
Abstract
We have previously shown that the chromogranin A (CgA)-derived peptide catestatin (CST: hCgA352–372) inhibits nicotine-induced secretion of catecholamines from the adrenal medulla and chromaffin cells. In the present study, we seek to determine whether CST regulates dense core (DC) vesicle (DCV) quanta (catecholamine and chromogranin/secretogranin proteins) during acute (0.5-h treatment) or chronic (24-h treatment) cholinergic (nicotine) or peptidergic (PACAP, pituitary adenylyl cyclase activating polypeptide) stimulation of PC12 cells. In acute experiments, we found that both nicotine (60 μM) and PACAP (0.1 μM) decreased intracellular norepinephrine (NE) content and increased 3H‐NE secretion, with both effects markedly inhibited by co-treatment with CST (2 μM). In chronic experiments, we found that nicotine and PACAP both reduced DCV and DC diameters and that this effect was likewise prevented by CST. Nicotine or CST alone increased expression of CgA protein and together elicited an additional increase in CgA protein, implying that nicotine and CST utilize separate signaling pathways to activate CgA expression. In contrast, PACAP increased expression of CgB and SgII proteins, with a further potentiation by CST. CST augmented the expression of tyrosine hydroxylase (TH) but did not increase intracellular NE levels, presumably due to its inability to cause post-translational activation of TH through serine phosphorylation. Co-treatment of CST with nicotine or PACAP increased quantal size, plausibly due to increased synthesis of CgA, CgB and SgII by CST. We conclude that CST regulates DCV quanta by acutely inhibiting catecholamine secretion and chronically increasing expression of CgA after nicotinic stimulation and CgB and SgII after PACAPergic stimulation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stimulation of the splanchnic nerve of the adrenal medulla causes release of both acetylcholine and pituitary adenylate cyclase-activating polypeptide (PACAP) at the splanchnicoadrenomedullary synapse, stimulating the release of catecholamines from chromaffin cells (Cheung et al. 1993; Eiden et al. 2018; Gayen et al. 2009a; Livett and Marley 1993; Mahata et al. 1997; Malhotra et al. 1989; Smith and Eiden 2012; Taupenot et al. 1998; Wakade 1988). It is becoming increasingly evident that acetylcholine and PACAP control catecholamine secretion at different levels of neuronal activity. Acetylcholine stimulates the secretion of norepinephrine (NE) and epinephrine (EPI) at a low frequency to supply catecholamines at a modest rate to set the homeostatic “rest and digest” state, whereas PACAP, the “stress transmitter,” predominantly stimulates the secretion of EPI at higher rates of splanchnic nerve stimulation (Guo and Wakade 1994; Smith and Eiden 2012; Stroth et al. 2013). Upon binding to the nicotinic acetylcholine receptor, acetylcholine activates voltage-dependent sodium channels. The entry of sodium ions causes depolarization of the cell membrane and opens voltage-gated calcium channels in primary chromaffin cells and adrenal medulla (Douglas et al. 1967; Douglas and Rubin 1961; Wakade and Wakade 1982). Entry of calcium ions results in calcium-dependent exocytosis of large dense-core vesicles containing catecholamines, dopamine β-hydroxylase (DBH), chromogranin A (CgA) (Eiden 1987; Winkler 1993; Winkler et al. 1986) and neuropeptides including enkephalins, neuropeptide Y (NPY), galanin and others (Anouar and Eiden 1995; Fischer-Colbrie et al. 1988; Waschek et al. 1987). In contrast, PACAP evokes catecholamine secretion from chromaffin cells through a rapid influx of extracellular Ca2+ (O'Farrell and Marley 1997; Tanaka et al. 1996) which does not depend upon the opening of voltage-gated sodium channels (Mustafa et al. 2007; Mustafa et al. 2010; Tanaka et al. 1996; Vandael et al. 2015).
When chromaffin cells secrete catecholamines and co-stored proteins/peptides, resynthesis mechanisms are activated to replenish secretory vesicles with the just-secreted components. Tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine synthesis, is transcriptionally activated by both cholinergic and peptidergic signaling to allow compensatory repletion of catecholamine stores (stimulus-secretion-synthesis coupling) in chromaffin cells and sympathetic neurons (Braas and May 1999; Corbitt et al. 1998; Gueorguiev et al. 2006; Gueorguiev et al. 2004; Gueorguiev et al. 2000; Haycock 2002a; Haycock 2002b; Hiremagalur et al. 1993; Ishiguro et al. 1997; Isobe et al. 1996; Stroth et al. 2013; Tonshoff et al. 1997). PACAP also activates synthesis of other catecholamine biosynthetic enzymes like DBH (Isobe et al. 1996) and phenylethanolamine N-methyl transferase (PNMT) (Tonshoff et al. 1997). Likewise, biosynthesis of secreted proteins and peptides, such as CgA (Tang et al. 1997; Tang et al. 1996), proenkephalin A (Suh et al. 1995) and NPY (Hiremagalur et al. 1994), can be activated by nicotinic stimulation. Similar to cholinergic stimulation, peptidergic stimulation also exhibits stimulus-secretion-synthesis coupling and induces synthesis of CgA (Taupenot et al. 1999; Taupenot et al. 1998), secretogranin II (SgII) (Turquier et al. 2001) and met-enkephalin (Fischer-Colbrie et al. 1988; Hahm et al. 1998).
Catestatin (CST: hCgA352–372) is a 21 amino acid peptide (human sequence: SSMKLSFRARAYGFRGPGPQL) proteolytically derived from the C-terminal region of CgA (Mahata et al. 2010; Mahata et al. 1997). CST is established as a pleiotropic peptide exhibiting diverse cardiovascular, metabolic and endocrine functions. These include inhibiting nicotine-evoked catecholamine secretion from the adrenal medulla and cultured cells (Mahata et al. 2003; Mahata et al. 1999; Mahata et al. 2000; Mahata et al. 2004; Mahata et al. 1997; Wen et al. 2004); decreasing blood pressure (Biswas et al. 2012; Fung et al. 2010; Kennedy et al. 1998; Mahapatra et al. 2005), improving baroreflex sensitivity (BRS) (Gayen et al. 2009b), increasing heart rate variability (HRV) (Dev et al. 2010) improving insulin sensitivity in diet-induced obese mice (Ying et al. 2018), providing cardioprotection by inhibiting inotropy and lusitropy (Angelone et al. 2008; Angelone et al. 2012; Imbrogno et al. 2010; Mazza et al. 2008), stimulating reperfusion injury salvage kinase (RISK) and survivor activating factor enhancement (SAFE) pathways (Bassino et al. 2015; Perrelli et al. 2013), preventing macrophage-driven atherosclerosis (Kojima et al. 2018) and modulating cardiomyocyte Ca2+ levels (Ottesen et al. 2017).
CST and PACAP are well-known modulators of DCV content and function as described above. However, ultrastructural evidence for correlative changes in the morphology of DCVs in CST/PACAP-treated chromaffin cells is not available and limited biochemical evidence prompted further studies. In the present study, we demonstrate in PC12 cells originating from rat pheochromocytoma, which are a model system for chromaffin cell secretion (Greene and Tischler 1976; Westerink and Ewing 2008), that acute CST treatment restores DCV quanta by inhibiting nicotine or PACAP-induced secretion of catecholamines. Further, chronic stimulation experiments to understand the resynthesis of DCV quanta provide novel ultrastructural and biochemical evidence for how CST and PACAP modulate DCV quanta by regulating the expression of major granulogenic proteins, contributing to secretory granule resynthesis.
Materials and methods
PC12 cell culture and treatments
Rat pheochromocytoma (PC12) cells were obtained from David R. Schubert (Salk Institute for Biological Studies, La Jolla, CA, USA) and grown in T25 flasks (Nunc, USA) in DMEM GlutaMAX (Invitrogen), supplemented with 10% heat-inactivated horse serum, 5% heat-inactivated fetal bovine serum, 100 units/ml penicillin G and 100 μg/ml streptomycin (Gibco, USA). Before all experiments, PC12 cells were treated with ascorbic acid (100 μM; to induce synthesis of norepinephrine) for 18–24-h (Schubert and Klier 1977; Tischler et al. 1983; Westerink and Ewing 2008). Cells were treated with secretion buffer containing either nicotine (60 μM), PACAP (0.1 μM), CST (2 μM), Nicotine+CST, or PACAP+CST for acute experiments for 0.5-h, whereas chronic treatments lasted for 18–24-h. For experiments where gene expression was analyzed, secretogogue treatments were carried out for ~ 18-h, while for experiments in which proteins and catecholamines were measured, treatments were carried out for ~ 24-h.
Intracellular norepinephrine (NE) measurements
PC12 cells grown to 80% confluence in poly-l-lysine coated 6-well plates were washed once with release buffer (150 mM NaCl, 5 mM KCl, 2 mM CaCl2, 10 mM HEPES pH 7) and incubated for 0.5-h at 37 °C in release buffer with none (basal) or with various secretogogues (nicotine, PACAP, CST, CST + nicotine, or CST + PACAP). In a second set of experiments, cells were treated with the above secretogogue stimuli in regular culture media for 24-h. After the treatments, cells were washed with PBS and lysed with lysis buffer (150 mM NaCl, 5 mM KCl, 2 mM CaCl2 and 10 mM HEPES pH 7, 0.1% (v/v) Triton X-100). The amounts of NE were quantified by a high sensitivity ELISA KIT (Rocky Mountain Diagnostics, Colorado Springs, Colorado, USA). In brief, NE was extracted using a cis-diol-specific affinity gel, acylated to N-acylnorepinephrine and eluted in 200 μl 0.025 M hydrochloric acid. The eluted samples were then converted enzymatically to N-acylnormetanephrine and detected by ELISA as per the manufacturer’s protocol. A series of standards were run along with the samples to estimate the absolute amounts of the catecholamines in the samples and the results were expressed as nM and normalized to the protein content estimated by BCA assays.
