
Abstract
Animal production units produce and store many contaminants on-site, including organic dust (OD) and hydrogen sulfide (H2S). Workers in these settings report various respiratory disease symptoms. Both OD and H2S have shown to induce lung inflammation. However, impact of co-exposure to both H2S and OD has not been investigated. Therefore, we tested a hypothesis that pre-exposure to H2S modulates the innate inflammatory response of the lungs to organic dust. In a mouse model of H2S and organic dust extract (ODE) exposure, we assessed lung inflammation quantitatively. We exposed human airway epithelial and monocytic cells to medium or H2S alone or H2S followed by ODE and measured cell viability, oxidative stress, and other markers of inflammation. Exposure to 10 ppm H2S followed by ODE increased the lavage fluid leukocytes. However, exposure to 10 ppm H2S alone resulted in changes in tight junction proteins, an increase in mRNA levels of tlr2 and tlr4 as well as ncf1, ncf4, hif1α, and nrf2. H2S alone or H2S and ODE exposure decreased cell viability and increased reactive nitrogen species production. ODE exposure increased the transcripts of tlr2 and tlr4 in both in vitro and in vivo models, whereas increased nfkbp65 transcripts following exposure to ODE and H2S was seen only in in vitro model. H2S alone and H2S followed by ODE exposure increased the levels of IL-1β. We conclude that pre-exposure to H2S modulates lung innate inflammatory response to ODE.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The concentrated animal feeding operations (CAFOs) achieve great efficiency in production but not without consequences (Charavaryamath and Singh 2006; Sethi et al. 2017). A higher density of animals and on-site storage of waste products (manure and urine) exposes animals and workers to the contaminants in the occupational settings. Ventilation in the winter is kept to a minimal to prevent higher heating costs. However, this could result in higher dust and gaseous contaminant levels in the barn (Park et al. 2013). Key on-site contaminants include airborne organic dust (OD), gases such as methane, ammonia, hydrogen sulfide (H2S), carbon dioxide, carbon monoxide, and other odiferous compounds (Iowa State University and University of Iowa 2002). Workers who are persistently exposed to OD and irritant gases report various respiratory symptoms including mucus membrane irritation, bronchitis, asthma-like symptoms, coughing, and the annual decline in the lung function (American Thoracic Society 1998; Nordgren and Charavaryamath 2018; Sethi et al. 2017).
Occupational exposure to OD and irritant gases is a risk factor for developing chronic respiratory symptoms (Christiani 1996; Vested et al. 2019). H2S gas is released as a byproduct of microbial degradation of the manure. While H2S concentrations of < 10 ppm are not known to be dangerous, the sudden release of H2S from stored manure pits (during pit agitation) resulting in higher concentration (> 500 ppm) inside the CAFOs is known to result in animal and human mortalities (Oesterhelweg and Puschel 2008). On the flip side, chronic exposure to concentrations < 100 ppm is known to cause various toxic symptoms (Costigan 2003; Iowa State University and University of Iowa 2002). However, there is a constant release of H2S at lower levels in CAFOs and other industrial occupational settings. Researchers have reported H2S levels of 221 to 1492 ppb in the swine confinement facilities (Ni et al. 2000), 0.1–5.7 ppm in shallow pit swine nursery room, and about 4.1 ppb H2S in the ambient air around the swine confinements (Pavilonis et al. 2013). According to the American Conference of Governmental Industrial Hygienists (ACGIH), the new recommendations for the threshold limit values (TLV) for airborne H S exposure (time-weighted average) TWA of 1 ppm for 8 h and a Short Term Exposure Limit (STEL) of 5 ppm (American Conference of Governmental Industrial Hygienists 2019). However, the levels of H2S in industrial settings can be in the range of 0.5–10 ppm (Richardson 1995). Occupational exposure to H2S has considerably increased with the development of CAFOs owing to the size of the operations and the health impacts of this exposure remain to be determined.
In recent years, the physiological significance of H2S is beginning to emerge (Giuffre and Vicente 2018,; Zhang et al. 2017). H2S is endogenously produced at lower levels (approximately 20 nM) (Linden et al. 2010) by the enzymes cystathionine-ɣ-lyase, cystathionine-β-synthetase, and by 3-mercaptopyruvate sulfur transferase from the amino acid cysteine (Chung 2014). Endogenous H2S gas is essential in many cellular processes including dietary restriction benefits (Hine et al. 2015; Li et al. 2011). However, the role of H2S in inflammation is controversial since it shows both cytoprotective and toxic properties at the cellular level in both in vivo and in vitro models. It has been shown to act as an anti-inflammatory agent in acute lung injury as well as preventive and therapeutic agents in the treatment of airway diseases (Esechie et al. 2008; Zhang et al. 2015). Previous results have shown that sodium hydrogen sulfide (NaHS), a donor of H2S, inhibits airway hyperresponsiveness in ozone-induced airway inflammation, emphysema (Chen et al. 2011; Zhang et al. 2013), and reduces airway inflammation and prevents emphysema in a cigarette smoke-induced COPD model (Han et al. 2011). These effects are true when H2S is given at low doses. However, chronic occupational exposure to low doses of H2S has been associated with several respiratory symptoms such as wheezing, shortness of breath, persistent cough, pneumonia, bronchitis, and other lung diseases (Legator et al. 2001; Lewis and Copley 2015). Therefore, mechanistic aspects of H2S exposure induced impacts on host innate responses warrant detailed investigation.
Endogenous H2S levels are usually decreased during an inflammatory process (Chen et al. 2009). Therefore, exogenous supplementation of H2S at physiological concentration will likely work as a cytoprotective and anti-inflammatory agent. H2S administered exogenously reduces nitric oxide (NO) production by inhibiting the expression of induced NO synthase (iNOS) and diminishes NF-κB via haem-oxygenase-1 expression in RAW264.7 macrophages (Hu et al. 2007; Oh et al. 2006). Further, exogenously administered H2S increased the anti-inflammatory cytokine (IL-10) and decreased the pro-inflammatory cytokines (IL-1β) in a burn and smoke-induced acute lung injury murine model (Esechie et al. 2008). Exogenous H2S reduces the production of reactive oxygen species (ROS) via p13k/AKT signaling in a ventilator-induced lung injury model (Spassov et al. 2017). While other studies have shown the pro-inflammatory attributes of H2S such as increasing pro-inflammatory cytokines via ERK/NF-κB pathways in U937 cell lines (IL-1β, IL-6, TNF-α) (Zhi et al. 2007), or substance P (SP)-neurokinin-1 receptor pathway in acute pancreatitis (Tamizhselvi et al. 2008). H2S exposure stimulates transient receptor potential (TRP) channels to release substance P (SP) and neurokinin A at sensory nerve terminals causing airway inflammation (Trevisani et al. 2005). Therefore, whether occupational exposure to 5–10 ppm of H2S would induce pro-inflammatory effects and if there will be any synergistic effect when the host is concurrently exposed to other contaminants such as OD remains elusive.
Based on several lines of evidence indicating the respiratory irritant nature of H2S gas, we hypothesize that prior exposure to H2S would modulate host innate inflammatory response to OD. Using human cells (in vitro) and mouse (in vivo) models of H2S and OD exposure, we show that prior exposure modulates host inflammatory response through changes in cell viability, production of reactive species, and inflammatory mediators.
Materials and methods
Cells, biological, and chemical reagents
Human monocytic cell line (THP-1) and immortalized human bronchial epithelial cells (BEAS-2B) were procured from ATCC (Manassas, VA). Lipopolysaccharide (LPS, Escherichia coli O127:B8) and sodium hydrosulfide hydrate (NaHS) were procured from Sigma. The breathing air tank was procured from Chemistry Store (Iowa State University), while 5 and 10 ppm H2S gas tanks were procured from a commercial vendor (Airgas, Radnor, PA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals, Flowery Branch, GA (cat# S11150H and lot # A17002).
Preparation of ODE
All experiments were performed as per the approved protocols from the Iowa State University’s Institution Biosafety Committee (IBC# 19-004). Settled dust samples from typical swine production units (representing organic dust) were collected into Ziploc bags and transported on ice and stored at − 80 °C until processed. A sterile organic dust extract was prepared as per a published manuscript (Romberger et al. 2002). Dust samples were weighed, and for every gram of dust, 10 mL of Hanks’ balanced salt solution without calcium (Gibco) was added, stirred, and allowed to stand at room temperature for an hour. The mixture was centrifuged (1365×g, 4 °C) for 20 min, supernatant recovered, and the pellet was discarded. The supernatant was centrifuged again with the same conditions, and the pellet discarded and recovered supernatant were filtered using 0.22 µm filter and stored at −80 °C until used. The filter-sterilized organic dust extract (ODE) samples were considered 100%. We used 1 or 5% ODE in our in vitro (THP-1 and BEAS-2B cells) and 12.5% (v/v) in our in vivo experiments, respectively. We routinely estimate lipopolysaccharide (LPS) levels in our ODE samples (Bhat et al. 2019).
