increase in slc2a6 in both ODE-exposed cells with or without HMGB1 was observed, whereas eno1 was upregulated in cells lacking HMGB1 (Fig. 9d, e). To corroborate this, we measured cellular glucose uptake potential and glycolytic activity in ODE-exposed NHBE cells by culturing cells with the glucose analog 2-NBDG. Here, a significant increase in the uptake of 2- NBDG was observed in ODE-exposed cells lacking HMGB1, while exposure to ODE alone significantly decreased 2-NBDG uptake (Fig. 9f).

Lack of HMGB1 initiates hypoxia response on acute OD exposure. Markers of hypoxia response in NHBE cells with or without siRNA-mediated HMGB1 knockdown and treated with ODE (1%) for 24 h were measured. Immunoblotting of whole-cell lysates of NHBE cells was performed to measure HIF1α (a, b). mRNA fold change of ubiquitin ligase, vhl (c), and slc2a6 (d) and eno1 (e), involved in glucose uptake and glycolysis, was measured by qPCR. 2-NBDG uptake to measure cellular glucose uptake was performed (f). For western blot, all the protein bands were normalized over β-actin (37 kD) and percentage intensity relative to control analyzed. Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 3–6/treatment (* indicates significant difference from control)
We compared the expression of the factors involved in hypoxia and glucose uptake in NHBE cells in the repeated ODE exposure model. mRNA fold change of hif1a and vhl was significantly decreased in ODE-exposed cells but remained unchanged in cells with HMGB1 neutralization (Fig. 10a, b). There was no significant change in the expression of slc2a6 in any of the treatments. However, the expression of eno1 was significantly downregulated in ODE-exposed cells (Fig. 10c, d). 2-NBDG uptake was decreased in ODE-exposed cells and was comparable to control in cells treated with HMGB1 neutralization antibody (Fig. 10e).

Normoxic conditions are maintained on HMGB1 neutralization during chronic OD exposure. Markers of hypoxia response in NHBE cells treated with medium or ODE (1%) followed by medium or HMGB1 neutralization antibody (10 µg/mL) for 8 h per day for 5 days were measured. mRNA fold change of hypoxia response factor, hif1a (a) and ubiquitin ligase, vhl (b), and Slc2a6 (c) and Eno1 (d), involved in glucose uptake and glycolysis was measured by qPCR. 2-NBDG uptake to measure cellular glucose uptake was performed (e). Samples for all assays were derived from the same experiment and were processed in parallel. Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 3–6/treatment (* indicates significant difference from control)
HMGB1 neutralization increased tight junction integrity in ALI cultures of NHBE cells
NHBE cells were seeded onto a semi-permeable membrane to develop an air–liquid interface (ALI) culture model and exposed to ODE, with or without HMGB1 neutralization for 1 h/ day for 5 days (Fig. 1c). mRNA fold change of forkhead box J1 (foxj1) and its downstream target cilia and flagella-associated protein 157 (cfap157) was measured (Fig. 11a, b). Exposure of cells to ODE alone showed a significant increase in fold change of both genes, while the expression was comparable to control in ODE-exposed cells with HMGB1 neutralization. Tight junction integrity was determined by measuring the mRNA fold change of occludin (ocln) and claudin 1 (cld1), and by quantifying the trans-epithelial electrical resistance (TEER). mRNA fold change of ocln and cldn1 was significantly downregulated in ODE-exposed cells, while on HMGB1, neutralization ocln levels were comparable to control, and cld1 was significantly decreased (Fig. 11c, d). TEER measurements were performed to identify the impact of ODE on tight junction integrity. A decrease in TEER values (electrical resistance) is proportional to decreased tight junction integrity. TEER values were significantly decreased on exposure to ODE as early as 12 h and continued to decrease with time compared to control. On the other hand, although a similar decrease was observed with the neutralization of HMGB1 at 12 h, TEER values remained steady after day 1 post-exposure and were not rescued significantly (Fig. 11e).