Tritiated norepinephrine (3H-NE) secretion experiments
3H-NE secretion experiments were carried out as previously described (Mahata et al. 1996; Sahu et al. 2017). PC12 cells grown to 80% confluence in poly-l-lysine–coated six-well plates were incubated with 1 μCi [3H] l-NE for 3-h, washed twice with release buffer (150 mM NaCl, 5 mM KCl, 2 mM CaCl2,10 mM HEPES pH 7) and incubated for 0.5-h at 37 °C in release buffer with none (basal) or with various secretogogues (nicotine, PACAP, CST, CST + nicotine or CST + PACAP). The supernatants were then collected and the cells were lysed in lysis buffer. Both the supernatants and the lysates were mixed with liquid scintillation cocktail and radioactivity was quantified using a Beckman liquid scintillation counter. Results were expressed as % of total.
Transmission electron microscopy
PC12 cells were fixed with 2.5% glutaraldehyde in 0.15 M cacodylate buffer and postfixed in 1% OsO4 in 0.1 M cacodylate buffer for 1-h on ice, followed by staining en bloc with 2–3% uranyl acetate for 1-h on ice as described previously (Patra et al. 2016). The cells were dehydrated in graded series of ethanol (20–100%) on ice followed by one wash with 100% ethanol and two washes with acetone (15 min each) and embedded with Durcupan. Ultrathin (50 ∼ 60 nm) sections were cut on a Leica UCT ultramicrotome and picked up on Formvar and carbon-coated copper grids. Sections were stained with 2% uranyl acetate for 5-min and Sato’s lead stain for 1-min. Grids were viewed using a JEOL JEM1400-plus TEM (JEOL, Peabody, MA) and photographed using a Gatan OneView digital camera with 4k × 4k resolution (Gatan, Pleasanton, CA).
Morphometric analysis
Samples were blinded and two people performed measurements randomly from different cells. The line segment tool in ImageJ was used to measure diameters of dense core vesicles (DCV) and the dense core (DC) inside the DCV. The free-hand tool was also used to measure areas by manually tracing around the DCV membrane and outer boundary of DC as described previously (Mahata et al. 2016; Pasqua et al. 2016).
Real-time RT-PCR assay for measurement of target mRNAs
After the respective treatments for 18-h, RNA was obtained from single wells of a 12‐well plate per condition by using an RNA isolation kit from Qiagen Inc. (Valencia, CA). Total RNA was digested with DNase I using a DNA removal kit water (Ambion, Austin, TX) and tested for the presence of DNA contamination using PCR. Total RNA concentration and purity were then determined by a spectrophotometer at 260 nm (NanoDrop 2000 UV–Vis Spectrophotometer, Thermo Scientific, USA). 500 ng of RNA was converted into cDNA using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA) and relative quantification of mRNAs was performed with 3.5 μl of cDNA used in each 11.5 μl real-time-RT-PCR reaction using a C1000 thermal cycler™ (Bio-Rad Laboratories, Hercules, CA). The PCR reactions were carried out using IQ™ SYBR Green Supermix (Bio-Rad). Thermal cycling parameters were as follows: an initial denaturing step (95 °C for 10 min), followed by 40 cycles of denaturing, annealing and extending (95 °C for 45 s, 58 °C for 45 sec and then 60 °C for 1 min, respectively) in a 96-well BioRad plate. cDNA samples were amplified by using PERFECTA SYBR FASTMIX L-ROX 1250 (Quantabio, Beverly, MA) and analyzed on an Applied Biosystems 7500 Fast Real-Time PCR system (ABI, Foster City, CA). All PCRs were normalized to Gapdh and Rplp0 and relative expression levels were determined by the ΔΔCt method. Primer sequences are provided in Table 1.
Western blotting
After the respective treatments for 24-h, cells were washed with PBS twice and harvested in RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS and 0.2% sodium azide). The following primary antibodies were used: anti-VS1 mouse monoclonal antibody (epitope hCgA54–57) (Ratti et al. 2000) at a dilution of 1:1000 that cross-reacts with murine CgA (Colombo et al. 2002); anti-CgA polyclonal antibodies including anti-pancreastatin (PST: rabbit polyclonal; epitope hCgA269–286) at a dilution of 1:1000; anti-WE14 (rabbit polyclonal; epitope hCgA324–337) at a dilution of 1:500, anti-CST (rabbit polyclonal made by a commercial vendor Sdix Inc., Newark, DE; epitope hCgA352–372) at a dilution of 1:1000 (Vaingankar et al. 2010); anti-CT-CST (rabbit polyclonal; epitope hCgA368–373) at a dilution of 1:500 (Bianco et al. 2016); anti-SPN (rabbit serpinin, rabbit polyclonal; epitope hCgA410–439) at a dilution of 1:500; anti-PE-11 (rabbit polyclonal; epitope rCgB552–562) (Kroesen et al. 1996) at a dilution of 1:1000; anti-secretoneurin (rabbit polyclonal; epitope rSgII154–186) (Kirchmair et al. 1993) at a dilution of 1:1000; anti-TH (rabbit polyclonal from Abcam, Cambridge, MA); anti-β-actin (mouse monoclonal from Sigma Aldrich, St. Louis, MO) at a dilution of 1:5000; anti-phospho-CREB (rabbit polyclonal from Fisher Scientific, Hampton, NH) at a dilution of 1:500 and anti-total CREB (mouse monoclonal from Fisher Scientific, Hampton, NH) at a dilution of 1:1000.
Data presentation and statistical analysis
Data are expressed as mean ± SEM. Statistical analyses were performed with GraphPad Prism 7 software. Data were analyzed by 1-way or 2-way ANOVA followed by multiple comparison tests where appropriate. Individual pair-wise comparisons were performed using unpaired Student’s t tests. Size distribution data were analyzed by Kolmogorov-Smirnov tests. Statistical significance was defined as p < 0.05.
Result
Acute stimulation experiments
We first addressed the effect of CST on NE release by PC12 cells. Both nicotine and PACAP decreased cellular (i.e., vesicular) NE content after 0.5-h of treatment. Although CST had no effect, it inhibited the nicotine and PACAP-induced decreases in vesicular NE content (Fig. 1a). Radiolabeled 3H-NE secretion experiments indicated that nicotine and PACAP-induced NE release was blocked by CST, without altering basal secretion (Fig. 1b). This blockade was also apparent from EM imaging. Under basal conditions, chromaffin vesicles in PC12 cells were distributed throughout the cytoplasm (Fig. 1c). Treatment with nicotine or PACAP for 0.5-h resulted in the movement of vesicles from the cytoplasm to the plasma membrane, where the majority of them were docked (Fig. 1d, e). The cytoplasmic location of the vesicles was comparable in CST-treated cells (Fig. 1c, f). Co-treatment with CST blocked the vesicle movement to the plasma membrane induced by nicotine or PACAP (Fig. 1g, h). These findings demonstrate that CST blocks NE release induced by both nicotine and PACAP.

Intracellular NE content, 3H-NE secretion and distribution of DCV in PC12 cells by TEM. a PC12 cells were treated with vehicle, nicotine, PACAP, CST, nicotine+CST or PACAP+CST for 0.5-h (n = 8). b 3H-NE-prelabeled PC12 cells were treated with none, nicotine, PACAP, CST, nicotine+CST and PACAP+CST for 0.5-h (n = 3). *p < 0.05; **p < 0.01; ***p < 0.001. c–h TEM photographs of PC12 cells after 0.5-h of treatment. c Basal. d Nicotine. e PACAP. f CST. g CST + Nicotine. h CST + PACAP. Inset shows a higher magnification of a single representative vesicle for the respective condition. Inset shows a higher magnification of a single representative vesicle for the respective condition. Dashed maroon lines indicate the plasma membrane (PM)
CST blocks the reduction in DCV and DC size after acute stimulation
Next, we determined whether CST would affect the reformation of DCV and DC following acute stimulation. In untreated PC12 cells, the median diameter of DCVs was 150 nm and that of DCs was 100 nm (Fig. 2a–f). Both nicotine and PACAP treatment for 0.5-h resulted in shifts in the DCV and DC size distributions toward smaller diameters (Fig. 2a–d). CST alone had no effect on either DCV or DC size distributions (Fig. 2e, f) but blocked the nicotine and PACAP-induced shift in DCV and DC diameter distribution (Fig. 2g–j). This effect was also apparent when the areas of the DCV and DC were used as a measure of DCV/DC size. Both the mean DCV and DC area and diameter were decreased after nicotine or PACAP treatment and both reductions were blocked by CST co-treatment (Fig. 2k–n).