Mouse model of H2S gas and ODE exposures
All animal experiments were performed as per the approved protocols (#10-17-8635-M) from the Iowa State University’s Institutional Animal Care and Use Committee (IACUC). About 6–8-week-old male (C57BL/6) mice were procured (Jackson Laboratories, Ann Arbor, MI) and allowed to acclimatize for up to 72 h. Mice were kept on a 12:12-h light/dark cycle with free access to food and water ad libitum.
A total of 60 mice were randomly divided into 6 groups (n = 10/group) as outlined in Table 1. Briefly, mice were subjected to whole-body exposure to breathing air (0 ppm H2S) or breathing air with 5 or 10 ppm H2S gas from gas cylinders for 60 min (treatment-1) by placing them in a transparent exposure chamber. The animal model of H2S exposure including the chamber, delivery of gases under pressure from tanks (H2S and breathing air), and safety measures used in our experiments have previously been described (Anantharam et al. 2017b). Following H2S exposure, treatment-2 was administered. Briefly, under isoflurane (2–3%) anesthesia, either saline or ODE (12.5%) were instilled intranasally (50 µL/mice) and mice were allowed to recover. Intra-nasal administration of ODE (12.5% v/v, 50 µL/mice) is known to induce lung inflammation, and this model has been well characterized to study innate inflammatory response to single or multiple day exposures to ODE (Poole et al. 2009). Following treatment-2, mice were returned to their cages. About 24 h later, mice were humanely euthanized by administering CO2 from a pressure cylinder as per the American Veterinary Medical Association (AVMA) Guidelines for Euthanasia of Animals.
BALF, tissue fixation, and staining
Following euthanasia, the trachea was exposed, and a small incision was made to cannulate a 20-gauge needle. Bronchoalveolar lavage fluid (BALF) was collected from the left lung lobe after ligating the right lung lobes. Each time 0.3–0.4 mL of ice-cold Hank’s balanced salt solution (HBSS) with 100 µM EDTA was instilled and collected for a total of three times. Total leukocytes in BALF were enumerated by counting the cells using a hemocytometer. About 20,000 leukocytes were spun, and a cytospin slide was prepared (800 rpm × 3 min) and stained with Shandon Kwik Diff Staining (Thermo Fisher Scientific) to enumerate the total and differential counts of the inflammatory cells recruited into the airways.
The right side non-lavaged lung lobes were snap-frozen by immersing in liquid nitrogen and stored at − 80 °C for western blotting and PCR. The left lung was fixed with 4% paraformaldehyde for 24 h, processed, and embedded, and 5-µm-thick tissue sections were cut using a microtome. The lung sections were processed for H&E staining (supplemental data) and immunofluorescence.
Quantification of histopathological changes in the lungs
Hematoxylin and eosin stained tissue sections were examined (n = 10 mice/group) by an investigator blinded to the treatment groups. An experienced researcher semi-quantified the specific morphological changes in the bronchioles, blood vessels, and alveolar septa using predetermined scoring criteria (outlined in Supplemental Table 1) and photographed the scored regions (Nikon Eclipse TE2000-U; SPOT Advanced imaging software, Michigan, USA).
Immunofluorescence
The lung sections were processed for staining with mouse monoclonal Claudin 1 antibody (1:1000, sc-81796, Santa Cruz Biotechnology, Dallas, TX). Lung sections were stained with claudin1 antibody for 12 h at 4° C followed by incubation with a biotinylated donkey anti-mouse biotin-conjugated secondary antibody (Jackson Immunoresearch, 1:300 for an hour at room temperature), and cy3 conjugated streptavidin (Jackson Immunoresearch, 1:400) for 30 min at room temperature. We quantified the staining intensity of the claudin 1 using a previously published method (Massey et al. 2019). Five randomly chosen fields per slide were processed, and a total number of pixels in the airways (bronchi and bronchioles) or alveolar septa were counted using ImageJ (NIH). Next, the total staining intensity from selected pixels (cy3) was measured using computer software (HC Image, Hamamatsu Corp, Sewickley, Pennsylvania). Total intensity (cy3) was divided by the number of pixels to obtain mean intensity (cy3) per pixel. The mean intensity (cy3) per pixel was analyzed and graphed.
In vitro models of H2S gas and ODE exposures
Human monocytic cell line (THP-1) was grown in RPMI media with 10% heat-inactivated fetal bovine serum (FBS), growth supplements, 100 U/mL of penicillin/streptomycin (Gibco), and 2 µg/mL of amphotericin B (Sigma) in a humidified chamber with 5% CO2 at 37 °C. The human bronchial epithelial cell line (BEAS-2B) was seeded onto type I bovine collagen (StemCell Technologies, Vancouver, BC, Canada) coated T-75 flasks. Cells were grown submerged in serum-free LHC-9 medium (Gibco) containing 100 U/mL of penicillin/streptomycin (Gibco) and 2 µg/mL of amphotericin B (Sigma) in a humidified chamber with 5% CO2 at 37 °C until approximately around 80% confluence was achieved. About 2–3 × 106 cells (THP-1 and BEAS-2B) were treated as outlined in the Table 2. Treatment-1 (H2S) was for a duration of 3 h followed by treatment-2 (ODE) for a duration of 24 h. Briefly, in treatment-1, cells were exposed to sodium hydrosulfide (NaHS, Sigma), a known H2S donor (Jiang et al. 2016; Zhao et al. 2014). According to the prior experiments, we measured NaHS amounts required to release 5, 10, and 50 ppm of H2S gas (Hu et al. 2007). We chose these H2S concentrations based on the fact that the National Institutes of Occupational Safety and Health (NIOSH) permissible exposure limits (PEL) for H2S are 10 ppm with 10 min ceiling and the current Occupational Safety and Health Act (OSHA) permissible exposure limits (PEL) for H2S are 20 ppm ceiling and 50 ppm with 10-min maximum peak (The National Institutes for Occupational Safety and Health (NIOSH) 2018). Three hours after treatment-1, cells were washed with culture medium and flask was flushed with medium to remove the remaining H2S gas traces before treatment with either media alone or with ODE 1% for THP-1 cells and 5% ODE for BEAS-2B cells for another 24 h (treatment-2). The 3-h exposure period was based on the fact that NaHS is suitable for shorter exposures (Jiang et al. 2016) due to the rapid release of H2S (Zhao et al. 2014). Subsequently, the cell pellets were collected to extract total protein or RNA. Cell culture supernatants were collected and immediately processed for quantification of secreted reactive nitrogen species (RNS) or stored at − 80 °C until used.
Quantification of cell viability
We performed MTT assay to quantify the viability of the cells after treatments. About 2 × 106 THP-1 cells were seeded/well in a 6-well plate. While BEAS-2B cells (5 × 104/well) were seeded in a 96-well cell culture plate and incubated in a 5% CO2 incubator until the cells reached 70–80% confluence. Cell treatments were performed as outlined in Table 1. Next, MTT dye ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Invitrogen) was loaded (5 ng/mL) into each well-containing cells and incubated at 37 °C for another 3–5 h. At the end of the incubation period, the supernatant was discarded and 100 µL DMSO (Sigma) was added for BEAS-2B cells. For THP1 cells, stained cells were collected and spun down, the pellet was dissolved in the required amount of DMSO, and dye absorbance (450 nm) was read in 96-well plates using SpectraMax M2 Gemini Microplate Reader (Molecular Devices, San Jose, CA).
Quantification of secreted RNS
Griess assay was performed as described (Gordon et al. 2011). Briefly, secreted RNS levels were indirectly quantified by measuring cell culture medium nitrite levels using Griess reagent (Sigma) and sodium nitrite standard curve, prepared using a stock solution of 200 µM. The assay was performed in a 96-well plate by adding 100-µL cell culture supernatants in duplicate wells (n = 5–6/group) along with Griess reagent (100 µL/well), and then absorbance was measured at 550 nm using SpectraMax M2 Gemini Microplate Reader (Molecular Devices, San Jose, CA).