HMGB1 neutralization upregulates tight junction integrity in ALI. NHBE cells differentiated on air–liquid interface (ALI) culture treated with medium or ODE (1%) followed by medium or HMGB1 neutralization antibody (10 µg/mL) for 1 h per day for 5 days. mRNA fold change of markers of motile cilia, foxj1 (a) and cfap157 (b), and tight junction, ocln (c) and cldn 1 (d), was measured by qPCR. Tight junction integrity was assessed by trans-epithelial electrical resistance (TEER) measurement (e). Samples for all assays were derived from the same experiment and were processed in parallel. Data was analyzed using one-way ANOVA with Tukey’s multiple comparison test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001) and represented as mean ± SEM with n = 3/treatment (* indicates significant difference from control)
Discussion
In the current study, we investigated the impact of secreted HMGB1 on ODE-induced mitochondrial dysfunction in NHBE cell models of single and repeated 5-day exposure. Overall, we demonstrate that blocking HMGB1 secretion induces mitochondrial fragmentation, mitochondrial biogenesis, and an increase in mitochondrial respiration. These responses were consistent across the single (acute) exposure, with siRNA knockdown of HMGB1, as well as in repeated (chronic) exposure to ODE, with antibody-mediated HMGB1 neutralization. In the present study, we used two in vitro models, a 24-h ODE exposure (single, acute) with siRNA-mediated knockdown of HMGB1 and a 5-day (8 h/day) ODE exposure (repeated, chronic) with antibody-mediated HMGB1 neutralization to examine exposure-induced mitochondrial dysfunction (Fig. 1a,b). The siRNA-mediated knockdown of HMGB1 is suitable for single (acute) exposure as the suppression of mRNA and protein lasts for about 72 h. Therefore, we used an antibody-mediated neutralization approach in our repeated (5-day) exposure model.
Our results from single exposure showed that ODE decreases the expression of MFN1 and MFN2 in cells with and without HMGB1, but OPA1 expression in cells lacking HMGB1 was significantly decreased. OPA1 is a dynamin-related GTPase that has been shown to mediate inner mitochondrial membrane (IMM) fusion, while MFN1/2 mediate the fusion of the outer mitochondrial membrane (OMM) (Song et al. 2009; MacVicar and Langer 2016). OPA1 in particular has been identified as a significant player in the regulation of various mitochondrial functions. Loss of OPA1 has been shown to induce DRP1-dependent mitochondrial fragmentation (Lee et al. 2004). This was observed in our results where siRNA-mediated HMGB1 knockdown increased the DRP1 expression in ODE-exposed cells. The fusion of mitochondria is dependent on dimerization of mitofusin proteins, MFN1 and MFN2. Thus, the decreased MFN1/2 expression in ODE-exposed cells lacking HMGB1 explains the increase in mitochondrial fragmentation (Chen et al. 2003; Song et al. 2009; Liesa et al. 2009; Palmer et al. 2011; Ranieri et al. 2013). Overall, our results highlight that lack of HMGB1 has an impact on OPA1 but not on MFN1/2.
Next, we used 2D fluorescence imaging and transmission electron microscopy (TEM) to evaluate mitochondrial morphology and ultrastructural changes in both acute and repeated exposure in vitro models. Interestingly, cells that lacked HMGB1 displayed increased fragmentation and globular mitochondria upon exposure to ODE. These results indicate that ODE exposure-induced mitochondrial changes become pronounced with the loss of nuclear HMGB1, underscoring the importance of nuclear HMGB1 in maintaining the health of the cellular mitochondria (Tang et al. 2011). Our TEM images confirmed that ODE exposure induces mitochondrial elongation, cristoysis, and hypertrophy, whereas HMGB1 neutralization reduced the ODE-induced cristolysis and mitochondria appeared healthy. Further, neutralization of HMGB1 induced a globular morphology of mitochondria with elongated cristae running parallel to the longitudinal axis of the mitochondria. Overall, ODE-induced mitochondrial fusion observed in our study could be a cellular stress response aimed at compensating for the loss of important mitochondrial functions (Chen et al. 2003; Landes et al. 2010; Westrate et al. 2014; Eisner et al. 2018; Tilokani et al. 2018). Our recent study using microglial model confirms the ODE exposure-induced cristolysis and the morphological basis of exposure-induced decrease in mt-bioenergetics (Massey et al. 2021).
ODE-exposed NHBE cells showed no significant change in PINK1 expression, while Parkin was significantly decreased and NHBE cells lacking HMGB1 showed reduced levels of both PINK1 and Parkin. These results indicate that the observed fragmentation of mitochondria in cells lacking HMGB1 could be due to active mitochondrial biogenesis (Ding and Yin 2012). We observed an increase in BNIP3 expression on ODE exposure, whereas, in cells lacking HMGB1, the BNIP3 levels return to baseline (control) levels. The increase in BNIP3 expression could be promoting permeability of the inner and outer mitochondrial membranes resulting in increased ROS production, release of cytochrome c, and mitochondrial depolarization, all of which together initiates cellular apoptosis (Kim et al. 2002; Ney 2015). Considering the increase in OPA1 levels in ODE-exposed cells, evidence suggests that BNIP3 interacts with OPA1 inhibiting OPA1-mediated fusion, in acute conditions, or disassembly of OPA1 complexes, in chronic conditions, leading to cellular death (Landes et al. 2010). This is further corroborated by the increase in cytosolic cytochrome C levels in 24 h ODE-exposed cells. On the contrary, cytochrome C levels were decreased in cells lacking HMGB1 following exposure. The release of cytochrome C in ODE-exposed cells could be promoting increased ROS generation, caspase activation (caspase 3 and caspase 9), eventually leading to apoptotic cell dismantling (Cai et al. 1998; Ott et al. 2002; Quinsay et al. 2010).
