
Abstract
Fetal onset hydrocephalus and abnormal neurogenesis are two inseparable phenomena turned on by a cell junction pathology first affecting neural stem/progenitor cells (NSPCs) and later the multiciliated ependyma. The neurological impairment of children born with hydrocephalus is not reverted by derivative surgery. NSPCs and neurosphere (NE) grafting into the cerebrospinal fluid (CSF) of hydrocephalic fetuses thus appears as a promising therapeutic procedure. There is little information about the cell lineages actually forming the NE as they grow throughout their days in vitro (DIV). Furthermore, there is no information on how good a host the CSF is for grafted NE. Here, we use the HTx rat, a model with hereditary hydrocephalus, with the mutation expressed in about 30% of the litter (hyHTx), while the littermates develop normally (nHTx). The investigation was designed (i) to establish the nature of the cells forming 4 and 6-DIV NE grown from NSPCs collected from PN1/nHTx rats and (ii) to study the effects on these NEs of CSF collected from nHTx and hyHTx. Immunofluorescence analyses showed that 90% of cells forming 4-DIV NEs were non-committed multipotential NSPCs, while in 6-DIV NE, 40% of the NSPCs were already committed into neuronal, glial and ependymal lineages. Six-DIV NE further cultured for 3 weeks in the presence of fetal bovine serum, CSF from nHTx or CSF from hyHTx, differentiated into neurons, astrocytes and βIV-tubulin+ multiciliated ependymal cells that were joined together by adherent junctions and displayed synchronized cilia beating. This supports the possibility that ependymal cells are born from subpopulations of NSC with their own time table of differentiation. As a whole, the findings indicate that the CSF is a supportive medium to host NE and that NE grafted into the CSF have the potential to produce neurons, glia and ependyma.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There is a growing evidence that stem cell transplantation represents a great opportunity for the treatment of many neurological diseases (Armstrong and Svendsen 2000; Buddensiek et al. 2010; Fuchs and Segre 2000; Neuhuber et al. 2008; Pluchino et al. 2003; Seaberg and van der Kooy 2002). Stem cells used for transplantation into the central nervous system include mesenchymal stem cells (Ahn et al. 2013; Satake et al. 2004), neural stem cells (Bai et al. 2003; Buddensiek et al. 2010) and neural progenitor cells. Neural stem cells and neural progenitor cells (in this text we will refer to both of them as NSPCs) can be obtained from fetal brain (Kim et al. 2004; Ohta et al. 2004; Wu et al. 2002) and from different regions of the adult brain such as the hippocampus and the neurogenic niche of the subventricular zone (Rietze et al. 2001). Cultured NSPCs grow to form neurospheres (NEs) that are able to generate neurons, astrocytes and oligodendrocytes (Bez et al. 2003; Gil-Perotín et al. 2013; Guerra et al. 2015a; Marshall et al. 2008; Reynolds and Weiss 1992).
In most of the early investigations, the stem cells were grafted in the vicinity of the injured or altered neural tissue. However, delivery of stem cells into the CSF is emerging as an alternative, particularly for those diseases with a broad distribution in the central nervous system, such as multiple sclerosis. NE grafted into the ventricular CSF of mice with experimental autoimmune encephalomyelitis generate neural precursors that enter into demyelinating areas, differentiate into mature brain cells and promote multifocal remyelination and functional recovery (Pluchino et al. 2003).
Fetal onset hydrocephalus affects 1 to 3 of 1000 live births and is characterized by abnormal CSF flow and dilatation of the ventricular system (Del Bigio 2001, 2010). Derivative surgery, either shunting or ventriculostomy, has been the prevailing treatment of hydrocephalic children for half a century. Although this treatment prevents further damage to the brain caused by intraventricular hypertension, it does not revert the neurological impairment of children born with hydrocephalus (McAllister 2012). This led Williams et al. (2007) to conclude that the most forward-looking research priorities for hydrocephalus research include the development of novel therapies that should emerge from improved understanding of the basic biology of hydrocephalus. These authors envisaged the need to determine the potential role of stem cell therapy. Investigations carried out during the past 10 years have further substantiated the view of Williams et al. (2007).