Intracellular ROS
Intracellular reactive oxygen species (ROS) production was measured using chloromethyl derivative of dichlorodihydrofluorescein diacetate (CM-H2DCFDA) (ThermoFisher Scientific, USA). A working solution of 10 µM of DCFDA in 1× HBSS was prepared. BEAS-2B cells (5 × 104/well) were seeded in a 96-well cell culture plate and incubated in a 5% CO2 incubator to reach confluence. The cells were incubated with H2DCFDA working solution at 37 °C for 60 min, then washed with PBS, and followed by treatments 1 and 2 (as outlined in Table 1). About 2 × 106 THP-1 cells were seeded in each well in 6-well plates and stained with 10 µM of CM-H2DCFDA dye for an hour at 37 \(^\circ \mathrm{C} \text{5\% CO}{2}\text{ incubator}\). Cells were centrifuged to remove the dye and were washed with 1× PBS followed by treatments-1 and -2 (Table 1). Cells suspended in 1X PBS were loaded onto 96-well plate (n = 6), and the fluorescence intensity of the oxidized form of H2DCFDA was measured at excitation/emission wavelengths of 488/535 nm, respectively (SpectraMax M2 Gemini Microplate Reader, Molecular Devices, San Jose, CA). The results were expressed as percentage fluorescence relative to control.
Western blotting
The cell pellets and lung tissue homogenates were lysed in RIPA buffer with a 1× protease inhibitor cocktail (Thermo Fisher Scientific, USA), and the resulting homogenates were centrifuged at 14,000 rpm for 20 min at 4 °C for whole-cell or tissue lysate preparation, respectively. Protein concentration was determined using a Bradford protein assay kit (Bio-Rad, USA). Equal amounts of protein were separated using different gel percentages: 7.5%, 10%, and 12% SDS-PAGE gels and electro transferred onto nitrocellulose membranes overnight using a constant voltage (23 V). Next, the membranes were incubated with the fluorescent blocking buffer (Rockland Immunochemicals, PA, USA) in PBS with 0.05% Tween-20 for an hour at room temperature to reduce non-specific binding followed by incubation with primary antibodies for staining overnight at 4 °C. Primary antibodies included rabbit anti-HMGB1 (1:1000, ab79823), rabbit anti-NF-κB, p-65 subunit (1:1000, ab16502, Mouse monoclonal VHL (1:1000, sc-17780), rabbit monoclonal anti-iNOS (1:1000, ab3523, mouse monoclonal Claudin 1 (1:1000, sc-81796), rabbit anti-B-actin (1:10,000, ab8227), or rabbit anti-Lamin B1 (1:1000, ab16048, Abcam, Cambridge, MA). The blots were washed in 1× PBS with 0.1% tween 20 (PBST) for 15–20 min twice and stained with secondary antibodies consisting of Alexa Fluor 680 Donkey anti-Rabbit IgG (1:10,000, Jackson) or anti-mouse 680 Alexa Fluor antibodies (1:10,000, ThermoFisher Scientific). Membranes were scanned using the Odyssey® CLx IR imaging system (LI-COR Biotechnology, Lincoln, NE). Following normalization using relevant housekeeping protein, densitometry analysis was performed using the ImageJ program (NIH).
qRT-PCR
The cell pellets and lung tissue homogenates were digested in TRIZOL (Sigma) for RNA extraction. The RNA concentration was measured using SpectraMax M2 Gemini Microplate Reader (Molecular Devices, San Jose, CA). Total RNA (0.5–2 µg) was used for the cDNA synthesis using the Superscript III first-strand synthesis kit (ThermoFisher Scientific, USA) as per the manufacturer’s protocol. For the qRT-PCR, 1 µL of cDNA per 10 µL of reaction volume including 1 µL of primers each, 5 µL of SYBR green and DPEC-treated water was used. Reactions were performed using forward and reverse primers. Sequences of all the primers (forward and reverse) listed in Supplemental Tables 2 and 3 were synthesized at Iowa State University’s DNA Facility. The housekeeping gene 18 S rRNA (ThermoFisher Scientific, USA) was used in all qPCR reactions. After 5 min of initial denaturation at 95 °C, reactions were carried out for 40 cycles, followed by a final elongation step at 72 °C for 10 min. All the reactions were run in a Bio-Rad CFX Connect™ detection system with CFX manager software. The mean threshold cycle (Ct) values were calculated and analyzed using the 2−ΔΔCt method (Rao et al. 2013).
Cytokine assay
Cytokines were quantified using commercially available kits from Thermo Fisher Scientific as per the manufacturer’s instructions. The lower limit of detection for all the cytokines was 2 pg/mL. Briefly, 96-well high binding plates (Nunc MaxiSorp, ThermoFisher Scientific) were coated with a capture antibody (100 µL/well) and incubated at 4 °C overnight. Wells were blocked with a blocking buffer (200 µL/well) for an hour at room temperature followed by washing with 1× PBST (three times) and then, incubated with 100 µL/well of recombinant standards or samples in duplicate wells (cell culture supernatants) for 2 h at room temperature. Next, plates were washed thrice with PBST and incubated with a detection antibody (100 µL/well) for an hour at room temperature. Finally, incubation was done with 3,3′,5,5′-tetramethylbenzidine (TMB) solution (100 µL/well) for 15 min (in dark) after removal of detection antibody and washing 5–7 times and the color development reaction was stopped by adding 50 µL/well of a stop solution (2 N H2SO4). The absorbance was read at 450 nm using SpectraMax M2 Gemini Molecular Device Microplate Reader.
Statistics
All data were expressed as mean ± SEM and analyzed by one-way and two-way ANOVA followed by Tukey’s post hoc comparison tests (GraphPad Prism, La Jolla, CA). A p ≤ 0.05 was considered statistically significant.
Results
LPS was used as a positive control pathogen-associated molecular pattern and a TLR4 agonist. Data from the LPS group is not included in this manuscript.
Airway inflammatory cell infiltration with H2S and ODE exposure
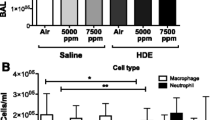
Animals exposed to 10 ppm H2S followed by ODE showed significantly higher total leukocyte counts in BALF compared with control and ODE (Fig. 1a). This increased leukocyte count was characterized by an increased infiltration of the macrophages and lymphocytes (Fig. 1c, d). However, neutrophils in the BALF were not significantly different among any of the groups (Fig. 1b).

H2S and ODE exposure induced recruitment of inflammatory cells into the BALF (broncho-alveolar lavage fluid). The cytospin preparations from BALF were enumerated for total leukocytes (a), neutrophils (b), macrophages (c), and lymphocytes (d). 10 ppm H2S followed by ODE increased the infiltration of leukocytes (a), macrophages (c), and lymphocytes (d) into BALF. Data (mean ± SEM, n = 8–10 mice/group) analyzed with two-way ANOVA followed by Tukey’s post hoc analysis is presented, and p ≤ 0.05 was considered significant (* compared with 0 ppm control, ɸ compared with 0 ppm ODE)
H2S and ODE exposure–induced histopathological changes in the lungs
Histopathological changes due to exposure to either H2S or ODE or co-exposure to H2S and ODE were not significantly different among any of the groups (supplemental Fig. S1, a-c).
H2S and ODE exposure–induced expression of TLR2 and TLR4 mRNA transcripts
Quantification of tlr2 and tlr4 gene transcripts revealed that 10 ppm H2S exposure significantly increased the fold change in the tlr2 and tlr4 transcripts when compared with 0 ppm control and 5 ppm H2S (Fig. 2a, b).

H2S-induced expression of TLR gene transcripts Rt-qPCR in the lung homogenates of the mice showed increased expression of tlr2 (a) and tlr4 (b) in 10 ppm H2S treatment group as compared with 0 ppm control and 5 ppm H2S. Data (mean ± SEM, 6–10 mice /group) analyzed with two-way ANOVA followed by Tukey’s post hoc analysis is presented, and p ≤ 0.05 was considered significant (* compared with 0 ppm control, # compared with 5 ppm control)
H2S and ODE exposure and expression of NF-кBp65
NF-кBp65 expression was quantified using western blotting and qRT PCR, and there was no difference in the protein and mRNA levels of nfкbp65 in any of the groups (Fig. 3a–c).