One of the major consequences of increased MPT during cellular stress is the release of mitochondrially derived molecules referred to as mtDAMPs (García and Chávez 2007; Zhang et al. 2010; Quinsay et al. 2010; Nakahira et al. 2011). Fragmented mtDNA is a critical mtDAMP, and when released from the damaged mitochondria, it regulates the innate immune responses (Lee and Wei 2005; West et al. 2015; Ploumi et al. 2017). Release of mitochondrial DNA into the cytoplasm and out into the extracellular milieu activates multiple pattern recognition receptors and innate immune responses, including cGAS‐STING, TLR9, and inflammasome formation leading to, among others, robust type I interferon responses (Cloonan and Choi 2012; West et al. 2015; Chen et al. 2016). The results of the present study show an increase in cytosolic and extracellular mitochondrial DNA copy number in NHBE cells exposed to ODE for 24 h, which is reduced in ODE-exposed cells lacking HMGB1 indicating how HMGB1 regulates mt-damage and release of mtDNA. Concurrently, we also observed an increase in the expression of TLR9, IFI16, IRF3, and type 1 interferon, which could be leading to cell death. On the other hand, during chronic ODE exposure, we document an increase in the cytosolic mtDNA on HMGB1 neutralization, but not extracellularly, and an increase in only ifna4 mRNA, indicating that the neutralization of HMGB1 could be promoting mitochondrial biogenesis.
Mitochondrial reactive oxygen species (mtROS) is the most universal and well-studied mtDAMP and is a by-product of mitochondrial respiration and ATP production (Zorov et al. 2014). The ODE-mediated increase in intracellular ROS is consistent with our previous findings, where it is known to initiate the inflammatory cascade (Bhat et al. 2019; Massey et al. 2019). The lack of increase in mtROS could be another indicator of mitochondrial biogenesis in play. Thus, the observed mitochondrial fragmentation in cells lacking HMGB1 or with antibody-mediated HMGB1 neutralization may reflect an important adaptive response to protect the mitochondria from an increase in ROS production and oxidative stress with ODE exposure. In addition to this, we also document an increase in mitochondrial calcium influx in ODE-treated cells lacking HMGB1 or with antibody-mediated HMGB1 neutralization. This increase in mitochondrial calcium influx could be promoting mitochondrial O2 consumption and ATP synthesis, as well as mitochondrial metabolism (Rizzuto et al. 2000; Parekh 2003; Brookes et al. 2004; Hajnóczky et al. 2007; Finkel et al. 2015).
The results of our study provide converging evidence that 24-h ODE exposure of cells with HMGB1 or cells without HMGB1 induces mitochondrial biogenesis. First, we observed an increase in PGC1α expression in cells exposed to ODE, with or without HMGB1. The role of PGC1α in regulating mitochondrial biogenesis has been well established (Wu et al. 1999; Handschin and Spiegelman 2006; Trian et al. 2007). We then measured the expression of NRF2 and observed a similar increase as that of PGC1α. NRF2 is known to regulate the expression of the electron transport chain subunits and binds to the promoter regions of genes involved in the transcription of mtDNA. NRF2 is known to be regulated by its transcriptional coactivator PGC1α, which together induces the expression of TFAM thus activating mitochondrial biogenesis. In line with this, we observe an increase in TFAM protein expression in mitochondria-free cytosolic fractions of 24-h ODE-exposed cells lacking HMGB1. However, we observed that a repeated 5-day ODE exposure resulted in significantly lower levels of the gene expression of PGC1α, NRF2, and TFAM when compared to the controls with and without HMGB1 neutralization.
In our study, we found that NHBE cells repeatedly exposed to ODE display a distinctive mitochondrial gene profile, where we observe a downregulation of genes associated with mitochondrial metabolism, citric acid (TCA) cycle, OXPHOS, and mitochondrial biogenesis. This is consistent with the increased levels of ROS, induction of MPT, and mtDNA release on ODE exposure. With the neutralization of HMGB1, we document an increase in the fold change of genes coding the protein subunits associated with mitochondrial electron transport chain (ETC), such as ATP, COX, NADUF, and UQCR. In line with this, we show evidence of ODE-mediated decrease in cellular glucose uptake and glycolytic ability in both acute and chronic exposure models, while lack of or neutralization of HMGB1 increased the cellular glucose uptake and glycolytic ability. This metabolic profile in NHBE cells may have a direct impact on the maintenance of overall lung homeostasis and function on exposure to ODE in vivo (Wu et al. 1999; Koopman et al. 2005; Dranka et al. 2011; Agrawal and Mabalirajan 2015). Our results provide a robust framework for the future in vivo experiments.