Morphometric measurements of DCV and DC after 0.5-h treatments with nicotine or PACAP in absence or presence of CST. a–j Histograms showing DCV and DC diameters (n = 650). Basal versus nicotine-treated DCV (a) and DC (b). Basal versus PACAP-treated DCV (c) and DC (d). Basal versus CST-treated DCV (e) and DC (f). Nicotine versus nicotine+CST-treated DCV (g) and DC (h). PACAP versus PACAP+CST-treated DCV (i) and DC (j). k–n Mean DCV and DC area and diameter (n = 650). (k), DCV area (l), DC area, (m), DCV diameter and (n), DC diameter. **p < 0.01; ***p < 0.001
CST prevented nicotine and PACAP-induced docking of chromaffin vesicles
We then examined the docking of vesicles at the plasma membrane. Consistent with previous reports (Mahata et al. 1997; Taupenot et al. 1998), both nicotine and PACAP caused profound increases (> 70%) in docking of DCVs (Fig. 3a–d, g, h). In contrast, co-treatment with CST profoundly (> 80%) blocked secretagogue-induced docking of DCVs for full fusion, which accounts for ~ 95% of docked vesicles (Fig. 3e, f, i, j). From these data, we conclude that CST blocks both nicotine and PACAP induced release of NE by inhibiting vesicle docking and fusion. The TEM also allowed us to differentiate different stages of secretion. Under basal conditions, the closest DCVs were located between 3.7 and 50 nm away from the plasma membrane (Fig. 4a, m). Nicotine treatment resulted in movement of DCVs toward the plasma membrane and DCVs could be seen between 10.2 and 22 nm away from the plasma membrane (Fig. 4b) or docked with the plasma membrane (Fig. 4c, d, m). We observed that in ~ 5% of the DCVs, the vesicle membrane opened with a diameter of 21 nm but the plasma membrane was still intact (Fig. 4e), possibly representing the first stage in membrane fusion leading to a “kiss-and-run” mechanism of exocytotic release (Albillos et al. 1997; Alvarez de Toledo et al. 1993; Henkel et al. 2001). This mechanism appeared to be completely blocked by CST. Figure 4(f) shows DCV with a 91 nm exocytotic pore where the vesicular quanta remained intact. Once the exocytotic pore reached a width of 106 nm, the release of vesicular quanta was observed (Fig. 4g, h). Following the release of vesicular quanta, the exocytotic pore is squeezed and the pore diameter narrowed successively (down 61 and 18 nm, Fig. 4i, j, respectively) before closure of the exocytotic pore and endocytic vesicle movement away from the plasma membrane (Fig. 4k, i) (Bittner et al. 2013; Creutz and Harrison 1984; Kakhlon et al. 2006). It should be noted that PC12 cells also contain a fraction of vesicles that do not contain a dense core (~10% of 100–150 nm-sized vesicles), so we cannot exclude the possibility that the empty vesicles in Fig. 4(j–l) are not DCV but indicate other exo- or endocytic events in this analysis.

TEM micrographs after 0.5-h treatment with nicotine or PACAP in absence or presence of CST. a, b Basal: a low magnification and b high magnification. c, d Nicotine: c low magnification and d high magnification of the area marked by dashed white rectangle in c. Note the docked vesicles. e, f CST + nicotine: e low magnification and f high magnification of the area marked by dashed white rectangle in e. Note vesicles are distributed throughout the cytoplasma and only a few vesicles are docked. g, h PACAP: g low magnification and h high magnification of the area marked by dashed white rectangle in g. Note docked vesicles. i, j CST + PACAP: i low magnification and j high magnification of the area marked by dashed white rectangle in (i). Note vesicles are distributed throughout the cytoplasm. Dashed maroon lines indicate the plasma membrane (PM)

High-magnification DCVs showing different stages of exocytosis and endocytosis under basal and after 0.5-h treatment with nicotine. a Basal showing vesicles are located 50 nm away from the plasma membrane. b Nicotine-treated PC12 cells. Vesicles moved closer to the plasma membrane (22 nm apart). c, d Docked vesicles after nicotine. e Tethering of vesicle membrane only (21 nm pore diameter) where plasma membrane remained intact. f Exocytotic pore (91 nm) with intact vesicular quanta. g Widened exocytotic pore (106 nm) with substantially released quanta. h Widened exocytotic pore (106 nm) with almost completely released quanta. i Shrinkage of exocytotic pore (61 nm). j Further shrinkage of exocytotic pore (18 nm). k Resealed endocytic vesicle. l Endocytic vesicle moved away (38 nm) from the plasma membrane. m Distribution of DCVs in the cytoplasm in basal and nicotine-treated (0.5-h of stimulation) cells with respect to the distance from the plasma membrane. Maroon arrows indicate the plasma membrane (PM)
CST inhibits chronic nicotine and PACAP-induced vesicular movement and NE release
In the following set of experiments, We next determined whether the effects of NE release by CST were also present following chronic stimulation with nicotine of PACAP. As observed in the acute experiments, nicotine and PACAP decreased vesicular NE content after 24-h of treatment and CST alone had no effect on vesicular NE content but it reversed nicotine and PACAP-induced decreases in vesicular NE content (Fig. 5a).

Intracellular NE content, TH expression, CREB signaling and intracellular DCV distribution. a Intracellular NE content. PC12 cells were treated with none, nicotine, PACAP, CST, nicotine+CST and PACAP+CST for 0.5-h (n = 8). b–g Western blot (n = 4) data showing expression of TH and CREB proteins after 24-h treatments with nicotine, PACAP, CST, nicotine + CST and PACAP + CST. b Western blot showing expression of TH with a polyclonal antibody and f the corresponding densitometric values. c Western blot with a monoclonal β-actin antibody. d Western blot of phospho-CREB at Ser133 with a polyclonal antibody and g the corresponding densitometric values. e Western blot with a monoclonal total CREB antibody. **p < 0.01; ***p < 0.001. h–m TEM micrographs after 24-h treatment with nicotine, PACAP in absence or presence of CST. h Basal. i Nicotine. j PACAP. k CST. l Nicotine+CST. m PACAP+CST. Inset shows a higher magnification of a single representative vesicle for the respective condition. Dashed maroon lines indicate the plasma membrane (PM)
Chronic stimulation of chromaffin cells results in transcriptional upregulation of of genes encoding proteins involved in biosynthesis of DC and DCV. In line with this, CST increased expression of TH protein (Fig. 5b, d). Since the Th gene has a cAMP-response element (CRE) in its proximal promoter region, we assessed CREB phosphorylation. Nicotine, PACAP or CST increased phosphorylation of CREB on Ser133 (Fig. 5e, g). The nicotine and PACAP effects are consistent with the known activation of cAMP signaling by these ligands; however, it is not known if CST activates this pathway. Co-treatment of CST with nicotine or PACAP did not show additive effects on CREB phosphorylation (Fig. 5g). Thus, CST may increase expression of TH protein through increased phosphorylation of CREB.
We also visualized the distribution of DCVs by TEM to elucidate the effects of CST on intracellular vesicle distribution. Nicotine and PACAP-treated DCVs appeared to be smaller than their basal counterparts and were localized away from the plasma membrane (Fig. 5h–m). CST-treated DCVs appeared larger than basal DCVs and were distributed throughout the cytoplasm when either nicotine or PACAP treatment was accompanied by co-treatment with CST (Fig. 5k–m).
CST blocks the reduction in DCV and DC size after chronic stimulus
We performed morphometric analyses to determine the effect of CST on DCV and DC size. Similar to the acute stimulation, both nicotine and PACAP treatment for 24-h resulted in a shift in the distribution of DCV and DC diameters to smaller sizes (Fig. 6a–d). No marked change in DCV or DC diameter was noticeable after CST treatment (Fig. 6e, f) but CST co-treatment blocked the reduction in DCV and DC size by chronic nicotine or PACAP stimulation (Fig. 6g–j). The average DCV area did not change after nicotine, PACAP, or CST treatments for 24-h (Fig. 6k). In contrast, CST, in combination with nicotine decreased the DCV area in comparison to either nicotine or CST (Fig. 6k) The mean DC area showed a decrease after nicotine or PACAP treatment (Fig. 6l), whereas CST, either alone or in combination with secretogogues, had no effect on DC area (Fig. 6l). The mean DCV and DC diameters were found to be decreased after nicotine or PACAP treatment (Fig. 6m, n). Although CST alone had no effects on those parameters per se, CST in combination with nicotine or PACAP inhibited decrease of DCV and DC diameters (Fig. 6m, n). Overall, these results show a similar effect of CST on DCV and DC size upon chronic nicotine or PACAP stimulation as we previously observed for acute stimulation and we conclude that CST blocks the PACAP or nicotine-mediated reduction of DCV and DC size.