H2S induced expression NF-кBp-65 Western blotting of lung homogenates followed by densitometry analysis revealed no significant changes in NF-кBp-65 expression among different treatment groups (a, b). Also, RT-qPCR of lung homogenates showed no significant differences in nf-кbp-65 mRNA expression among different treatment groups (c). Data (mean ± SEM, n = 6–10 mice/group) analyzed with two-way ANOVA followed by Tukey’s post hoc analysis is presented, and p ≤ 0.05 was considered significant
H2S and ODE exposure–induced oxidative stress in mice
Oxidative stress was measured by quantifying oxidative stress (NOS2, VHL, HIF1-α, NCF1, NCF2, and NCF4) and anti-oxidant (NRF2) markers using western blotting and qRT-PCR. Compared with saline-treated control mice, none of the treatment groups showed any significant changes in NOS2 protein expression and inos mRNA fold change (Fig. 4 (a–a′, c)). Similarly, VHL protein and vhl1 gene expression did not differ significantly among any of the treatment groups (Fig. 4 (b–b′, d)). However, 10 ppm H2S exposure induced significantly higher expression of transcripts for ncf1, ncf4, hif1α, and nrf2 compared with 0 ppm control and 5 ppm H2S (Fig. 4 (e, g, h, and i), respectively). However, 10 ppm H2S followed by ODE exposure significantly decreased ncf1, ncf4, and nrf2 as compared with 10 ppm H2S alone (Fig. 4 (e, g, and i), respectively). Transcripts of ncf2 did not change significantly in any of the groups (Fig. 4 (f)).

H2S and OD exposure induced oxidative stress in the mouse lung tissue Western blotting analysis and RT-qPCR of the mice lungs showed no significant difference in expression of NOS2 (a-a’) and inos (c) and vhl (b-b’, d) among the different treatment groups. However, fold change in inos gene was higher in 10 ppm H2S and 10 ppm H2S followed by ODE exposure as compared with control (d). About 10 ppm H2S induced significantly higher fold change expression of ncf1 (e), ncf4 (g), hif1a (h), and nrf2 (i) as compared with 0 ppm control and 5 ppm H2S. 10 ppm H2S with ODE significantly decreased ncf1 (e), ncf4 (g), and nrf2 (i) when compared with 10 ppm H2S. Data (mean ± SEM, n = 6–10 mice/group) analyzed with two-way ANOVA followed by Tukey’s post hoc analysis is presented, and p ≤ 0.05 was considered significant (* compared with 0 ppm control, # compared with 5 ppm control, $ compared with 10 ppm control)
H2S and ODE exposure–induced expression of HMGB1
HMGB1 protein and mRNA fold changes were measured, and compared with control mice, no significant differences in the expression of HMGB1 protein were seen among any of the groups (Fig. 5a, b).

H2S and ODE exposure induced expression of HMGB1. Cell homogenates processed for western blotting (a, b) and qRT-PCR (c) to quantify HMGB1 protein and mRNA fold changes. Densitometry values showed no significant difference of HMGB1 protein expression among the groups (b). Data (mean ± SEM, n = 6–10 mice/group) analyzed with two-way ANOVA followed by Tukey’s post hoc test is presented, and p ≤ 0.05 was considered significant
H2S and ODE exposure–induced expression of cytokine transcripts
Pro and anti-inflammatory cytokines were measured by quantifying fold changes in mRNA transcript using qRT-PCR. Compared with 5 ppm H2S exposed mice, 10 ppm H2S exposure significantly increased the tnfα and il-1β transcripts (Fig. 6a, c). Compared with 0 ppm control and 5 ppm H2S, 10 ppm H2S exposure increased the fold change in the expression of il-6, cxcl2, il-10, and tgfβ (Fig. 6 b, d, e, and f, respectively). Whereas, 10 ppm H2S followed by ODE exposure significantly decreased the fold change in the expression of il-6, cxcl2, il-10, and tgfβ as compared with 10 ppm H2S exposure (Fig. 6 b, d, e, and f, respectively). However, 10 ppm H2S followed by ODE exposure significantly increased the fold change in the expression of tgfβ as compared with ODE alone (Fig. 6f).

H2S and ODE exposure induced expression of cytokine mRNA transcripts Rt-qPCR of the lung homogenates from 10 ppm H2S exposed mice showed higher fold change in both inflammatory (a–d) and anti-inflammatory (e, f) cytokines mRNA transcripts as compared with 0 ppm control, 5 ppm H2S, and 10 ppm H2S with ODE. Data (mean ± SEM, n = 6–10 mice/group) analyzed with one-way ANOVA followed by Tukey’s post hoc test is represented, and p ≤ 0.05 was considered significant (* compared with 0 ppm control, # compared with 5 ppm control, $ compared with 10 ppm control, ɸ compared with 0 ppm ODE)
Impact of H2S and ODE exposure on the tight junction proteins/mRNAs of airway epithelium
Claudin 1 immunofluorescence staining revealed expression in bronchioles (Fig. 7a) and alveolar septa (data not shown) within the lung sections. The quantification of the staining intensity revealed that 10 ppm H2S alone (a‴) when compared with 0 (a′) and 5 ppm (a″) controls (H2S), and ODE (a″″) alone when compared with 0 ppm control significantly decreased the expression of claudin 1 protein in the bronchi and bronchioles (Fig. 7b). The staining intensity of claudin 1 protein in the alveolar septa did not differ between any of the groups (data not shown). Next, we used western blotting analysis and qRT-PCR to quantify the protein and mRNAs of claudin 1 and found no difference among any of the treatment groups (Fig. 7 (c-c′, d)). However, 10 ppm H2S exposure did increase claudin3 mRNA fold changes compared with 5 ppm H2S alone (Fig. 7 (e)). Whereas, 10 ppm H2S followed by ODE exposure significantly decreased the fold change in the expression of claudin3 as compared with 10 ppm H2S exposure (Fig. 7 (e)). Claudin18 mRNA fold change was significantly higher for 5 ppm H2S with ODE as compared with 5 ppm H2S alone (Fig. 7 (g)). Interestingly, for claudin5 mRNA, no significant differences were observed among any of the different treatment groups (Fig. 7 (f)).

Impact of H2S and ODE exposure on tight junction protein and mRNA expression Immunofluorescence staining on five-micron thick lung tissue sections showed that the expression of claudin1 in bronchioles decreased significantly in 10 ppm H2S (a‴) and ODE (a″″) exposed mice (n = 6–10) as compared with control (a′) and 5 ppm H2S(a″) (a, b). Western blotting and Rt-qPCR showed no change in expression of claudin 1 among different treatment groups (c–c′, d). 10 ppm H2S significantly increased the fold change in claudin3 as compared with 5 ppm H2S (e). claudin5 mRNA showed no significant differences among the treatment groups (f). claudin18 mRNA expression was significantly increased in 5 ppm H2S followed by ODE as compared with 5 ppm H2S (g). (Micrometer bar = 100 µm). Data (mean ± SEM, n = 6–10 mice/group) analyzed with two-way ANOVA followed by Tukey’s post hoc analysis is presented, and p ≤ 0.05 was considered significant (* compared with 0 ppm control, # compared with 5 ppm control, $ compared with 10 ppm control)
Cell viability following exposure to H2S alone or H2S and ODE exposure
Cellular metabolic activity and viability were assessed using the MTT assay. Exposure to H2S alone or H2S and ODE exposure significantly decreased the cell viability in THP-1 cells. Compared with 0 ppm control, 5, 10, and 50 ppm H2S exposure alone or H2S followed by ODE or ODE alone significantly decreased the viability of THP-1 cells (Fig. 8a). Within the ODE + H2S exposed groups, compared with 0 ppm H2S exposed cells, 5, 10, and 50 ppm H2S exposure significantly decreased the viability of THP-1 cells (Fig. 8a). Neither H2S alone nor H2S and ODE exposure altered the cell viability significantly in BEAS-2B cells (Fig. 8b).