Reduced oxygen availability elicited by hypoxia and mitochondrial dysfunction leads to increased production of ROS by the electron transport chain (ETC) (Baik and Jain 2020). The observed ODE-exposure-induced hypoxia response could be well explained based on the increased intracellular ROS production and downregulation of ETC. Interestingly, we observed a decrease in HIF1α expression in 24 h ODE-exposed NHBE cells while it increased in cells lacking HMGB1, indicative of hypoxia. Thus, lack of HMGB1 acts as protective through activation of HIF1a. This is supported by the cellular studies showing that HIF1α activation could rescue the effects of ETC deficiency in mitochondrial diseases (Jain et al. 2016). Hypoxia may be triggering a HIF-dependent transcriptional program activating key pathways promoting glycolysis, ATP production, and decreased ROS generation. This would also be reducing the levels of O2 being taken up by the cell which would, in turn, decrease the availability of substrate for free radical production and aberrant cellular signaling. We observed that HMGB1 neutralization resulted in the normoxic conditions in the repeated ODE exposure model. Hence, we can theorize that short-term hypoxic conditions may be beneficial in reversing mitochondrial dysfunction.
In addition, using an ALI model of ODE exposure, we investigated the impact of HMGB1 on tight junction integrity. Previously, we have shown the impact of 24-h ODE exposure on tight junction expression in vivo, but the role of HMGB1 remains to be investigated (Shrestha et al. 2021). In this study, we observe a significant decrease in transepithelial electrical resistance (TEER) with repeated ODE exposure as early as 12-h post-exposure. Although HMGB1 neutralization does not reverse the effect of ODE on tight junction integrity, the TEER values plateau and remains unchanged 1-day post-exposure. The neutralization of HMGB1 does not significantly rescue ODE-induced decrease in TEER. However, there is a certain amount of protection of epithelial integrity. This was corroborated by the measurement of claudin-1 and occludin, indicating that ODE exposure directly affects the tight junction integrity. Although this is valid preliminary evidence on the impact of ODE on tight junction integrity, thorough in vivo work needs to be performed to confirm this finding.
In summary, the results of the present study indicate that targeting HMGB1 in ODE-induced inflammatory process promotes mitochondrial biogenesis and metabolism by promoting hypoxic conditions and decreasing ROS production (Fig. 12). It is likely that blocking HMGB1 release following ODE exposure leads to a coordinated adaptive response in NHBE cells involving mitochondrial fragmentation, increased mitochondrial biogenesis, and mitochondrial metabolism. As a result, the maximum O2 consumption and ROS production per mitochondrion are reduced, while the demand for ATP synthesis is still met. Future studies using HMGB1 airway epithelial cell-specific conditional knockout mice or systemic neutralization of HMGB1 may unravel further mechanistic basis of targeting HMGB1 in rescuing ODE-induced mitochondrial deficits.

An overview of impact of HMGB1 on ODE exposure-induced mitochondrial dysfunction in NHBE cells. HMGB1 increases autophagy, inhibits apoptosis, and regulates mitochondria functions. ODE exposure promoted mitochondrial fusion and mtDNA release promoting a pro-inflammatory response through TLR9, NFκB, and IRF3 signaling pathways resulting in pro-inflammatory cytokine release. On the other hand, HMGB1 knockdown or use of Anti-HMGB1 antibody rescues ODE-induced mtDNA release and promotes mitochondrial fission. ODE exposure also induces a decrease in OXPHOS and glucose uptake while increasing ROS generation and mitochondrial membrane permeability. HMGB1 knockdown or use of Anti-HMGB1 antibody treatment decreases ROS generation, promotes mitochondrial biogenesis, and upregulates markers associated with OXPHOS (Created with BioRender.com)
Abbreviations
- OD:
-
Organic dust
- ODE:
-
Organic dust extract
- NHBE:
-
Normal human bronchial epithelial
- BEGM:
-
Bronchial epithelial growth medium
- ALI:
-
Air liquid interface
- HMGB1:
-
High mobility group box 1
- RAGE:
-
Receptor for advanced glycation end products
- TLR:
-
Toll-like receptor
- NOD2:
-
Nucleotide-binding oligomerization domain-containing 2
- ATP:
-
Adenosine triphosphate
- OXPHOS:
-
Oxidative phosphorylation
- TFAM:
-
Mitochondrial transcription factor A
- COX:
-
Cytochrome C oxidase
- mtND1:
-
Mitochondrial NADH dehydrogenase 1