Numerous studies carried out in human hydrocephalic fetuses and in animal models of congenital hydrocephalus have led to the conclusion that fetal onset hydrocephalus and abnormal neurogenesis are two inseparable phenomena that are turned on early in development by a cell junction pathology of the NSPCs (Guerra et al. 2015a; Rodríguez et al. 2012). This would explain both, the neurological impairment of children born with hydrocephalus and that such an impairment is not reversed by derivative surgery (Ortega et al. 2016; Rodríguez and Guerra 2017). Thus, the grafting of stem cells into hydrocephalic fetuses appears as a valid therapeutic procedure. However, there are no reports yet on the use of NSPC grafting in animal models with inherited hydrocephalus.
Although grafting of NE into the CSF has been used in animal models of multiple sclerosis or induced brain injuries (see above), minimal information has been provided concerning the nature of the cells forming the transplanted NE. It is generally assumed that NEs are exclusively formed by NSPCs and that grafting of NE is equivalent to grafting both cell types. Is this really the case? In the NE assay described initially by Reynolds and Weiss (1992), a single NSPC under a serum-free condition and supplemented with mitogenic factors proliferate to form growing cell clusters called NE (Reynolds and Weiss 1992). After 6 DIV, NEs are formed by a heterogeneous population of neural cells, including NSPCs (Guerra et al. 2015a). Nevertheless, there is no information about the cell lineages, forming the NE, as they grow from 1 DIV to several DIV. Gaining insight about the cell biology of NE will allow the design of more reliable and potentially more efficient grafting strategies. Indeed, it seems quite necessary to know what has actually been transplanted.
On the other hand, when grafting NE into the brain ventricles, would CSF be a supportive host? The CSF is a complex milieu containing a few hundred different proteins, several of them being growth factors (Parada et al. 2006; Zappaterra et al. 2007). How would the cells forming the NE react when exposed to wealth of proteins present in the CSF, either normal or hydrocephalic?
The aims of the present investigation are threefold. (1) To identify the cell lineages that progressively form the NE as they grow through several days in vitro. (2) To establish the multipotential capacity of 4- and 6-DIV NE when processed for the differentiation assay. (3) To investigate the behavior of NE exposed to normal or hydrocephalic CSF. NEs were obtained from NSPCs from the ventricular and subventricular zones (VZ/SVZ) of PN1 normal, non-hydrocephalic HTx rats. In this rat strain, about 30% of the pups express the hydrocephalic phenotype (hyHTx), while about 70% do not (non-hydrocephalic rats, nHTx). HTx rat is a unique model to study congenital hydrocephalus because (i) it is the only rat strain with inherited congenital hydrocephalus and, (ii) the neuropathological alterations occurring early in development, namely, a cell junction pathology of neural stem cells, disruption of the ventricular zone and onset of hydrocephalus, are similar to those observed in human fetuses with hydrocephalus (Rodríguez and Guerra 2017). Furthermore, this rat strain is a good candidate for transplantation experiments by grafting NE obtained from nHTx (as reported presently) into the CSF of hyHTx.
The findings of the present investigation, i.e., the potentiality of NE to generate neurons and multiciliated ependyma in particular, are a solid base to design well-substantiated grafting protocols for repairing/diminishing the early disruption of the ventricular zone in fetal onset hydrocephalus (Rodríguez and Guerra 2017).
Material and methods
Animals
The HTx rat represents the only rat model with hereditary hydrocephalus; the mutation is expressed in about 30% of the litter (hyHTx), while the littermates develop normally (nHTx). The HTx strain (Jones and Bucknall 1987) was obtained from the laboratory of Dr. Hazel Jones (University of Florida, Gainesville, FL) in 2002 and bred into a colony in the Animal Facility at the Instituto de Anatomía Histología y Patología, Universidad Austral de Chile, Valdivia, Chile. Housing, handling, care and processing of animals were carried out according to regulations approved by the National Research Council of Chile (CONICYT). The ethics committees of Universidad Austral de Chile approved the experimental protocol. The hydrocephalic phenotype was identified from an overtly domed head and by transillumination of the head of newborns.