H2S and ODE exposure and cell viability MTT assay was used to access the cell viability and showed that the cell viability was decreased with H2S exposure alone or ODE alone or H2S with ODE as compared with control in THP-1 cells (a) but not in BEAS-2B cells (b). Data (mean ± SEM, n = 6/group) analyzed with two-way ANOVA followed by Tukey’s post hoc test is shown, and p ≤ 0.05 was considered significant (* compared with 0 ppm control, ɸ compared with 0 ppm ODE),
Quantification of oxidative stress using in vitro models of H2S alone or co-exposure to H2S and ODE
Oxidative stress was measured using Griess and DCFDA assay by way of quantifying secreted nitrite (representing RNS) and intracellular ROS, respectively. Compared with 0 ppm control, 10 and 50 ppm H2S exposure alone or ODE alone or ODE with 5 ppm, 10 ppm, and 50 ppm H2S exposure significantly increased the secreted nitrite levels in THP-1 cells (Fig. 9a). However, ODE with 50 ppm H2S significantly increased the nitrite levels compared with 5, 10, and 50 ppm H2S exposure and ODE alone (Fig. 9a). Similarly, 50 ppm H2S followed by ODE significantly increased the nitrite levels as compared with 50 ppm H2S and ODE alone (Fig. 9a). However, the nitrite levels were significantly decreased with 10 ppm H2S exposure alone in BEAS-2B cells (Fig. 9b). Next, in BEAS-2B cells, significantly higher secreted nitrite levels were seen in ODE groups as compared with respective H2S treatments in control group and 0 ppm control (Fig. 9b). Within ODE groups, 5 and 10 ppm H2S exposure significantly decreased the secreted nitrite levels compared with 0 ppm. Next, inos mRNA was measured as a source of secreted nitrite. The fold change in inos mRNA in H2S alone groups did not change significantly in both the cell lines (Fig. 9c, d). However, in THP-1 cells, ODE alone when compared with 0 ppm control and ODE with 50 ppm H2S when compared with 0 ppm control and 50 ppm H2S significantly induced inos mRNA (Fig. 9c). Whereas in the ODE group, H2S (5, 10 ppm) and ODE co-exposure significantly decreased inos mRNA transcription as compared 0 ppm ODE (Fig. 9c).

Quantification of H2S and ODE exposure induced oxidative stress using in vitro models Pre-exposure to H2S followed by ODE and ODE alone significantly increased the oxidative stress as compared with control and H2S exposed groups in THP-1 cells measured in terms of RNS (Griess assay), inos (RT-qPCR), ROS (DCFDA assay), and nrf2 (RT-qPCR). In BEAS-2B cells, pre-exposure to H2S followed by ODE and ODE alone significantly increased RNS secretion as compared with control and H2S exposed groups (b). RT-qPCR showed increased fold change in expression of nrf2 mRNA in both the cell lines treated with ODE and ODE with H2S as compared with control group (g, h). Data (mean ± SEM, n = 3/group, in ROS and RNS, n = 5–6/group) analyzed with two-way ANOVA followed by Tukey’s post hoc is represented, and p ≤ 0.05 was considered significant (* compared with 0 ppm control, # compared with 5 ppm control, $ compared with 10 ppm control, + compared with 50 ppm control and ɸ compared with 0 ppm ODE)
Compared with treatments in control group, ODE alone or H2S with ODE-treated THP-1 cells produced significantly higher amounts of intracellular ROS cells (Fig. 9e). Interestingly, exposure to H2S in both control and ODE + H2S group significantly decreased the intracellular ROS levels when compared with 0 ppm and ODE, respectively (Fig. 9e). However, H2S exposure followed by ODE significantly increased the ROS level as compared with respective treatments in the control group (Fig. 9e). In BEAS-2B cells, ODE alone significantly increased the intracellular ROS levels when compared with 0 ppm controls, whereas 10 ppm H2S and ODE co-exposure significantly decreased the intracellular ROS levels when compared with ODE alone (Fig. 9f).
Next, nrf2 mRNA was quantified as readout of the anti-oxidant signaling. Compared with the control group in THP-1 cells, ODE alone or ODE with 5, 10, and 50 ppm H2S co-exposure significantly increased the fold change in the expression of nrf2 transcript as compared with 0 ppm and respective treatments in the control group (Fig. 9g). Similarly, in BEAS-2B cells, ODE alone when compared with 0 ppm control or ODE with 50 ppm H2S compared with 0 ppm control and 50 ppm H2S, significantly increased fold change expression of nrf2 transcript (Fig. 9h). However, 5 and 10 ppm H2S and ODE co-exposure in BEAS-2B cells significantly decreased the fold change in the nrf2 transcript when compared with 0 ppm ODE, but the fold change was higher compared with control group (Fig. 9h).
H2S and ODE exposure and expression of tlr2 and tlr4 genes
Fold changes in the expression of tlr2 and tlr4 genes were quantified to examine the impact of pre-exposure to H2S on host response to ODE-induced inflammation signaling. H2S exposure alone did not significantly alter the expression of tlr2 transcripts in both THP-1 and BEAS-2B cells (Fig. 10 a and b, respectively). Compared with 0 ppm control, H2S (0, 5, 10, and 50 ppm) and ODE co-exposure significantly increased the fold change in the tlr2 transcript. Compared with 5, ppm controls, 5 ppm H2S and ODE co-exposure significantly increased the fold change in the tlr2 transcript (Fig. 10a). Interestingly, compared with ODE alone, 10 and 50 ppm H2S and ODE co-exposure significantly decreased the fold change in the tlr2 transcript in THP-1 cells (Fig. 10a). In BEAS-2B cells, compared with 0 ppm control, ODE with 10 ppm H2S significantly increased the fold change in the tlr2 transcript (Fig. 10b). Compared with 0 ppm control, H2S (0, 5, and 10 ppm) and ODE co-exposure significantly increased the fold change in the tlr4 transcript (Fig. 10c). Compared with 5 ppm H2S, 5 ppm H2S and ODE co-exposure significantly increased the fold change in the tlr4 transcript (Fig. 10c). In BEAS-2B cells, 10 ppm H2S with ODE significantly increased the expression of the tlr4 gene compared with 0 ppm control, 10 ppm H2S and ODE alone, (Fig. 10d).

H2S and ODE exposure induced expression of TLR gene transcripts RT-qPCR of THP-1 cells showed increased fold change of tlr2 and tlr4 mRNA following exposure to H2S followed by ODE as compared with control (a, c). In BEAS-2B cells, 10 ppm H2S followed by ODE significantly increased tlr2 mRNA folds as compared with control (b). Also, 10 ppm H2S followed by ODE exposure significantly increased tlr4 mRNA folds as compared with 0 ppm control, 10 ppm H2S, and ODE alone in BEAS-2B cells (d). Data (mean ± SEM, n = 3/group) analyzed with two-way ANOVA followed by Tukey’s post hoc analysis is presented, and p ≤ 0.05 was considered significant (* compared with 0 ppm control, # compared with 5 ppm control, ɸ compared with 0 ppm ODE, $ compared to 10 ppm control)
H2S-induced expression of NF-κBp65
mRNA expression of NF-κBp65 was quantified to assess if H2S exposure alone or H2S and ODE co-exposure predominantly use this pathway for inflammatory cytokine signaling (Fig. 11a–d). When BEAS-2B cells were exposed to either H2S or ODE alone for 10 and 15 min, the nuclear levels of NF-κBp65 significantly increased when compared with respective cytoplasmic levels (Fig. 11 a and b). Exposure of BEAS-2B cells with 50 ppm H2S alone significantly increased the nfkbp65 mRNA fold change compared with 0 and 5 ppm H2S controls (Fig. 11c). However, in THP-1 cells, compared with 0 ppm H2S control, 10 and 50 ppm H2S exposure and different treatments in the ODE group significantly increased the fold change in the expression of nfkbp65 transcript (Fig. 11d). Within the ODE exposure groups, 5 ppm H2S co-exposure increased the fold change in the expression of nfkbp65 transcript in comparison to 5 ppm H2S alone in THP-1 cells. Within the ODE groups, co-exposure to H2S (5, 10 or 50 ppm) decreased the fold change in the transcript of nfkbp65 (Fig. 11d).

H2S and ODE exposure induced expression NF-κBp-65 Western blotting analysis of the BEAS-2B cells (a) followed by densitometry analysis (b) showed increased nuclear translocation of NF-κBp-65 at shorter time points following exposure to 10 ppm H2S and ODE. Rt-qPCR showed increased fold change expression of nf-кbp65 gene in BEAS-2B (n = 3/group) following exposure to 50 ppm H2S as compared with control and 5 ppm H2S exposure (c). In THP-1 cells (n = 3/group), compared with control, 10 ppm H2S, 50 ppm H2S, and H2S with ODE showed significantly higher nf-кbp65 gene folds (d). Compared with ODE exposed THP-1 cells, H2S followed by ODE exposure decreased nf-кbp65 gene folds (d). Data (mean ± SEM, n = 3/group) analyzed with two-way ANOVA followed by Tukey’s post hoc analysis is presented and p ≤ 0.05 was considered significant (* compared with 0 ppm control, # compared with 5 ppm control, ɸ compared with 0 ppm ODE, δ between the cytoplasmic and nuclear fraction)
Pro-inflammatory and anti-inflammatory cytokines with H2S and ODE exposure
H2S exposure induced the secretion of pro and anti-inflammatory cytokines in THP-1 and BEAS-2B cell lines. Compared with 0 ppm H2S control, H2S alone or H2S followed by ODE exposure significantly increased IL-1β, IL-6, IL-8, and IL-10 levels in both the cell lines (Fig. 12a–h).