Morphometric measurements of DCV and DC after 24-h treatments with nicotine or PACAP in absence or presence of CST. a–j Size distribution of DCV and DC diameters (n = 650). Basal versus nicotine-treated DCV a and DC b Basal versus PACAP-treated DCV c and DC d Basal versus CST-treated DCV e and DC f Nicotine versus nicotine+CST-treated DCV g and DC h PACAP versus PACAP+CST-treated DCV i and DC j. k–n Mean DCV and DC area and diameter (n = 650). k DCV area, l DC area, m DCV diameter and n DC diameter. **p < 0.01; ***p < 0.001
Effect of CST on expression of genes associated with secretory granules
In order to identify the molecular mechanism underlying the effect of CST on DCV and DC secretion, we investigated the expression of proteins associated with secretory granules. First, we focused on genes involved in catecholamine biosynthesis. Since Th and Dbh are the predominant catecholamine biosynthetic enzymes in PC12 cells, we looked at the expression of those genes. Nicotine, PACAP or CST increased mRNA levels of Th (Fig. 7a). CST exerted no effect on nicotine or PACAP-induced increase in Th mRNA levels. Likewise, Dbh mRNA levels were increased by nicotine, PACAP, or CST, and CST exerted no effect on increased Dbh mRNA levels induced by nicotine or PACAP (Fig. 7a). We also investigated CgA, CgB and SgII, as these are the major vesicular proteins in the DCV of chromaffin cells. Nicotine, PACAP or CST augmented steady-state mRNA levels of Chga (Fig. 7b). CST when co-treated with either nicotine or PACAP exhibited no additional mRNA expression of the Chga gene (Fig. 7b). In contrast, Chgb and Scg2 mRNA levels were increased by PACAP, which did not show any additive expression in the presence of CST (Fig. 7b). Finally, we tested for the levels of SNARE proteins involved in the fusion of secretory granules with the plasma membrane. Although there were no changes in steady-state mRNA levels of Vamp1, nicotine and PACAP increased mRNA levels of Vamp2 (Fig. 7c). While CST alone had no effect on the expression of Vamp2, it inhibited nicotine or PACAP-induced increase (Fig. 7c). PACAP increased Snap25 mRNA levels, which was inhibited by CST (Fig. 7c). In contrast, nicotine is the only secretogogue that caused an increase of Stx1a mRNA levels and such an increase was inhibited by CST (Fig. 7c). Overall, we conclude that CST increases the mRNA expression levels of certain catecholamine biosynthesis and secretory proteins (especially Chga), whereas it blocks the increase in SNARE expression upon PACAP/nicotine stimulation.

Effect of CST on expression of genes involved in DCV and DC secretion by qRT-PCR. Cells were stimulated for 18-h with nicotine or PACAP in absence or presence of CST. a Expression of genes (n = 6) encoding catecholamine biosynthetic enzymes: Th and Dbh. b Expression of genes encoding vesicular proteins: Chga, Chgb and Scg2. c Expression of genes encoding SNARE proteins: Vamp1, Vamp2, Snap25 and Stx1a. *p < 0.05; **p < 0.01; ***p < 0.001
We further investigated the effect on CST on CgA protein and CgA-derived peptide expression by Western blot. In contrast to qPCR, Western blot allows resolution of different bioactive cleavage products of CgA. Consistent with increased Chga mRNA, Western blot data with α-VS1, α-WE14 and α-CST revealed an increase in CgA protein after 24-h of treatment with nicotine (Fig. 8a, j, c, i, d, m). Although PACAP increased Chga mRNA levels, it did not alter expression of full-length CgA protein (Fig. 8a–f, j–o). We also studied expression of CgB and SgII. As opposed to the expression of CgA protein, neither nicotine nor CST changed expression of the CgB (Fig. 8g, p) and SgII proteins (Fig. 8h, q). Consistent with increased mRNA levels, PACAP increased expressions of CgB and SgII proteins, that were unaffected by the presence of CST (Fig. 8g, h, p, q). Chga, Chgb and Scg2 genes also contain CRE in their proximal promoter regions. It is noteworthy that the pattern of pCREB activation corroborates with the increased mRNA and protein levels of granins suggesting an important role for CREB in regulating granulogenesis.

Effect of CST on cellular levels of CgA and cleavage products by Western blot. Cells were stimulated for 24-h with nicotine or PACAP in the absence or presence of CST. a Western blot with a monoclonal antibody against VS1 (hCgA54–57) detecting full-length CgA and j the corresponding densitometry values (n = 4). b Western blot with a polyclonal antibody against PST (hCgA269–286) detecting processed CgA and k the corresponding densitometry values. c Western blot with a polyclonal antibody against WE14 (hCgA324–337) detecting full-length CgA and l the corresponding densitometry values. d Western blot with a polyclonal antibody against full-length human CST (hCgA352–372) detecting full-length CgA and m the corresponding densitometry values. e Western blot with a polyclonal antibody against the C-terminal CST domain (hCgA368–373) detecting processed CgA and n the corresponding densitometry values. f Western blot with a polyclonal antibody against serpinin domain (hCgA410–439) detecting full-length CgA and o the corresponding densitometry values. g Western blot with a polyclonal antibody against PE-11 (rCgB552–562) detecting full-length CgB and p the corresponding densitometric values. h Western blot with a polyclonal antibody against secretoneurin (rSgII154–186) and q the corresponding densitometric values. i Western blot with a monoclonal antibody against β-Actin
Accumulation of dense cores in the trans-Golgi apparatus upon CST treatment
In the final set of experiments, we studied the effects of CST on the morphology of the Golgi apparatus by EM, as this compartment plays a critical role in the biosynthesis of DC and DCV. Golgi apparatuses are abundant in PC12 cells (Fig. 9a). Surprisingly, dense cores (possibly representing accumulation of proteins) were observed in the middle (Fig. 9b) as well as at the tip (Fig. 9c) of the trans-Golgi cisterna. This was particularly prominent after 24-h of CST treatment, where dense cores in Golgi cisternae were observed in ~63% of cells. Although we are not in a position to explain this interesting observation, it does suggest that CST may play a role in efficient packaging of secretory proteins in the trans-Golgi. A DCV budded off from the trans-Golgi cisterna is shown in Fig. 9(d).

Golgi apparatus in PC12 cells treated with CST for 24-h showing a dense core in the trans-Golgi apparatus. a Golgi apparatus in control cell. CCV: clathrin-coated vesicles. b Golgi apparatus in CST-treated cells showing a dense core (red arrow) in the middle of a trans-Golgi cisterna. c Golgi apparatus in CST-treated cells showing a dense core at the tip of a trans-Golgi cisterna. d Golgi apparatus in CST-treated cells showing budded off DCV
Discussion
The present study uncovers an important role for CST in the regulation of vesicular quanta, especially upon interaction with cholinergic and peptidergic stimulation of PC12 cells. Our previous studies focused on the role of CST in regulating catecholamine secretion induced by nicotine (cholinergic agonist), KCl (membrane depolarization), BaCl2 (alkaline earth), A23187 (calcium ionophore), chloroquine (vesicle core alkalinizing agent) and ATP (P2x purinergic agonist) where CST inhibited catecholamine secretion induced by nicotine only (Mahata et al. 1997). Decreased vesicular NE content after 0.5-h of treatment with nicotine corresponds to increased secretion as shown by 3H-NE secretion studies. Co-treatment of nicotine and CST did not change vesicular NE content, in agreement with our previous secretion studies (Mahata et al. 2000; Mahata et al. 1997). Although we did not perform secretion assays after 24-h of treatment, vesicular NE content remained low even after 24-h of secretogogue treatment, indicating prolonged secretion in response to nicotine or PACAP (Taupenot et al. 1999).
Although several studies implicate CST action through a G protein-coupled receptor (GPCR) (Angelone et al. 2008; Angelone et al. 2015; Angelone et al. 2012), the effects of CST on PACAP-induced catecholamine secretion has not previously been examined. In the present study, we found that CST also inhibited PACAP-induced decrease of vesicular quanta as reflected by decreased 3H-NE secretion after 0.5-h of treatment. We have previously shown that CST inhibited nicotine action by inhibiting the first and second steps in nicotinic signaling cascade, i.e., uptake of Na+ (inhibition of membrane depolarization) and Ca2+ ions (inhibition of exocytosis) (Mahata et al. 1997). Since PACAP acts through the PAC1 receptor in PC12 cells, as in chromaffin cells (Eiden et al. 2018; Taupenot et al. 1998), we suggest that CST inhibited PACAP action by inhibiting PACAP’s actions immediately downstream of the PAC1 receptor. Like nicotine, PACAP treatment for 24-h also exhibited decreased vesicular NE content, indicating prolonged secretion, which is consistent with our previous findings.
It has been reported previously that, as with nicotine or PACAP-induced secretion of catecholamines from adrenal medullary chromaffin cells, increased splanchnic nerve activity results in compensatory pathways to replenish endogenous catecholamine stores diminished by secretion (Haycock 1996). Increased TH activity is the biochemical endpoint of these pathways. While brief or intermittent increases in sympathetic activity have been known to produce episodic increases in TH activity, presumably via phosphorylation/dephosphorylation of existing TH molecules, more sustained sympathetic activation leads to a persistent increase in TH activity via an increase in the number of TH molecules through increased transcription and translation (Haycock 1996; Zigmond et al. 1989). Previous reports revealed that TH is transcriptionally activated by both the cholinergic and peptidergic signals to chromaffin cell exocytosis including PC12 cells (Braas and May 1999; Corbitt et al. 1998; Gueorguiev et al. 2006; Gueorguiev et al. 2004; Gueorguiev et al. 2000; Haycock 2002a, b; Hiremagalur et al. 1993; Ishiguro et al. 1997; Isobe et al. 1996; Stroth et al. 2013; Tonshoff et al. 1997). Consistent with the above reports, we found increased expression of Th gene after cholinergic or peptidergic stimulations. However, the increased Th mRNA level after nicotine or PACAP was not translated into protein. It is quite possible that both nicotine and PACAP can still increase TH activity by phosphorylating one or more serine residues in TH protein, as the phosphorylation of serine residues 8, 19, 31 and 40 are critical for TH activity. Although CST increased expression of both Th gene and TH protein, it did not increase synthesis of catecholamines. One possible explanation could be that CST does not phosphorylate TH protein at serine residues. Of note, Ser8, Ser19, Ser31 and Ser40 residues of TH protein are phosphorylated by cell cycle-associated protein kinase cdc2 (Hall et al. 1991), type II calcium/calmoduin-dependent protein kinase (Ca/CaM-PKII) (Haycock et al. 1998; Vulliet et al. 1985), the mitogen-activated protein kinases ERK1/2 (Haycock et al. 1992) and numerous protein kinases (cAMP-dependent protein kinase PKA, protein kinase C and even Ca/CaM-PKII) (Halloran and Vulliet 1994; Haycock et al. 1992; Haycock and Wakade 1992; Lindgren et al. 2002; Sutherland et al. 1993; Zigmond et al. 1989), respectively. It remains to be determined whether CST activates cdc2, CaM-PKII, ERK, or PKA and the findings will either confirm or refute our proposed hypothesis. The increased DCV area and diameter that we observed at the ultrastructural level after 24-h treatment with CST, CST + nicotine or CST + PACAP are presumably not due to increased synthesis of catecholamines but probably due to the increased synthesis of CgA in case of CST + nicotine; CgB and SgII in case of CST + PACAP.