- TEM:
-
Transmission electron microscopy
- DMSO:
-
Dimethyl sulfoxide
References
Aghapour M, Remels AHV, Pouwels SD et al (2019) Mitochondria: at the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 318:L149–L164. https://doi.org/10.1152/ajplung.00329.2019
Agrawal A, Mabalirajan U (2015) Rejuvenating cellular respiration for optimizing respiratory function: targeting mitochondria. Am J Physiol Lung Cell Mol Physiol 310:L103–L113. https://doi.org/10.1152/ajplung.00320.2015
An J Jr, H, Aj N, et al (2020) MyD88 regulates a prolonged adaptation response to environmental dust exposure-induced lung disease. Respir Res 21:97–97. https://doi.org/10.1186/s12931-020-01362-8
Andersson U, Tracey KJ (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol 29:139–162. https://doi.org/10.1146/annurev-immunol-030409-101323
Baik AH, Jain IH (2020) Turning the oxygen dial: balancing the highs and lows. Trends Cell Biol 30:516–536. https://doi.org/10.1016/j.tcb.2020.04.005
Bailey KL, Poole JA, Mathisen TL et al (2008) Toll-like receptor 2 is upregulated by hog confinement dust in an IL-6-dependent manner in the airway epithelium. Am J Physiol Lung Cell Mol Physiol 294:L1049–L1054. https://doi.org/10.1152/ajplung.00526.2007
Bhat SM, Massey N, Karriker LA et al (2019) Ethyl pyruvate reduces organic dust-induced airway inflammation by targeting HMGB1-RAGE signaling. Respir Res 20:27. https://doi.org/10.1186/s12931-019-0992-3
Brookes PS, Yoon Y, Robotham JL et al (2004) Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 287:C817–C833. https://doi.org/10.1152/ajpcell.00139.2004
Cai J, Yang J, Jones Dean P (1998) Mitochondrial control of apoptosis: the role of cytochrome c. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1366:139–149. https://doi.org/10.1016/S0005-2728(98)00109-1
Cantaert T, Baeten D, Tak PP, van Baarsen LG (2010) Type I IFN and TNFα cross-regulation in immune-mediated inflammatory disease: basic concepts and clinical relevance. Arthritis Res Ther 12:219. https://doi.org/10.1186/ar3150
Chang K, Marran K, Valentine A, Hannon GJ (2012) RNAi in cultured mammalian cells using synthetic siRNAs. Cold Spring Harb Protoc 2012:pdb.prot071076. https://doi.org/10.1101/pdb.prot071076
Charavaryamath C, Juneau V, Suri SS et al (2008) Role of toll-like receptor 4 in lung inflammation following exposure to swine barn air. Exp Lung Res 34:19–35. https://doi.org/10.1080/01902140701807779
Charavaryamath C, Singh B (2006) Pulmonary effects of exposure to pig barn air. J Occup Med Toxicol 1:10. https://doi.org/10.1186/1745-6673-1-10
Chaung WW, Wu R, Ji Y et al (2012) Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int J Mol Med 30:199–203. https://doi.org/10.3892/ijmm.2012.959
Chen H, Detmer SA, Ewald AJ et al (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200. https://doi.org/10.1083/jcb.200211046
Chen Q, Sun L, Chen ZJ (2016) Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat Immunol 17:1142–1149. https://doi.org/10.1038/ni.3558
Cloonan SM, Choi AM (2012) Mitochondria: commanders of innate immunity and disease? Curr Opin Immunol 24:32–40. https://doi.org/10.1016/j.coi.2011.11.001
Cloonan SM, Choi AMK (2016) Mitochondria in lung disease. J Clin Invest 126:809–820. https://doi.org/10.1172/JCI81113
Cloonan SM, Kim K, Esteves P et al (2020) Mitochondrial dysfunction in lung ageing and disease. Eur Respir Rev. https://doi.org/10.1183/16000617.0165-2020
Csonka C, Páli T, Bencsik P et al (2015) Measurement of NO in biological samples. Br J Pharmacol 172:1620–1632. https://doi.org/10.1111/bph.12832
Ding W-X, Yin X-M (2012) Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 393:547–564. https://doi.org/10.1515/hsz-2012-0119
Dranka BP, Benavides GA, Diers AR et al (2011) Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radical Biol Med 51:1621–1635. https://doi.org/10.1016/j.freeradbiomed.2011.08.005
Eisner V, Picard M, Hajnóczky G (2018) Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 20:755–765. https://doi.org/10.1038/s41556-018-0133-0
Fetterman JL, Sammy MJ, Ballinger SW (2017) Mitochondrial toxicity of tobacco smoke and air pollution. Toxicology 391:18–33. https://doi.org/10.1016/j.tox.2017.08.002
Finkel T, Menazza S, Holmström KM et al (2015) The ins and outs of mitochondrial calcium. Circ Res 116:1810–1819. https://doi.org/10.1161/CIRCRESAHA.116.305484
García N, Chávez E (2007) Mitochondrial DNA fragments released through the permeability transition pore correspond to specific gene size. Life Sci 81:1160–1166. https://doi.org/10.1016/j.lfs.2007.08.019