H2S and ODE exposure induced secretion of cytokines and chemokines ELISA was done to measure the cytokines released in the supernatants of both THP-1 (a–d) and BEAS-2B (e–h) cells. Pre-exposure to H2S followed by ODE further increased the secretion of both inflammatory and anti-inflammatory cytokines in both the cell lines as compared with control group. Data (mean ± SEM, n = 5–6/group) analyzed by two-way ANOVA followed by Tukey’s post hoc analysis is presented, and p ≤ 0.05 was considered significant (* compared with 0 ppm control, # compared with 5 ppm control, $ compared with 10 ppm control, + compared with 50 ppm control and ɸ compared with 0 ppm ODE)
Compared with 0 ppm control, H2S alone or H2S with ODE co-exposure produced significantly higher amounts of IL-1β (Fig. 12a). Compared with H2S exposure alone or ODE alone, H2S with ODE further accentuated IL-1β secretion, in THP-1 cells (Fig. 12a). In BEAS-2B cells, compared with 0 ppm control and 5 ppm H2S, 50 ppm H2S alone significantly increased the IL-1β production. Similarly, compared with different H2S treatments in the control group, respective H2S treatments in the ODE group significantly increased the production of IL-1β. Within the ODE groups, compared with ODE alone, 50 ppm H2S and ODE co-exposure significantly increased the IL-1β production in BEAS-2B cells (Fig. 12e).
Compared with 0 ppm controls, 5 ppm H2S alone as well as H2S (0, 5, 10, and 50 ppm) and ODE co-exposure significantly increased the IL-6 production in THP-1 cells. Compared with H2S alone, pre-exposure to H2S followed by ODE significantly increased the IL-6 production in THP-1 cells (Fig. 12b). Interestingly, compared with ODE alone, H2S and ODE co-exposure significantly increased the IL-6 production in THP-1 cells (Fig. 12b). In BEAS-2B cells, compared with 0 ppm control, 5 and 50 ppm H2S exposure significantly decreased the IL-6 production. Different H2S treatments within the ODE group significantly increased the IL-6 production when compared with 0 ppm control and respective H2S treatments in control group (Fig. 12f).
In THP-1 cells, compared with 0 ppm control, H2S alone, as well as H2S and ODE co-exposure significantly increased the production of IL-8 (Fig. 12c). In BEAS-2B cells, compared with 0 ppm controls, H2S and ODE co-exposure significantly increased the IL-8 levels. Pre-exposure to H2S followed by ODE further exaggerated the IL-8 secretion in both the cell lines (Fig. 12 c and g).
In both the cell lines, no significant elevation in IL-10 secretion was seen in H2S treatment alone as compared with 0 ppm control (Fig. 12d, h). In THP-1 cells, IL-10 secretion in the ODE group was significantly elevated when compared with 0 ppm control and ODE alone and respective H2S treatments in control group (Fig. 12d). Similarly, different treatments in the ODE group significantly increased the production of IL-10 in BEAS-2B cells when compared with 0 ppm control and respective H2S treatments in the control group except for the 50 ppm H2S (Fig. 12h).
Discussion
The physiological role of endogenously produced H2S is beginning to emerge (Zhang et al. 2017), while the toxic nature of supra-physiological concentrations of H2S on respiratory, nervous, and other body systems is being studied (Anantharam et al. 2018, 2017a, 2017b; Kim et al. 2018).
In the current manuscript, we examined the impact of pre-exposure to H2S on the innate inflammatory response of the lung to OD. Using both in vitro and in vivo models of H2S followed by ODE exposure, we show that pre-exposure to H2S alters the innate inflammatory response of the lungs to ODE. There may be concentration dependent effects of exposure to H2S and OD.
In general, exposure to H2S alone or H2S followed by ODE exposure decreased the cell viability, increased the secretion of RNS and ROS suggesting the induction of oxidative stress whereas ODE exposure alone increased the expression of Pattern Recognition Receptors (PRRs) namely tlr2 and tlr4 as well as nfkbp65 mRNAs. H2S or ODE exposure increased the protein levels of IL-1β, IL-6, IL-8 and IL-10. Interestingly, in a mouse model of H2S and ODE exposure, significant changes were observed in animals exposed to 10 ppm H2S but ODE exposure suppressed the H2S-induced pro-inflammatory cytokine gene expression possibly due to interaction of multiple signaling pathways (Nath Neerukonda et al. 2018).
Exposure to H2S alone or H2S followed by ODE or ODE alone in human monocytic (THP-1) cells reduced the cell viability, whereas the viability of human bronchial epithelial cells (BEAS-2B) remained unaffected. This difference in the sensitivity of THP-1 cells in comparison to BEAS-2B is possibly due to the inherent sensitivity and pro-inflammatory nature of the monocytes (Basic et al. 2017).
Our data indicate that both H2S and ODE exert toxic effects on THP-1 cells and influence innate inflammatory responses in the lung. The decrease in the viability of THP-1 cells could partly be explained by the fact that exposure to either H2S or ODE or both significantly increased the production of secreted nitrite levels (representing RNS). Exposure of THP-1 cells to 50 ppm H2S followed by ODE produced much higher levels of RNS than all other groups indicating synergistic effects of combined exposure. Surprisingly in BEAS-2B cells ODE alone or H2S followed by ODE exposure significantly increased the RNS production without affecting the cell viability. These results indicate the fundamental differences in the cell viability between THP-1 (monocytic) and BEAS-2B (bronchial epithelial) cells. We used these two (THP-1 and BEAS-2B) cell lines to model the exposure of alveolar macrophages and bronchial epithelial cells in the lungs respectively. However, we are aware of the limitation of modeling alveolar macrophages using THP-1 (monocytic cells) (Bosshart and Heinzelmann 2016). Upon exposure to ODE alone or H2S followed ODE, THP-1 cells significantly upregulated the expression of inos mRNA but not the BEAS-2B cells to indicate the inherent pro-inflammatory nature of the monocytic cells (Basic et al. 2017) when compared with the airway epithelial cells indicating that oxidative stress was more severe in THP-1 cells. Despite not using an alveolar macrophage cell line or primary alveolar macrophages, we provide data to show that pre-exposure to H2S could increase oxidant-induced lung injury following exposure to OD. Our results are in line with another study that demonstrated that swine dust extract exposure of human primary airway epithelial primary cells increases the nitrite levels which could be abrogated using sorrel extract (Gerald et al. 2019).
THP-1 and BEAS-2B cells show different reactive species production responses upon pre-exposure to H2S alone indicating cell-specific mechanisms of H2S impact. Further, THP-1 cells exhibit higher sensitivity when exposed to H2S followed ODE indicating that targeting lung macrophages with anti-inflammatory strategies could be beneficial. Another study has documented that swine dust exposure of THP-1 cells leads to a concentration-dependent increase in ROS production (Pender et al. 2014). Interestingly, ODE exposure groups in both THP-1 and BEAS-2B cells showed a significant increase in nrf2 gene expression indicating a possible role for NRF2-mediated anti-oxidant response as studied before (McGovern et al. 2019).
Exposure to ODE alone or H2S (5 ppm) followed by ODE resulted in the upregulation of both tlr2 and tlr4 genes in THP-1 cells, whereas only tlr2 gene expression was increased in ODE alone groups in the BEAS-2B cells. Interestingly, in THP-1 cells pre-exposure to 10 or 50 ppm H2S followed by ODE led to a decrease in the expression of tlr2 and tlr4, respectively. These results are in line with the previous published work wherein we (Charavaryamath et al. 2008) and others (Poole et al. 2011) had made similar observations that both TLR2 and TLR4 play roles in OD-induced lung inflammation. Despite lack of TLR2 or 4 transcriptional responses to H2S exposure, TLR2 and 4 antagonisms may be beneficial in curtailing the effect of co-exposure to H2S and ODE. Our results show an upregulation of nfkbp65 mRNA but not the protein levels following H2S exposure or ODE alone or H2S followed by ODE in our in vitro models. In BEAS-2B cells upon exposure to H2S or ODE for 10 and 15 min resulted in a significant increase in the mRNA levels of nfkbp65 in the nuclear fractions when compared with cytoplasmic fractions. These observations are in line with our previous published work (Bhat et al. 2019).