Nicotinic or peptidergic stimulation increases expression of Th, Dbh, Chga, Chgb and Scg2 genes via cAMP signaling. These genes contain a CRE site (TGACGTCA or TGACGTAA) in their proximal promoter and are activated with increased phosphorylation of CREB (Gueorguiev et al. 2006; Gueorguiev et al. 2004; Hiremagalur et al. 1993; Mahapatra et al. 2000; Wu et al. 1995). Thus, in the present study, the increased phosphorylation of CREB at Ser133 by nicotine or PACAP confirms the previous findings in PC12 cells. The induction of CgB and SgII proteins and mRNA by PACAP alone was consistent with this receptor being a very strong inducer of cAMP signaling and CREB phosphorylation. Nicotine is a weaker Gnas agonist and therefore produces a less robust CREB phosphorylation, which might be sufficient to activate the CREs in the Chgb and Scg2 genes. In contrast, Chga is induced by all ligands in correspondence with threshold activation/phosphorylation of CREB.
CREB is a target for cAMP signaling through PKA but other kinases also phosphorylate and activate CREB (Delghandi et al. 2005) and transcription factors oher than CREB may activate the CRE (Hai and Curran 1991). CST may modulate transcription of CRE/CREB activated genes through these additional pathways. In addition, promoter elements in the Chga gene other than the CRE may strengthen the response to other pathways, such as Ca2+ or ERK, which contribute to differential regulation at the gene level. These factors may explain why CgA protein regulation seems most sensitive to CST, whereas a relatively pure Gs-coupled activator of cAMP elevation, such as PACAP, does not induce CgA protein. The induction of CgB and SgII protein and their cognate mRNAs under various conditions of secretogogue stimulation and co-treatment with CST are highly correlated, and PACAP is the only ligand that causes induction of Chgb and Scg2. PACAP is a very strong inducer of cAMP signaling and causes strong phosphorylation of CREB (Ser133). Nicotine is a weaker Gs agonist and CREB phosphorylation is correspondingly weaker and, again, likely insufficient to activate the CREs in the Chgb and Scg2 genes. In contrast, Chga is induced by all ligands and its induction correlates well with the activation/phosphorylation of CREB.
Decreased presence of 100, 150 and 200 nm DC diameter vesicles and increased presence of 50 nm vesicles after 0.5-h of nicotine treatment indicate release of vesicular contents. Likewise, decreased presence of 150 and 200 nm DC vesicles and increased presence of 50 nm vesicles after 0.5-h of PACAP treatment also indicate increased exocytosis. Co-treatment of nicotine with CST inhibited nicotine-induced decrease of 100, 150 and 200 nm DC diameter vesicles, which fits with the catecholamine data as well as with our previous findings. Unlike nicotine, co-treatment of PACAP with CST resulted in an increased presence of 150 and 200 nm diameter DC vesicles. It is unclear why PACAP alone or in combination with CST had no effect on 100 nm DC diameter vesicles. Consistent with the major stimulators at the splanchnicoadrenomedullary synapse, the vast majority (> 80%) of the vesicles were shown to be docked after 0.5-h treatments with nicotine or PACAP. While most vesicles appear to fuse fully with the plasma membrane (Burgoyne et al. 2001; Dhara et al. 2018; Holz and Axelrod 2008; Lopez-Font et al. 2010; Sugita 2008), we observed that in ~5% of cases, the DCV membrane opened first before opening of the plasma membrane. This may represent an intermediate stage or possibly the “kiss-and-run” method of exocytosis (Albillos et al. 1997; Alvarez de Toledo et al. 1993; Henkel et al. 2001). While the fusion pore in the “kiss-and-run” method of exocytosis has been reported to be < 3 nm in diameter (Albillos et al. 1997), we observed here that the widest exocytotic pore in full fusion in PC12 DCV is ~ 106 nm. We reported that CST inhibited nicotine-induced uptake of Ca2+ (Mahata et al. 1997) and high intracellular Ca2+ has been reported to be reported to be responsible for shifting the “kiss-and-run” exocytosis to “full fusion” exocytosis (Elhamdani et al. 2006). Our present observation of reduced full fusion (> 70%) and increased “kiss-and-run” exocytosis by CST are consistent with these previous reports (Biswas et al. 2012; Mahata et al. 1997).
The term “vesicular cocktail” coined by Winkler and colleagues (Winkler and Fischer-Colbrie 1992; Winkler et al. 1987) refers to the high concentrations of catecholamines (0.5-1 M), ATP (~150 mM), Ca2+ (~ 40 mM) and CgA (~ 1.8 mM) and the modulation of secretory component ratios under various physiological conditions (Winkler and Westhead 1980). Acute stimulation of PC12 cells with nicotine and PACAP restored the vesicular quanta as evident by electron microscopy and supported by intracellular catecholamine and 3H-NE secretion experiments. It is interesting to note that CST increased expression of steady-state mRNA as well as protein levels of CgA, the precursor of CST. This effect also should lead to an increase in vesicular quanta. The additional increase in CgA protein after co-treatments of nicotine and CST indicate that nicotine and CST signal through divergent pathways to activate expression of CgA protein leading to an additional increase in vesicular quantal size. The increased DCV area and diameter that we have observed at the ultrastructural level after 24-h treatment of CST + nicotine strengthens and supports the above hypothesis. Increased CgA expression by nicotine and CST is observed with various anti-CgA antibodies, except for the anti-PST and CT-CST. Considering that the anti-CT-CST antibody can recognize CgA cleaved at residues 372/373 but not the full-length CgA (Bianco et al. 2016), these data suggest that nicotine and CST induce the expression of CgA but not its fragmentation at the 372/373 residues. Thus, it is unlikely that nicotine induced CST production in these conditions. In contrast, although PACAP increased expression of the Chga mRNA level, such increased mRNA was not translated into more CgA protein. Interestingly, PACAP increased expression of both Chgb and Scg2 mRNA levels as well as CgB and SgII protein levels, indicating a differential regulation of CgB and SgII proteins by cholinergic and peptidergic stimulation (vide supra). The increased CgB and SgII levels by PACAP may also lead to an increase in vesicular quanta. Increased DCV area and diameter as documented by our ultrastructural findings after 24-h of CST+PACAP strengthens and supports the above hypothesis. Thus, the increased DCV area and diameter seen in CST+nicotine treatment is due to an increase in CgA protein induced both by CST and nicotine; the increased DCV area and diameter seen after PACAP is due to an increase in synthesis of CgB and SgII proteins. Figure 10 depicts the overall proposed model for the role of CST in regulation of vesicular quanta. In summary, our results suggest that CST modulated DCV quanta both in response to chronic and acute stimulations of cholinergic and PACAPergic nature. Acutely, CST appears to inhibit augmented exocytosis by nicotine and PACAP. Chronically CST appears to modulate expression of DCV proteins including CgA, CgB and SgII. In future studies, we plan to follow up our observations and dissect the molecular events contributing to the cholinergic- and PACAPergic regulation of vesicular quanta, and modulation by CST in both adrenal and other endocrine organs.