Gordon R, Hogan CE, Neal ML et al (2011) A simple magnetic separation method for high-yield isolation of pure primary microglia. J Neurosci Methods 194:287–296. https://doi.org/10.1016/j.jneumeth.2010.11.001
Hajnóczky G, Saotome M, Csordás G et al (2007) Calcium signalling and mitochondrial motility. In: Mitochondrial Biology: New Perspectives. John Wiley & Sons, Ltd, pp 105–121
Handschin C, Spiegelman BM (2006) Peroxisome proliferator-activated receptor γ coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 27:728–735. https://doi.org/10.1210/er.2006-0037
Jain IH, Zazzeron L, Goli R et al (2016) Hypoxia as a therapy for mitochondrial disease. Science 352:54–61. https://doi.org/10.1126/science.aad9642
Julian MW, Shao G, Vangundy ZC et al (2013) Mitochondrial transcription factor A, an endogenous danger signal, promotes TNFα release via RAGE- and TLR9-responsive plasmacytoid dendritic cells. PLoS One 8
Kelly KJ, Poole JA (2019) Pollutants in the workplace: effect on occupational asthma. Journal of Allergy and Clinical Immunology 143:2014–2015. https://doi.org/10.1016/j.jaci.2019.04.013
Kim J-Y, Cho J-J, Ha J, Park J-H (2002) The carboxy terminal C-tail of BNip3 is crucial in induction of mitochondrial permeability transition in isolated mitochondria. Arch Biochem Biophys 398:147–152. https://doi.org/10.1006/abbi.2001.2673
Koopman WJH, Visch H-J, Verkaart S et al (2005) Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol 289:C881–C890. https://doi.org/10.1152/ajpcell.00104.2005
Landes T, Emorine LJ, Courilleau D et al (2010) The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep 11:459–465. https://doi.org/10.1038/embor.2010.50
Latchoumycandane C, Anantharam V, Kitazawa M et al (2005) Protein kinase Cdelta is a key downstream mediator of manganese-induced apoptosis in dopaminergic neuronal cells. J Pharmacol Exp Ther 313:46–55. https://doi.org/10.1124/jpet.104.078469
Lee H-C, Wei Y-H (2005) Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol 37:822–834. https://doi.org/10.1016/j.biocel.2004.09.010
Lee Y, Jeong S-Y, Karbowski M et al (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. MBoC 15:5001–5011. https://doi.org/10.1091/mbc.e04-04-0294
Liesa M, Palacín M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89:799–845. https://doi.org/10.1152/physrev.00030.2008
Liu J, Fang H, Chi Z et al (2015) XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage. Nucleic Acids Res 43:5476–5488. https://doi.org/10.1093/nar/gkv472
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
MacVicar T, Langer T (2016) OPA1 processing in cell death and disease – the long and short of it. J Cell Sci 129:2297–2306. https://doi.org/10.1242/jcs.159186
Mahadev Bhat S, Shrestha D, Massey N et al (2021) Organic dust exposure induces stress response and mitochondrial dysfunction in monocytic cells. Histochem Cell Biol. https://doi.org/10.1007/s00418-021-01978-x
Massey N, Puttachary S, Bhat SM et al (2019) HMGB1-RAGE signaling plays a role in organic dust-induced microglial activation and neuroinflammation. Toxicol Sci 169:579–592. https://doi.org/10.1093/toxsci/kfz071
Massey N, Shrestha D, Bhat SM et al (2021) Organic dust-induced mitochondrial dysfunction could be targeted via cGAS-STING or cytoplasmic NOX-2 inhibition using microglial cells and brain slice culture models. Cell Tissue Res. https://doi.org/10.1007/s00441-021-03422-x
May S, Romberger DJ, Poole JA (2012) Respiratory health effects of large animal farming environments. J Toxicol Environ Health B Crit Rev 15:524–541. https://doi.org/10.1080/10937404.2012.744288
Nakahira K, Haspel JA, Rathinam VAK et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–230. https://doi.org/10.1038/ni.1980
Nath Neerukonda S, Mahadev-Bhat S, Aylward B et al (2018) Kinome analyses of inflammatory responses to swine barn dust extract in human bronchial epithelial and monocyte cell lines. Innate Immun 24:366–381. https://doi.org/10.1177/1753425918792070
Ney PA (2015) Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1853:2775–2783. https://doi.org/10.1016/j.bbamcr.2015.02.022
Nordgren TM, Charavaryamath C (2018) Agriculture occupational exposures and factors affecting health effects. Curr Allergy Asthma Rep 18:65. https://doi.org/10.1007/s11882-018-0820-8
Ott M, Robertson JD, Gogvadze V et al (2002) Cytochrome c release from mitochondria proceeds by a two-step process. PNAS 99:1259–1263. https://doi.org/10.1073/pnas.241655498
Palmer CS, Osellame LD, Stojanovski D, Ryan MT (2011) The regulation of mitochondrial morphology: Intricate mechanisms and dynamic machinery. Cell Signal 23:1534–1545. https://doi.org/10.1016/j.cellsig.2011.05.021