Interestingly, H2S exposure followed by ODE augmented the production of inflammatory cytokines to indicate the priming effect of pre-exposure to H2S. In order to explore if DAMPs such as HMGB1 could be involved in priming the lung innate inflammatory response, we quantified the HMGB1 protein in both cytoplasmic and nuclear fractions and found no significant changes. It might be possible that some of the secreted HMGB1 could have formed complexes with other molecules and these complexes were not detected by our antibody (Willis et al. 2018). Alternatively, HMGB1 may be secreted into the cell culture supernatants. We were able to measure secreted free-HMGB1 in the mouse lung BALF (data not shown).
Results from our mouse experiments primarily indicated a synergistic effect of pre-exposure to H2S (10 ppm) followed by ODE. Compared with all other groups, animals exposed to 10 ppm H2S followed by ODE showed a significantly higher number of leukocytes in the BALF which was characterized by an increase in the macrophage numbers but not neutrophils and lymphocytes. However, H and E stained lung tissue sections showed no significant difference in the pathology scores. Our study did not explore the mechanistic basis of influx of macrophages in those mice exposed to H2S and OD. It is likely that, combined exposure resulted in CCL2 (MCP1) and CCL3 (MIP1α) mediated recruitment and influx of monocytes into the airways (Shi and Pamer 2011).
Previously, we have shown that exposure to the barn environment containing OD could result in airway epithelial damage independent of TLR4 (Charavaryamath et al. 2008). Therefore, we mechanistically addressed if H2S or ODE exposure would affect the airway epithelial tight junctions and showed that compared with all other groups, mice exposed to 10 ppm H2S significantly upregulated the expression of claudin3 gene but not claudin1, claudin5, and claudin18. However, claudin18 was significantly upregulated when compared with 5 ppm H2S alone exposure. The upregulation of claudin18 is an indication of alveolar type II cell-mediated repair response (Kotton 2018), whereas an increase in claudin3 is known to be pro-carcinogenic (Che et al. 2018; de Souza et al. 2013). Interestingly, compared with all other groups, mice exposed to 10 ppm H2S significantly upregulated the HMGB1 mRNA in comparison to ODE exposure alone but not the protein levels. Previously we have shown that ODE exposure leads to nucleocytoplasmic translocation of HMGB-1 and HMGB-1 could be targeted to curtail ODE induced inflammation (Bhat et al. 2019; Massey et al. 2019). Interestingly, exposure to 10 ppm H2S alone when compared with other groups leads to an increase in the transcripts of tlr2 and tlr4 without any significant change in the mRNA or protein levels of NF-κB p65.
Next, we explored the mechanistic basis of oxidative stress-induced inflammation in H2S or ODE or both exposure groups by quantifying NOS2 (protein) or inos mRNA expression. Compared with controls, ODE exposed mice showed an increase in NOS2 protein without any change in inos gene expression. NOS2 has been shown to be important in carcinogenesis and inflammation (Wang et al. 2018). Interestingly, compared with all other groups, mice exposed to 10 ppm H2S showed an upregulation of ncf1, ncf4, hif1α, and nrf2 but not ncf2. Further, compared with all other groups, mice exposed to 10 ppm H2S alone showed an upregulation of mRNA for tnf-α, il-6, il-1β, cxcl-2, il-10, and tgf-β to indicate that TLR2 and TLR4, HIF1-α, and NRF-2 mediated responses result in inflammatory cytokine production. Addition of ODE to the H2S exposed animals resulted in a dampened inflammatory response through downregulation of mRNA for tnf-α, il-6, il-1β, cxcl-2, il-10, and tgf-β possibly due to complex and multiple overlapping signaling events (Nath Neerukonda et al. 2018).
Overall, our study explored if pre-exposure to H2S would modulate lung innate response to OD exposure through the use of in vitro and in vivo models. There was a clear disconnect between some of the results observed in our in vitro models to our mouse (in vivo) model. This could be due to different sensitivities of human airway epithelial (BEAS-2B) and monocytic (THP-1) cell lines to that of the mouse strain used. Another possible weakness in our study is the use of cell lines instead of primary human cells. Future studies using mouse precision cut lungs slices (PCLS) and mouse models may provide improved translation between in vitro and in vivo models.
Conclusions
Based on the results from our in vivo and in vitro experiments, we conclude that H2S exposure effects appear to modulate the inflammatory signals induced by ODE. To bring relevance to the real-world exposures, future studies examining the impact of multiple exposures to H2S alone and H2S followed by ODE and targeting TLR2/4, NRF2, and oxidative stress pathways to abrogate inflammation would be valuable. The scientific community would benefit from these studies related to agriculture-exposure associated lung diseases since exposure to H2S, and organic dust may be seen in other settings.
References
American Conference of Governmental Industrial Hygienists (2019) Monitoring H2S to meet new exposure standards—Occupational Health & Safety. vol 2019. Occupational Health and Safety, ohsonline.com
American Thoracic Society (1998) Respiratory health hazards in agriculture. Am J Respir Crit Care Med 158:S1–S76
Anantharam P, Kim DS, Whitley EM et al (2018) Midazolam efficacy against acute hydrogen sulfide-induced mortality and neurotoxicity. J Med Toxicol 14:79–90
Anantharam P, Whitley EM, Mahama B et al (2017a) Characterizing a mouse model for evaluation of countermeasures against hydrogen sulfide-induced neurotoxicity and neurological sequelae. Ann N Y Acad Sci 1400:46–64
Anantharam P, Whitley EM, Mahama B et al (2017b) Cobinamide is effective for treatment of hydrogen sulfide-induced neurological sequelae in a mouse model. Ann N Y Acad Sci 1408:61–78
Basic A, Alizadehgharib S, Dahlén G et al (2017) Hydrogen sulfide exposure induces NLRP3 inflammasome-dependent IL-1β and IL-18 secretion in human mononuclear leukocytes in vitro. Clin Exp Dent Res 3:115–120
Bhat SM, Massey N, Karriker LA, Singh B, Charavaryamath C (2019) Ethyl pyruvate reduces organic dust-induced airway inflammation by targeting HMGB1-RAGE signaling. Respir Res 20:27
Bosshart H, Heinzelmann M (2016) THP-1 cells as a model for human monocytes. Ann Transl Med 4:438–438
Charavaryamath C, Juneau V, Suri SS et al (2008) Role of toll-like receptor 4 in lung inflammation following exposure to swine barn air. Exp Lung Res 34:19–35
Charavaryamath C, Singh B (2006) Pulmonary effects of exposure to pig barn air. J Occup Med Toxicol 1:10
Che J, Yue D, Zhang B et al (2018) Claudin-3 inhibits lung squamous cell carcinoma cell epithelial-mesenchymal transition and invasion via suppression of the wnt/beta-catenin signaling pathway. Int J Med Sci 15:339–351
Chen YH, Wang PP, Wang XM et al (2011) Involvement of endogenous hydrogen sulfide in cigarette smoke-induced changes in airway responsiveness and inflammation of rat lung. Cytokine 53:334–341
Chen YH, Wu R, Geng B et al (2009) Endogenous hydrogen sulfide reduces airway inflammation and remodeling in a rat model of asthma. Cytokine 45:117–123
Christiani DC (1996) Organic dust exposure and chronic airway disease. Am J Respir Crit Care Med 154:833–834
Chung KF (2014) Hydrogen sulfide as a potential biomarker of asthma. Expert Rev Respir Med 8:5–13
Costigan MG (2003) Hydrogen sulfide: UK occupational exposure limits. Occup Environ Med 60:308–312
de Souza WF, Fortunato-Miranda N, Robbs BK et al (2013) Claudin-3 overexpression increases the malignant potential of colorectal cancer cells: roles of ERK1/2 and PI3K-Akt as modulators of EGFR signaling. PLoS ONE 8:e74994
Esechie A, Kiss L, Olah G et al (2008) Protective effect of hydrogen sulfide in a murine model of acute lung injury induced by combined burn and smoke inhalation. Clin Sci (Lond) 115:91–97
Gerald CL, McClendon CJ, Ranabhat RS et al (2019) Sorrel extract reduces oxidant production in airway epithelial cells exposed to swine barn dust extract in vitro. Mediators Inflamm 2019:7420468
Giuffre A, Vicente JB (2018) Hydrogen sulfide biochemistry and interplay with other gaseous mediators in mammalian physiology. Oxid Med Cell Longev 2018:6290931
Gordon R, Hogan CE, Neal ML et al (2011) A simple magnetic separation method for high-yield isolation of pure primary microglia. J Neurosci Methods 194:287–296