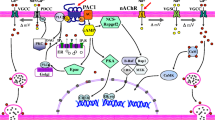
Schematic diagram showing a model for CST’s role in the regulation of vesicular quanta in response to acute and chronic cholinergic/peptidergic stimuli. Nicotine, after binding with nicotinic-acetylcholine receptor (nAChR), activates influx of sodium ions into the cytoplasm causing depolarization of PC12 cell membrane, which results in influx of calcium ions through voltage-gated calcium channels. The increased calcium ion concentration inside the cytosol results in exocytosis and phosphorylate CREB through activation of PKC and ERK. Phospho-CREB then enters the nucleus and binds with the Chga-CRE site (TGACGTAA) and phospho-CBP to stimulate transcription of the Chga gene followed by translation and trafficking to catecholamine storage vesicles. Upon binding to PAC1 receptor, PACAP induces exocytosis as well as phosphorylates CREB through Gsα → AC → cAMP → PKA → CREB. Phospho-CREB enters the nucleus and binds with the Chgb-CRE (TGACGTCA) and phospho-CBP to stimulate transcription of the Chgb gene followed by translation and trafficking to catecholamine storage vesicles. Upon binding to a putative GPCR, CST can phosphorylate CREB through Gsα → AC → cAMP → PKA → CREB. Phospho-CREB enters the nucleus and binds with the Chga-CRE (TGACGTAA) and phospho-CBP to stimulate transcription of the Chga gene followed by translation and trafficking to catecholamine storage vesicles
References
Albillos A, Dernick G, Horstmann H, Almers W, Alvarez de Toledo G, Lindau M (1997) The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 389:509–512
Alvarez de Toledo G, Fernandez-Chacon R, Fernandez JM (1993) Release of secretory products during transient vesicle fusion. Nature 363:554–558
Angelone T, Quintieri AM, Brar BK, Limchaiyawat PT, Tota B, Mahata SK, Cerra MC (2008) The antihypertensive chromogranin a peptide catestatin acts as a novel endocrine/paracrine modulator of cardiac inotropism and lusitropism. Endocrinology 149:4780–4793
Angelone T, Quintieri AM, Pasqua T, Gentile S, Tota B, Mahata SK, Cerra MC (2012) Phosphodiesterase type-2 and NO-dependent S-nitrosylation mediate the cardioinhibition of the antihypertensive catestatin. Am J Phys Heart Circ Phys 302:H431–H442
Angelone T, Quintieri AM, Pasqua T, Filice E, Cantafio P, Scavello F, Rocca C, Mahata SK, Gattuso A, Cerra MC (2015) The NO stimulator, Catestatin, improves the Frank-Starling response in normotensive and hypertensive rat hearts. Nitric Oxide 50:10–19
Anouar Y, Eiden LE (1995) Rapid and long-lasting increase in galanin mRNA levels in rat adrenal medulla following insulin-induced reflex splanchnic nerve stimulation. Neuroendocrinology 62:611–618
Bassino E, Fornero S, Gallo MP, Gallina C, Femmino S, Levi R, Tota B, Alloatti G (2015) Catestatin exerts direct protective effects on rat cardiomyocytes undergoing ischemia/reperfusion by stimulating PI3K-Akt-GSK3beta pathway and preserving mitochondrial membrane potential. PLoS One 10:e0119790
Bianco M, Gasparri AM, Colombo B, Curnis F, Girlanda S, Ponzoni M, Bertilaccio MT, Calcinotto A, Sacchi A, Ferrero E, Ferrarini M, Chesi M, Bergsagel PL, Bellone M, Tonon G, Ciceri F, Marcatti M, Caligaris-Cappio F, Corti A (2016) Chromogranin a is preferentially cleaved into proangiogenic peptides in the bone marrow of multiple myeloma patients. Cancer Res 76:1781–1791
Biswas N, Gayen J, Mahata M, Su Y, Mahata SK, O'Connor DT (2012) Novel peptide isomer strategy for stable inhibition of catecholamine release: application to hypertension. Hypertension 60:1552–1559
Bittner MA, Aikman RL, Holz RW (2013) A nibbling mechanism for clathrin-mediated retrieval of secretory granule membrane after exocytosis. J Biol Chem 288:9177–9188
Braas KM, May V (1999) Pituitary adenylate cyclase-activating polypeptides directly stimulate sympathetic neuron neuropeptide Y release through PAC(1) receptor isoform activation of specific intracellular signaling pathways. J Biol Chem 274:27702–27710
Burgoyne RD, Fisher RJ, Graham ME, Haynes LP, Morgan A (2001) Control of membrane fusion dynamics during regulated exocytosis. Biochem Soc Trans 29:467–472
Cheung NS, Basile S, Livett BG (1993) Identification of multiple tachykinins in bovine adrenal medulla using an improved chromatographic procedure. Neuropeptides 24:91–97
Colombo B, Curnis F, Foglieni C, Monno A, Arrigoni G, Corti A (2002) Chromogranin A expression in neoplastic cells affects tumor growth and morphogenesis in mouse models. Cancer Res 62:941–946
Corbitt J, Vivekananda J, Wang SS, Strong R (1998) Transcriptional and posttranscriptional control of tyrosine hydroxylase gene expression during persistent stimulation of pituitary adenylate cyclase-activating polypeptide receptors on PC12 cells: regulation by protein kinase A-dependent and protein kinase A-independent pathways. J Neurochem 71:478–486
Creutz CE, Harrison JR (1984) Clathrin light chains and secretory vesicle binding proteins are distinct. Nature 308:208–210
Delghandi MP, Johannessen M, Moens U (2005) The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells. Cell Signal 17:1343–1351
Dev NB, Gayen JR, O'Connor DT, Mahata SK (2010) Chromogranin A and the autonomic system: decomposition of heart rate variability by time and frequency domains, along with non-linear characteristics during chromogranin A ablation, with “rescue” by its catestatin. Endocrinology 151:2760–2768
Dhara M, Mohrmann R, Bruns D (2018) v-SNARE function in chromaffin cells. Pflugers Arch 470:169–180
Douglas WW, Rubin RP (1961) Mechanism of nicotinic action at the adrenal medulla: calcium as a link in stimulus-secretion coupling. Nature 192:1087–1089
Douglas WW, Kanno T, Sampson SR (1967) Effects of acetylcholine and other medullary secretagogues and antagonists on the membrane potential of adrenal chromaffin cells: an analysis employing techniques of tissue culture. J Physiol 188:107–120
Eiden LE (1987) Is chromogranin a prohormone? [news]. Nature 325:301
Eiden LE, Emery AC, Zhang L, Smith CB (2018) PACAP signaling in stress: insights from the chromaffin cell. Pflugers Arch 470:79–88
Elhamdani A, Azizi F, Artalejo CR (2006) Double patch clamp reveals that transient fusion (kiss-and-run) is a major mechanism of secretion in calf adrenal chromaffin cells: high calcium shifts the mechanism from kiss-and-run to complete fusion. J Neurosci 26:3030–3036
Fischer-Colbrie R, Iacangelo A, Eiden LE (1988) Neural and humoral factors separately regulate neuropeptide Y, enkephalin, and chromogranin A and B mRNA levels in rat adrenal medulla. Proc Natl Acad Sci U S A 85:3240–3244
Fung MM, Salem RM, Mehtani P, Thomas B, Lu CF, Perez B, Rao F, Stridsberg M, Ziegler MG, Mahata SK, O'Connor DT (2010) Direct vasoactive effects of the chromogranin A (CHGA) peptide catestatin in humans in vivo. Clin Exp Hypertens 32:278–287
Gayen JR, Gu Y, O'Connor DT, Mahata SK (2009a) Global disturbances in autonomic function yield cardiovascular instability and hypertension in the chromogranin a null mouse. Endocrinology 150:5027–5035
Gayen JR, Gu Y, O’Connor DT, Mahata SK (2009b) Global disturbances in autonomic function yield cardiovascular instability and hypertension in the chromogranin A null mouse. Endocrinology 150:5027–5035
Greene LA, Tischler AS (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci U S A 73:2424–2428
Gueorguiev VD, Zeman RJ, Meyer EM, Sabban EL (2000) Involvement of alpha7 nicotinic acetylcholine receptors in activation of tyrosine hydroxylase and dopamine beta-hydroxylase gene expression in PC12 cells. J Neurochem 75:1997–2005
Gueorguiev VD, Frenz CM, Ronald KM, Sabban EL (2004) Nicotine and epibatidine triggered prolonged rise in calcium and TH gene transcription in PC12 cells. Eur J Pharmacol 506:37–46
Gueorguiev VD, Cheng SY, Sabban EL (2006) Prolonged activation of cAMP-response element-binding protein and ATF-2 needed for nicotine-triggered elevation of tyrosine hydroxylase gene transcription in PC12 cells. J Biol Chem 281:10188–10195
Guo X, Wakade AR (1994) Differential secretion of catecholamines in response to peptidergic and cholinergic transmitters in rat adrenals. J Physiol Lond 475:539–545
Hahm SH, Hsu CM, Eiden LE (1998) PACAP activates calcium influx-dependent and -independent pathways to couple met-enkephalin secretion and biosynthesis in chromaffin cells. J Mol Neurosci 11:43–56
Hai T, Curran T (1991) Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proceedings of the National Academy of Sciences 88(9):3720–3724
Hall FL, Braun RK, Mihara K, Fung YK, Berndt N, Carbonaro-Hall DA, Vulliet PR (1991) Characterization of the cytoplasmic proline-directed protein kinase in proliferative cells and tissues as a heterodimer comprised of p34cdc2 and p58cyclin A. J Biol Chem 266:17430–17440