Parekh AB (2003) Mitochondrial regulation of intracellular Ca2+ signaling: more than just simple Ca2+ buffers. Physiology 18:252–256. https://doi.org/10.1152/nips.01458.2003
Piantadosi CA, Suliman HB (2017) Mitochondrial dysfunction in lung pathogenesis. Annu Rev Physiol 79:495–515. https://doi.org/10.1146/annurev-physiol-022516-034322
Ploumi C, Daskalaki I, Tavernarakis N (2017) Mitochondrial biogenesis and clearance: a balancing act. FEBS J 284:183–195. https://doi.org/10.1111/febs.13820
Poole JA, Barnes CS, Demain JG et al (2019) Impact of weather and climate change with indoor and outdoor air quality in asthma: A Work Group Report of the AAAAI Environmental Exposure and Respiratory Health Committee. J Allergy Clin Immunol 143:1702–1710. https://doi.org/10.1016/j.jaci.2019.02.018
Poole JA, Burrell AM, Wyatt TA et al (2010) NOD2 negatively regulates organic dust-induced inflammation in monocytes/macrophages. J Allergy Clin Immunol 125:AB118. https://doi.org/10.1016/j.jaci.2009.12.467
Poole JA, Romberger DJ (2012) Immunological and inflammatory responses to organic dust in agriculture. Curr Opin Allergy Clin Immunol 12:126–132. https://doi.org/10.1097/ACI.0b013e3283511d0e
Poole JA, Wyatt TA, Essen SGV et al (2007) Repeat organic dust exposure–induced monocyte inflammation is associated with protein kinase C activity. J Allergy Clin Immunol 120:366–373. https://doi.org/10.1016/j.jaci.2007.04.033
Poole JA, Wyatt TA, Kielian T et al (2011) Toll-like receptor 2 regulates organic dust-induced airway inflammation. Am J Respir Cell Mol Biol 45:711–719. https://doi.org/10.1165/rcmb.2010-0427OC
Prakash YS, Pabelick CM, Sieck GC (2017) Mitochondrial Dysfunction in Airway Disease CHEST 152:618–626. https://doi.org/10.1016/j.chest.2017.03.020
Qi L, Sun X, Li F-E et al (2015) HMGB1 promotes mitochondrial dysfunction–triggered striatal neurodegeneration via autophagy and apoptosis activation. PLoS One. https://doi.org/10.1371/journal.pone.0142901
Qin S, Wang H, Yuan R et al (2006) Role of HMGB1 in apoptosis-mediated sepsis lethality. J Exp Med 203:1637–1642. https://doi.org/10.1084/jem.20052203
Quinsay MN, Lee Y, Rikka S et al (2010) Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J Mol Cell Cardiol 48:1146–1156. https://doi.org/10.1016/j.yjmcc.2009.12.004
Ranieri M, Brajkovic S, Riboldi G et al (2013) Mitochondrial fusion proteins and human diseases. In: Neurology Research International. https://www.hindawi.com/journals/nri/2013/293893/. Accessed 27 Jan 2021
Rizzuto R, Bernardi P, Pozzan T (2000) Mitochondria as all-round players of the calcium game. J Physiol 529:37–47. https://doi.org/10.1111/j.1469-7793.2000.00037.x
Romberger DJ, Bodlak V, Von Essen SG et al (2002) Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J Appl Physiol 93:289–296. https://doi.org/10.1152/japplphysiol.00815.2001
Rubartelli A, Lotze MT (2007) Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol 28:429–436. https://doi.org/10.1016/j.it.2007.08.004
Schumacker PT, Gillespie MN, Nakahira K et al (2014) Mitochondria in lung biology and pathology: more than just a powerhouse. Am J Physiol Lung Cell Mol Physiol 306:L962–L974. https://doi.org/10.1152/ajplung.00073.2014
Seo J, Ottesen EW, Singh RN (2014) Antisense methods to modulate pre-mRNA splicing. In: Hertel KJ (ed) Spliceosomal Pre-mRNA Splicing: Methods and Protocols. Humana Press, Totowa, NJ, pp 271–283
Sethi RS, Schneberger D, Charavaryamath C, Singh B (2017) Pulmonary innate inflammatory responses to agricultural occupational contaminants. Cell Tissue Res 367:627–642. https://doi.org/10.1007/s00441-017-2573-4
Shrestha D, Bhat SM, Massey N et al (2021) Pre-exposure to hydrogen sulfide modulates the innate inflammatory response to organic dust. Cell Tissue Res. https://doi.org/10.1007/s00441-020-03333-3
Song Z, Ghochani M, McCaffery JM et al (2009) Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. MBoC 20:3525–3532. https://doi.org/10.1091/mbc.e09-03-0252
Srinivasan B, Kolli AR, Esch MB et al (2015) TEER measurement techniques for in vitro barrier model systems. J Lab Autom 20:107–126. https://doi.org/10.1177/2211068214561025
Tang D, Kang R, Livesey KM et al (2011) High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab 13:701–711. https://doi.org/10.1016/j.cmet.2011.04.008
Tilokani L, Nagashima S, Paupe V, Prudent J (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem 62:341–360. https://doi.org/10.1042/EBC20170104
Trian T, Benard G, Begueret H et al (2007) Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med 204:3173–3181. https://doi.org/10.1084/jem.20070956