Neurospheres from the brain of nHTx rats
At postnatal day 1, nHTx rats were euthanized by decapitation. The dorsolateral walls of both lateral ventricles containing the ventricular/subventricular zone (VZ/SVZ) were excised under a microscope and placed in a 2-ml tube containing 1 ml of NE culture medium (NeuroCult NS-A Proliferation Medium-Rat), supplemented with 20 ng/ml epidermal growth factor and 2 μg/ml heparin (StemCell Technologies, Vancouver, CA) and 100 μg/ml penicillin/streptomycin (Sigma). The tissue was disaggregated mechanically and 1 ml of fresh culture medium was added. The cell suspension was centrifuged for 10 min at 110 g. The viable cells were counted using the trypan blue cell viability assay by a hemocytometer and 60,000 cells/ml were seeded in a non-adherent plate dish (Techno Plastic Products AG, Trasadingen, Switzerland). The cells were cultured for 4 or 6 DIV and monitored once a day by phase contrast microscopy. Ten micromolars of BrdU was added for the last 3, 12, or 24 h of culture.
Quantitative analysis of BrdU labeling
NSPC of nHTx were cultured for 6 days. The resulting NEs were exposed to 10 μM BrdU for 3 h (group 1) and 24 h (group 2). In each group, the experiment was performed three times. NEs were spun down, fixed in Bouin fixative, embedded in paraffin and serially cut. About 100 sections, 8 μm thick, were obtained from each pellet. Every tenth section of the series, a pair of adjacent sections were processed for hematoxylin-eosin stain and immunocytochemistry using anti-BrdU (see below); they were used to measure NE diameter and to count the proliferative cells, respectively.
Analysis
NEs were grouped into small (20- to 80-μm diameter) and large (100- to 160-μm diameter). In each of the experimental groups (3 and 24 h labeling), sections of 90 small NEs and 90 large NEs were analyzed. In each section, the total number of cells and the number of BrdU+ cells was recorded. The findings were expressed as percentage of BrdU+ cells versus total number of cells. Within each population of NE (large and small), the relative number of BrdU+ cells after 3 and 24 h was compared using the Student’s t test, before the determination of the data normality using the D’Agostino-Pearson omnibus test. Data were expressed as average SE with p < 0.0001 for statistical significance. Statistical analysis and normality test of the data were made using Prism GraphPad 6.0 (GraphPad Software, Inc., San Diego, CA).
Colchicine experiment
NEs of 6 DIV were cultured in the presence of different concentrations of colchicine diluted in PBS pH 7.4 (1, 2, 5, 10 and 20 μM) for 4 h and monitored by phase contrast microscopy. The vehicle (PBS pH 7.4) was used as control. These experiments were performed three times for each condition.
Horseradish peroxidase experiment
Six-DIV NEs were exposed for 1 min to 1% horseradish peroxidase (Sigma, Grade VI) diluted in PBS pH 7.4; then, they were fixed in Bouin fixative and processed for conventional histology and immunocytochemistry using anti-horseradish peroxidase. This experiment was performed four times.
Light microscopy histology
NEs were collected and spun down; the resulting pellet was fixed in Bouin fixative and embedded in paraffin. Serial sections 5 μm thick were obtained.
Immunohistochemistry
Sections from NE were processed for immunohistochemistry by the streptavidin/biotin method (Vectastain kit; Vector, Serva, Heidelberg, Germany), with diaminobenzidine (3,3-diaminobenzidine tetrahydrochloride; Sigma, St. Louis, MO, USA) as the electron donor. Antibodies against mouse anti-bromodeoxyuridine (BrdU), monoclonal (Developmental Studies Hybridoma Bank, Iowa, IA, USA) and horseradish peroxidase, polyclonal developed in rabbits (Instituto de Histología y Patología, Universidad Austral de Chile) were used. Sections to be immunoreacted with anti-BrdU were incubated in 0.02 M citrate buffer, pH 6.0, followed by microwave irradiation, three sessions, 4 min each. Incubation in the primary antibody was for 18 h at room temperature.