Han W, Dong Z, Dimitropoulou C et al (2011) Hydrogen sulfide ameliorates tobacco smoke-induced oxidative stress and emphysema in mice. Antioxid Redox Signal 15:2121–2134
Hine C, Harputlugil E, Zhang Y et al (2015) Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160:132–144
Hu L-F, Wong PT-H, Moore PK et al (2007) Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation by inhibition of p38 mitogen-activated protein kinase in microglia. J Neurochem 100:1121–1128
Iowa State University and University of Iowa (2002) IOWA CONCENTRATED ANIMAL FEEDING OPERATIONS AIR QUALITY STUDY. Final Report., vol 2018. Iowa State University and University of Iowa, Iowa
Jiang J, Chan A, Ali S et al (2016) Hydrogen sulfide-mechanisms of toxicity and development of an antidote. Sci Rep 6:20831–20831
Kim DS, Anantharam P, Hoffmann A et al (2018) Broad spectrum proteomics analysis of the inferior colliculus following acute hydrogen sulfide exposure. Toxicol Appl Pharmacol 355:28–42
Kotton DN (2018) Claudin-18: unexpected regulator of lung alveolar epithelial cell proliferation. J Clin Invest 128:903–905
Legator MS, Singleton CR, Morris DL et al (2001) Health effects from chronic low-level exposure to hydrogen sulfide. Archives of Environmental Health: An International Journal 56:123–131
Lewis RJ, Copley GB (2015) Chronic low-level hydrogen sulfide exposure and potential effects on human health: a review of the epidemiological evidence. Crit Rev Toxicol 45:93–123
Li L, Rose P, Moore PK (2011) Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol 51:169–187
Linden DR, Levitt MD, Farrugia G et al (2010) Endogenous production of H2S in the gastrointestinal tract: still in search of a physiologic function. Antioxid Redox Signal 12:1135–1146
Massey N, Puttachary S, Mahadev-Bhat S et al (2019) HMGB1-RAGE Signaling plays a role in organic dust-induced microglial activation and neuroinflammation. Toxicol Sci 169(2):579–592
McGovern T, Farahnak S, Chen M, Larsson K, Martin JG, Adner M (2019) Organic dust, causing both oxidative stress and Nrf2 activation, is phagocytized by bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 317:L305–l316
Nath Neerukonda S, Mahadev-Bhat S, Aylward B et al (2018) Kinome analyses of inflammatory responses to swine barn dust extract in human bronchial epithelial and monocyte cell lines. Innate Immun 24:366–381
Ni J-Q, Heber AJ, Diehl CA et al (2000) SE—structures and environment: ammonia, hydrogen sulphide and carbon dioxide release from pig manure in under-floor deep pits. J Agric Eng Res 77:53–66
Nordgren TM, Charavaryamath C (2018) Agriculture occupational exposures and factors affecting health effects. Curr Allergy Asthma Rep 18:65
Oesterhelweg L, Puschel K (2008) Death may come on like a stroke of lightening”: phenomenological and morphological aspects of fatalities caused by manure gas. Int J Legal Med 122:101–107
Oh GS, Pae HO, Lee BS et al (2006) Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic Biol Med 41:106–119
Park JH, Peters TM, Altmaier R et al (2013) Simulation of air quality and cost to ventilate swine farrowing facilities in winter. Comput Electron Agric 98:136–145
Pavilonis BT, O’Shaughnessy PT, Altmaier R et al (2013) Passive monitors to measure hydrogen sulfide near concentrated animal feeding operations. Environ Sci Process Impacts 15:1271–1278
Pender RL, Minor RC, Hurley SL et al (2014) Exposure to swine housing dust modulates macrophage morphology and function. Am J Immunol 10:35–45
Poole JA, Wyatt TA, Kielian T et al (2011) Toll-like receptor 2 regulates organic dust-induced airway inflammation. Am J Respir Cell Mol Biol 45:711–719
Poole JA, Wyatt TA, Oldenburg PJ et al (2009) Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am J Physiol Lung Cell Mol Physiol 296:L1085-1095
Rao X, Huang X, Zhou Z et al (2013) An improvement of the 2ˆ(–delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostatistics, bioinformatics and biomathematics 3:71–85
Richardson DB (1995) Respiratory effects of chronic hydrogen sulfide exposure. Am J Ind Med 28:99–108
Romberger DJ, Bodlak V, Von Essen SG et al (1985) Wyatt TA (2002) Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J Appl Physiol (1985) 93:289–296
Sethi RS, Schneberger D, Charavaryamath C et al (2017) Pulmonary innate inflammatory responses to agricultural occupational contaminants. Cell Tissue Res 367:1–16
Shi C, Pamer EG (2011) Monocyte recruitment during infection and inflammation. Nat Rev Immunol 11:762–774
Spassov SG, Donus R, Ihle PM et al (2017) Hydrogen sulfide prevents formation of reactive oxygen species through PI3K/Akt signaling and limits ventilator-induced lung injury. Oxid Med Cell Longev 2017:3715037
Tamizhselvi R, Moore PK, Bhatia M (2008) Inhibition of hydrogen sulfide synthesis attenuates chemokine production and protects mice against acute pancreatitis and associated lung injury. Pancreas 36:e24-31
The National Institutes for Occupational Safety and Health (NIOSH) (2018) CDC - Immediately Dangerous to Life or Health Concentrations (IDLH): hydrogen sulfide - NIOSH Publications and Products. vol 2020. The Centers for Disease Control and Prevention
Trevisani M, Patacchini R, Nicoletti P et al (2005) Hydrogen sulfide causes vanilloid receptor 1-mediated neurogenic inflammation in the airways. Br J Pharmacol 145:1123–1131
Vested A, Basinas I, Burdorf A et al (2019) A nationwide follow-up study of occupational organic dust exposure and risk of chronic obstructive pulmonary disease (COPD). Occup Environ Med 76:105–113
Wang X, Gray Z, Willette-Brown J et al (2018) Macrophage inducible nitric oxide synthase circulates inflammation and promotes lung carcinogenesis. Cell Death Discovery 4:46
Willis WL, Wang L, Wada TT et al (2018) The proinflammatory protein HMGB1 is a substrate of transglutaminase-2 and forms high-molecular weight complexes with autoantigens. J Biol Chem 293(22):8394–8409
Zhang G, Wang P, Yang G et al (2013) The inhibitory role of hydrogen sulfide in airway hyperresponsiveness and inflammation in a mouse model of asthma. Am J Pathol 182:1188–1195
Zhang JY, Ding YP, Wang Z et al (2017) Hydrogen sulfide therapy in brain diseases: from bench to bedside. Med Gas Res 7:113–119
Zhang P, Li F, Wiegman CH et al (2015) Inhibitory effect of hydrogen sulfide on ozone-induced airway inflammation, oxidative stress, and bronchial hyperresponsiveness. Am J Respir Cell Mol Biol 52:129–137
Zhao Y, Biggs TD, Xian M (2014) Hydrogen sulfide (H2S) releasing agents: chemistry and biological applications. Chem Commun (Camb) 50:11788–11805
Zhi L, Ang AD, Zhang H et al (2007) Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-kappaB pathway. J Leukoc Biol 81:1322–1332
Acknowledgments
We would like to thank Dr. Locke A. Karriker (VDPAM, Iowa State University) for supplying us with OD samples. We would like to thank Dr. Jacek Koziel (Department of Agricultural and Biosystems Engineering, Iowa State University) for initial help with the measurement of H2S concentrations and the Department of Biomedical Sciences for providing access to the core laboratory facilities.
Funding
C.C. laboratory is currently funded through a startup grant from Iowa State University and seed grants from the College of Veterinary Medicine, Iowa State University. This manuscript was supported in part by Grant Number 5 U54 OH007548 from CDC—NIOSH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. This article does not contain any studies with human participants performed by any of the authors.
Informed consent
Since this manuscript does not contain any studies with human participants, it is not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
441_2020_3333_MOESM1_ESM.tif
Supplementary file1: Fig. S1 H2S and ODE exposure induced histopathological changes in the lungs H&E images are shown in the left and inflammatory score is shown in the right. Histopathological changes due to exposure to either H2S or ODE or co-exposure to H2S and ODE were not significantly different among any of the groups. Compared with controls (a-c), ODE exposure of mice did not induce any morphological changes in the blood vessels, bronchioles and septa except for the number of club cells (b). (Micrometer bar = 100 µm). Data (mean ± SEM, n = 5–6/group) analyzed with one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons as compared with 0 ppm (TIF 92 MB)
Rights and permissions
About this article

Cite this article
Shrestha, D., Bhat, S.M., Massey, N. et al. Pre-exposure to hydrogen sulfide modulates the innate inflammatory response to organic dust. Cell Tissue Res 384, 129–148 (2021). https://doi.org/10.1007/s00441-020-03333-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-020-03333-3