Halloran SM, Vulliet PR (1994) Microtubule-associated protein kinase-2 phosphorylates and activates tyrosine hydroxylase following depolarization of bovine adrenal chromaffin cells. J Biol Chem 269:30960–30965
Haycock JW (1996) Short- and long-term regulation of tyrosine hydroxylase in chromaffin cells by VIP and PACAP. Ann N Y Acad Sci 805:219–230 discussion 230-211
Haycock JW (2002a) Peptide substrates for ERK1/2: structure-function studies of serine 31 in tyrosine hydroxylase. J Neurosci Methods 116:29–34
Haycock JW (2002b) Species differences in the expression of multiple tyrosine hydroxylase protein isoforms. J Neurochem 81:947–953
Haycock JW, Wakade AR (1992) Activation and multiple-site phosphorylation of tyrosine hydroxylase in perfused rat adrenal glands. J Neurochem 58:57–64
Haycock JW, Ahn NG, Cobb MH, Krebs EG (1992) ERK1 and ERK2, two microtubule-associated protein 2 kinases, mediate the phosphorylation of tyrosine hydroxylase at serine-31 in situ. Proc Natl Acad Sci U S A 89:2365–2369
Haycock JW, Lew JY, Garcia-Espana A, Lee KY, Harada K, Meller E, Goldstein M (1998) Role of serine-19 phosphorylation in regulating tyrosine hydroxylase studied with site- and phosphospecific antibodies and site-directed mutagenesis. J Neurochem 71:1670–1675
Henkel AW, Kang G, Kornhuber J (2001) A common molecular machinery for exocytosis and the ‘kiss-and-run’ mechanism in chromaffin cells is controlled by phosphorylation. J Cell Sci 114:4613–4620
Hiremagalur B, Nankova B, Nitahara J, Zeman R, Sabban EL (1993) Nicotine increases expression of tyrosine hydroxylase gene. Involvement of protein kinase A-mediated pathway. J Biol Chem 268:23704–23711
Hiremagalur B, Kvetnansky R, Nankova B, Fleischer J, Geertman R, Fukuhara K, Viskupic E, Sabban EL (1994) Stress elicits trans-synaptic activation of adrenal neuropeptide Y gene expression. Brain Res Mol Brain Res 27:138–144
Holz RW, Axelrod D (2008) Secretory granule behaviour adjacent to the plasma membrane before and during exocytosis: total internal reflection fluorescence microscopy studies. Acta Physiol (Oxf) 192:303–307
Imbrogno S, Garofalo F, Cerra MC, Mahata SK, Tota B (2010) The catecholamine release-inhibitory peptide catestatin (chromogranin A344-364) modulates myocardial function in fish. J Exp Biol 213:3636–3643
Ishiguro H, Ichino N, Yamada K, Nagatsu T (1997) Nicotine regulates mRNA level of tyrosine hydroxylase gene but not that of nicotinic acetylcholine receptor genes in PC12 cells. Neurosci Lett 228:37–40
Isobe K, Yukimasa N, Nakai T, Takuwa Y (1996) Pituitary adenylate cyclase-activating polypeptide induces gene expression of the catecholamine synthesizing enzymes, tyrosine hydroxylase and dopamine beta hydroxylase, through 3′,5′-cyclic adenosine monophosphate- and protein kinase C-dependent mechanisms in cultured porcine adrenal medullary chromaffin cells. Neuropeptides 30:167–175
Kakhlon O, Sakya P, Larijani B, Watson R, Tooze SA (2006) GGA function is required for maturation of neuroendocrine secretory granules. EMBO J 25:1590–1602
Kennedy BP, Mahata SK, O'Connor DT, Ziegler MG (1998) Mechanism of cardiovascular actions of the chromogranin A fragment catestatin in vivo. Peptides 19:1241–1248
Kirchmair R, Hogue-Angeletti R, Gutierrez J, Fischer-Colbrie R, Winkler H (1993) Secretoneurin—a neuropeptide generated in brain, adrenal medulla and other endocrine tissues by proteolytic processing of secretogranin II (chromogranin C). Neuroscience 53:359–365
Kojima M, Ozawa N, Mori Y, Takahashi Y, Watanabe-Kominato K, Shirai R, Watanabe R, Sato K, Matsuyama TA, Ishibashi-Ueda H, Koba S, Kobayashi Y, Hirano T, Watanabe T (2018) Catestatin prevents macrophage-driven atherosclerosis but not arterial injury-induced neointimal hyperplasia. Thromb Haemost 118:182–194
Kroesen S, Marksteiner J, Leitner B, Hogue-Angeletti R, Fischer-Colbrie R, Winkler H (1996) Rat brain: distribution of immunoreactivity of PE-11, a peptide derived from chromogranin B. Eur J Neurosci 8:2679–2689
Lindgren N, Goiny M, Herrera-Marschitz M, Haycock JW, Hokfelt T, Fisone G (2002) Activation of extracellular signal-regulated kinases 1 and 2 by depolarization stimulates tyrosine hydroxylase phosphorylation and dopamine synthesis in rat brain. Eur J Neurosci 15:769–773
Livett BG, Marley PD (1993) Noncholinergic control of adrenal catecholamine secretion. J Anat 183:277–289
Lopez-Font I, Torregrosa-Hetland CJ, Villanueva J, Gutierrez LM (2010) t-SNARE cluster organization and dynamics in chromaffin cells. J Neurochem 114:1550–1556
Mahapatra NR, Mahata M, Datta A, Gerdes H-H, Huttner WB, O’Connor DT, Mahata SK (2000) Neuroendocrine cell type-specific and inducible expression of the chromogranin B gene: crucial role of the proximal promoter. Endocrinology 141:3668–3678
Mahapatra NR, O'Connor DT, Vaingankar SM, Hikim AP, Mahata M, Ray S, Staite E, Wu H, Gu Y, Dalton N, Kennedy BP, Ziegler MG, Ross J, Mahata SK (2005) Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J Clin Invest 115:1942–1952
Mahata M, Mahata SK, Parmer RJ, O'Connor DT (1996) Vesicular monoamine transport inhibitors. Novel action at calcium channels to prevent catecholamine secretion. Hypertension 28:414–420
Mahata SK, O'Connor DT, Mahata M, Yoo SH, Taupenot L, Wu H, Gill BM, Parmer RJ (1997) Novel autocrine feedback control of catecholamine release. A discrete chromogranin A fragment is a noncompetitive nicotinic cholinergic antagonist. J Clin Invest 100:1623–1633
Mahata SK, Mahata M, Parmer RJ, O'Connor DT (1999) Desensitization of catecholamine release: the novel catecholamine release-inhibitory peptide catestatin (chromogranin A344-364) acts at the receptor to prevent nicotinic cholinergic tolerance. J Biol Chem 274:2920–2928
Mahata SK, Mahata M, Wakade AR, O'Connor DT (2000) Primary structure and function of the catecholamine release inhibitory peptide catestatin (chromogranin A344-364): identification of amino acid residues crucial for activity. Mol Endocrinol 14:1525–1535
Mahata SK, Mahapatra NR, Mahata M, Wang TC, Kennedy BP, Ziegler MG, O'Connor DT (2003) Catecholamine secretory vesicle stimulus-transcription coupling in vivo. Demonstration by a novel transgenic promoter/photoprotein reporter and inhibition of secretion and transcription by the chromogranin A fragment catestatin. J Biol Chem 278:32058–32067
Mahata SK, Mahata M, Wen G, Wong WB, Mahapatra NR, Hamilton BA, O'Connor DT (2004) The catecholamine release-inhibitory “catestatin” fragment of chromogranin A: naturally occurring human variants with different potencies for multiple chromaffin cell nicotinic cholinergic responses. Mol Pharmacol 66:1180–1191
Mahata SK, Mahata M, Fung MM, O'Connor DT (2010) Catestatin: a multifunctional peptide from chromogranin A. Regul Pept 162:33–43
Mahata SK, Zheng H, Mahata S, Liu X, Patel KP (2016) Effect of heart failure on catecholamine granule morphology and storage in chromaffin cells. J Endocrinol 230:309–323
Malhotra RK, Wakade TD, Wakade AR (1989) Cross-communication between acetylcholine and VIP in controlling catecholamine secretion by affecting cAMP, inositol triphosphate, protein kinase C, and calcium in rat adrenal medulla. J Neurosci 9:4150–4157
Mazza R, Gattuso A, Mannarino C, Brar BK, Barbieri SF, Tota B, Mahata SK (2008) Catestatin (chromogranin A344-364) is a novel cardiosuppressive agent: inhibition of isoproterenol and endothelin signaling in the frog heart. Am J Physiol Heart Circ Physiol 295:H113–H122
Mustafa T, Grimaldi M, Eiden LE (2007) The hop cassette of the PAC1 receptor confers coupling to Ca2+ elevation required for pituitary adenylate cyclase-activating polypeptide-evoked neurosecretion. J Biol Chem 282:8079–8091
Mustafa T, Walsh J, Grimaldi M, Eiden LE (2010) PAC1hop receptor activation facilitates catecholamine secretion selectively through 2-APB-sensitive Ca(2+) channels in PC12 cells. Cell Signal 22:1420–1426
O'Farrell M, Marley PD (1997) Multiple calcium channels are required for pituitary adenylate cyclase- activating polypeptide-induced catecholamine secretion from bovine cultured adrenal chromaffin cells. Naunyn Schmiedeberg's Arch Pharmacol 356:536–542
Ottesen AH, Carlson CR, Louch WE, Dahl MB, Sandbu RA, Johansen RF, Jarstadmarken H, Bjoras M, Hoiseth AD, Brynildsen J, Sjaastad I, Stridsberg M, Omland T, Christensen G, Rosjo H (2017) Glycosylated chromogranin A in heart failure: implications for processing and cardiomyocyte calcium homeostasis. Circ Heart Fail 10:e003675. https://doi.org/10.1161/CIRCHEARTFAILURE.116.003675