Ugrinova I, Pasheva E (2017) HMGB1 protein: a therapeutic target inside and outside the cell. Adv Protein Chem Struct Biol 107:37–76. https://doi.org/10.1016/bs.apcsb.2016.10.001
Warren KJ, Dickinson JD, Nelson AJ et al (2019) Ovalbumin-sensitized mice have altered airway inflammation to agriculture organic dust. Respir Res 20:51. https://doi.org/10.1186/s12931-019-1015-0
West AP, Khoury-Hanold W, Staron M et al (2015) Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520:553–557. https://doi.org/10.1038/nature14156
Westrate LM, Drocco JA, Martin KR et al (2014) Mitochondrial morphological features are associated with fission and fusion events. PLoS One 9
Whitsett JA, Alenghat T (2015) Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol 16:27–35. https://doi.org/10.1038/ni.3045
Wu Z, Puigserver P, Andersson U et al (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98:115–124. https://doi.org/10.1016/S0092-8674(00)80611-X
Wunschel J, Poole JA (2016) Occupational agriculture organic dust exposure and its relationship to asthma and airway inflammation in adults. J Asthma 53:471–477. https://doi.org/10.3109/02770903.2015.1116089
Yang H, Wang H, Chavan SS, Andersson U (2015) High mobility group box protein 1 (HMGB1): the prototypical endogenous danger molecule. Mol Med 21(Suppl 1):S6–S12. https://doi.org/10.2119/molmed.2015.00087
Ye C, Choi J-G, Abraham S et al (2012) Human macrophage and dendritic cell-specific silencing of high-mobility group protein B1 ameliorates sepsis in a humanized mouse model. PNAS 109:21052–21057. https://doi.org/10.1073/pnas.1216195109
Zhang Q, Raoof M, Chen Y et al (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464:104–107. https://doi.org/10.1038/nature08780
Zhou H, Wang Y, Wang W et al (2009) Generation of monoclonal antibodies against highly conserved antigens. PLoS One 4
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94:909–950. https://doi.org/10.1152/physrev.00026.2013
Zou C, Wang Y, Shen Z (2005) 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J Biochem Biophys Methods 64:207–215. https://doi.org/10.1016/j.jbbm.2005.08.001
Acknowledgements
We would like to thank Tracey Stewart at Iowa State University’s Roy J. Carver High-Resolution Microscopy Facility for assistance with transmission electron microscopy. We would like to thank Dr. Kevin Tracey (Feinstein Institutes for Medical Research, Northwell Health, NY) for providing us with the anti-HMGB1 neutralization antibody and Dr. Y.S. Prakash (Mayo Clinic, Rochester, MN) and Dr. K. L. Bailey (UNMC, Omaha, NE) for providing us with normal human bronchial epithelial cells when needed. We would also like to thank the department of biomedical sciences and Dr. M. Cho at Iowa State University for providing us with access to necessary equipment. We would like to thank Drs. Thippeswamy and Kanthasamy (Biomedical Sciences) for access to equipment and facilities.
Funding
C.C. laboratory is funded through startup grant through Iowa State University, a pilot grant (5 U54 OH007548) from CDC-NIOSH (Centers for Disease Control and Prevention-The National Institute for Occupational Safety and Health) and a seed grant through CVM (College of Veterinary Medicine) at the Iowa State University.
Author information
Authors and Affiliations
Contributions
S.M. Bhat participated in the design of experiments, performed the experiments, analyzed the data, and wrote the manuscript. N. Massey performed organic dust extraction. D. Shrestha assisted in culturing of NHBE cells. L. Karriker collected the organic dust samples and edited the manuscript. T. Jelesijević provided the technical expertise required to perform fluorescent microscopy and participated in editing the manuscript. C. Charavaryamath conceptualized the study, participated in the design of the experiments, performed dust extraction, participated in the interpretation of data, and edited the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The terms of this arrangement have been reviewed and approved by Iowa State University per its conflict-of-interest policies. All other authors have declared no potential conflicts of interest. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bhat, S.M., Massey, N., Shrestha, D. et al. Transcriptomic and ultrastructural evidence indicate that anti-HMGB1 antibodies rescue organic dust-induced mitochondrial dysfunction. Cell Tissue Res 388, 373–398 (2022). https://doi.org/10.1007/s00441-022-03602-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-022-03602-3