Pasqua T, Mahata S, Bandyopadhyay GK, Biswas A, Perkins GA, Sinha-Hikim AP, Goldstein DS, Eiden LE, Mahata SK (2016) Impact of chromogranin A deficiency on catecholamine storage, catecholamine granule morphology and chromaffin cell energy metabolism in vivo. Cell Tissue Res 363:693–712
Patra M, Mahata SK, Padhan DK, Sen M (2016) CCN6 regulates mitochondrial function. J Cell Sci 129:2841–2851
Perrelli MG, Tullio F, Angotti C, Cerra MC, Angelone T, Tota B, Alloatti G, Penna C, Pagliaro P (2013) Catestatin reduces myocardial ischaemia/reperfusion injury: involvement of PI3K/Akt, PKCs, mitochondrial KATP channels and ROS signalling. Pflugers Arch 465:1031–1040
Ratti S, Curnis F, Longhi R, Colombo B, Gasparri A, Magni F, Manera E, Metz-Boutigue MH, Corti A (2000) Structure-activity relationships of chromogranin A in cell adhesion. Identification of an adhesion site for fibroblasts and smooth muscle cells. J Biol Chem 275:29257–29263
Sahu BS, Manna PT, Edgar JR, Antrobus R, Mahata SK, Bartolomucci A, Borner GHH, Robinson MS (2017) Role of clathrin in dense core vesicle biogenesis. Mol Biol Cell 28:2676–2685
Schubert D, Klier FG (1977) Storage and release of acetylcholine by a clonal cell line. Proc Natl Acad Sci U S A 74:5184–5188
Smith CB, Eiden LE (2012) Is PACAP the major neurotransmitter for stress transduction at the adrenomedullary synapse? J Mol Neurosci 48:403–412
Stroth N, Kuri BA, Mustafa T, Chan SA, Smith CB, Eiden LE (2013) PACAP controls adrenomedullary catecholamine secretion and expression of catecholamine biosynthetic enzymes at high splanchnic nerve firing rates characteristic of stress transduction in male mice. Endocrinology 154:330–339
Sugita S (2008) Mechanisms of exocytosis. Acta Physiol (Oxf) 192:185–193
Suh HW, Hudson PM, McMillian MK, Das KP, Wilson BC, Wu GC, Hong JS (1995) Long-term stimulation of nicotinic receptors is required to increase proenkephalin A mRNA levels and the delayed secretion of [Met5]-enkephalin in bovine adrenal medullary chromaffin cells. J Pharmacol Exp Ther 275:1663–1670
Sutherland C, Alterio J, Campbell DG, Le Bourdelles B, Mallet J, Haavik J, Cohen P (1993) Phosphorylation and activation of human tyrosine hydroxylase in vitro by mitogen-activated protein (MAP) kinase and MAP-kinase-activated kinases 1 and 2. Eur J Biochem 217:715–722
Tanaka K, Shibuya I, Nagamoto T, Yamashita H, Kanno T (1996) Pituitary adenylate cyclase-activating polypeptide causes rapid Ca2+ release from intracellular stores and long lasting Ca2+ influx mediated by Na+ influx-dependent membrane depolarization in bovine adrenal chromaffin cells. Endocrinology 137:956–966
Tang K, Wu H, Mahata SK, Taupenot L, Rozansky DJ, Parmer RJ, O'Connor DT (1996) Stimulus-transcription coupling in pheochromocytoma cells. Promoter region-specific activation of chromogranin A biosynthesis. J Biol Chem 271:28382–28390
Tang K, Wu H, Mahata SK, Mahata M, Gill BM, Parmer RJ, O'Connor DT (1997) Stimulus coupling to transcription versus secretion in pheochromocytoma cells. Convergent and divergent signal transduction pathways and the crucial roles for route of cytosolic calcium entry and protein kinase C. J Clin Invest 100:1180–1192
Taupenot L, Mahata SK, Wu H, O'Connor DT (1998) Peptidergic activation of transcription and secretion in chromaffin cells. cis and trans signaling determinants of pituitary adenylyl cyclase-activating polypeptide (PACAP). J Clin Invest 101:863–876
Taupenot L, Mahata M, Mahata SK, O’Connor DT (1999) Time-dependent effects of the neuropeptide PACAP on catecholamine secretion. Stimulation and desensitization. Hypertension 34:1152–1162
Tischler AS, Perlman RL, Morse GM, Sheard BE (1983) Glucocorticoids increase catecholamine synthesis and storage in PC12 pheochromocytoma cell cultures. J Neurochem 40:364–370
Tonshoff C, Hemmick L, Evinger MJ (1997) Pituitary adenylate cyclase activating polypeptide (PACAP) regulates expression of catecholamine biosynthetic enzyme genes in bovine adrenal chromaffin cells. J Mol Neurosci 9:127–140
Turquier V, Yon L, Grumolato L, Alexandre D, Fournier A, Vaudry H, Anouar Y (2001) Pituitary adenylate cyclase-activating polypeptide stimulates secretoneurin release and secretogranin II gene transcription in bovine adrenochromaffin cells through multiple signaling pathways and increased binding of pre-existing activator protein-1-like transcription factors. Mol Pharmacol 60:42–52
Vaingankar SM, Li Y, Corti A, Biswas N, Gayen JR, O'Connor DT, Mahata SK (2010) Long human CHGA flanking chromosome 14 sequence required for optimal BAC transgenic "rescue" of disease phenotypes in the mouse Chga knockout. Physiol Genomics 41:91–101
Vandael DH, Ottaviani MM, Legros C, Lefort C, Guerineau NC, Allio A, Carabelli V, Carbone E (2015) Reduced availability of voltage-gated sodium channels by depolarization or blockade by tetrodotoxin boosts burst firing and catecholamine release in mouse chromaffin cells. J Physiol 593:905–927
Vulliet PR, Woodgett JR, Ferrari S, Hardie DG (1985) Characterization of the sites phosphorylated on tyrosine hydroxylase by Ca2+ and phospholipid-dependent protein kinase, calmodulin-dependent multiprotein kinase and cyclic AMP-dependent protein kinase. FEBS Lett 182:335–339
Wakade AR (1988) Noncholinergic transmitter(s) maintains secretion of catecholamines from rat adrenal medulla for several hours of continuous stimulation of splanchnic neurons. J Neurochem 50:1302–1308
Wakade AR, Wakade TD (1982) Secretion of catecholamines from adrenal gland by a single electrical shock: electronic depolarization of medullary cell membrane. Proc Natl Acad Sci U S A 79:3071–3074
Waschek JA, Pruss RM, Siegel RE, Eiden LE, Bader MF, Aunis D (1987) Regulation of enkephalin, VIP, and chromogranin biosynthesis in actively secreting chromaffin cells. Multiple strategies for multiple peptides. Ann N Y Acad Sci 493:308–323
Wen G, Mahata SK, Cadman P, Mahata M, Ghosh S, Mahapatra NR, Rao F, Stridsberg M, Smith DW, Mahboubi P, Schork NJ, O'Connor DT, Hamilton BA (2004) Both rare and common polymorphisms contribute functional variation at CHGA, a regulator of catecholamine physiology. Am J Hum Genet 74:197–207
Westerink RH, Ewing AG (2008) The PC12 cell as model for neurosecretion. Acta Physiol (Oxf) 192:273–285
Winkler H (1993) The adrenal chromaffin granule: a model for large dense core vesicles of endocrine and nervous tissue. J Anat 183:237–252
Winkler H, Fischer-Colbrie R (1992) The chromogranins a and B: the first 25 years and future perspectives. Neuroscience 49:497–528
Winkler H, Westhead E (1980) The molecular organization of adrenal chromaffin granules. Neuroscience 5:1803–1823
Winkler H, Apps DK, Fischer-Colbrie R (1986) The molecular function of adrenal chromaffin granules: established facts and unresolved topics. Neuroscience 18:261–290
Winkler H, Sietzen M, Schober M (1987) The life cycle of catecholamine-storing vesicles. Ann N Y Acad Sci 493:3–19
Wu H, Mahata SK, Mahata M, Webster NJ, Parmer RJ, O'Connor DT (1995) A functional cyclic AMP response element plays a crucial role in neuroendocrine cell type-specific expression of the secretory granule protein chromogranin A. J Clin Invest 96:568–578
Ying W, Mahata S, Bandyopadhyay GK, Zhou Z, Wollam J, Vu J, Mayoral R, Chi NW, Webster NJG, Corti A, Mahata SK (2018) Catestatin inhibits obesity-induced macrophage infiltration and inflammation in the liver and suppresses hepatic glucose production, leading to improved insulin sensitivity. Diabetes 67:841–848
Zigmond RE, Schwarzschild MA, Rittenhouse AR (1989) Acute regulation of tyrosine hydroxylase by nerve activity and by neurotransmitters via phosphorylation. Annu Rev Neurosci 12:415–461
Acknowledgments
The electron micrographs were taken in the Cellular and Molecular Medicine Electron microscopy core facility at UCSD, which is supported in part by National Institutes of Health Award number S10OD023527. This research was supported by a grant from the Department of Veterans Affairs (I01BX000323 to S.K.M.; I01BX002709 and an SRCS award to N.J.G.W) and the National Institutes of Health (NIH/NIDDK DK102496 to A.B. and MH002386 to L.E.E.). JCSTF-180217 travelling research fellowship to B.S.S. from company of biologists (Cambridge, UK). The Noland Scholarship from the California Institute of Technology supported S.M.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Sahu, B.S., Mahata, S., Bandyopadhyay, K. et al. Catestatin regulates vesicular quanta through modulation of cholinergic and peptidergic (PACAPergic) stimulation in PC12 cells. Cell Tissue Res 376, 51–70 (2019). https://doi.org/10.1007/s00441-018-2956-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-018-2956-1