All antibodies were diluted in 0.1 M Tris buffer, pH 7.8, containing 0.7% non-gelling seaweed gelatin lambda carrageenan and 0.5% Triton X-100 (both from Sigma, St. Louis, MO, USA). Omission of the primary antibody during incubation provided the control for the immunoreactions.
Immunofluorescence
Sections from neurospheres were processed for immunofluorescence using the following antibodies: (i) BrdU, monoclonal (Developmental Studies Hybridoma Bank, USA), supernatant 1:500 dilution; (ii) Rabbit anti-BrdU (Rockland antibodies and assay, Limerick, PA, USA); (iii) glial fibrillary acidic protein (GFAP, astrocyte marker), polyclonal raised in rabbit (Sigma, St. Louis, MO, USA), 1:750 dilution; (iv) βIII-tubulin (neuronal marker), monoclonal (Sigma, St. Louis, MO, USA), 1:750 dilution; (v) nestin (NSPC marker), monoclonal (Developmental Studies Hybridoma Bank, Iowa, USA), supernatant, 1:20 dilution; (vi) connexin 43 (gap junction), polyclonal raised in rabbit (gift from Dr. Juan Carlos Sáez, Universidad Católica de Chile, Santiago, Chile), 1:750 dilution; (vii) N-cadherin (adherent junctions) polyclonal raised in rabbit (Santa Cruz Biotechnology Inc., CA, USA), dilution 1:50; (viii) β-catenin (adherent junctions), polyclonal raised in rabbit (BD Biosciences, USA), 1:100 dilution; (ix) MAP2 (microtubule-associated protein 2; neuronal marker) monoclonal (Abcam, Cambridge, UK), 1:100 dilution; (x) rabbit anti-AQP4 (astrocyte marker) (Abnova, Taipei, Taiwan), 1:750 dilution; and (xi) mouse anti-βIV-tubulin (multiciliated ependyma marker) (Abcam, Cambridge, UK), 1:50 dilution. Antibodies were diluted in a buffer containing 0.1 M Tris buffer, pH 7.8, 0.7% non-gelling seaweed gelatin lambda carrageenan and 0.5% Triton-X 100. Appropriate secondary antibodies conjugated with Alexa Fluor 488 or 594 (1:500, Invitrogen, Carlsbad, CA) were used. In some cases, the samples were incubated with a DAPI solution (4′, 6-Diamidino-2-Phenylindole, Dihydrochloride, Molecular ProbesTM, Thermo Fisher scientific) for 10 min. Slides were coverslipped by using Vectashield mounting medium (Dako, Santa Clara, CA, USA) and inspected under an epifluorescence microscope to study colocalization by using the multidimensional acquisition software AxioVision Rel version 4.6 (Zeiss, Aalen, Germany) or a confocal microscope (LSM700/Axio Imager Z2m). Incubation was carried out for 18 h at RT. Omission of the primary antibody during incubation provided the control for the immunoreaction.
Whole mount immunofluorescence
The NEs (from three experiments) were fixed in 2% paraformaldehyde in PBS, pH 7.4, for 15 min at RT and then post-fixed in 4% paraformaldehyde, in PBS pH 7.4, for 15 min. Finally, NEs were incubated with anti-nestin/anti-caveolin 1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), 1:20 and 1:50 dilutions, respectively.
Differentiation assay
After 4 or 6 DIV (number of experiments: n = 16 and n = 42, respectively), NE obtained from NSPCs of VZ/SVZ of nHTx rats were collected, plated on a poly-l-lysine coated coverslip and cultured in a 24-well culture dish containing basal medium (NeuroCult NS-A basal medium, StemCell Technologies), without growth factors and supplemented with 5% FBS. The cultures were maintained for 7 or 21 days and monitored by phase contrast microscopy. Finally, the cells were fixed with 4% paraformaldehyde and processed for double immunofluorescence analysis using anti-βIII-tubulin/anti-GFAP, anti-βIV-tubulin/anti-GFAP, anti-βIV-tubulin/anti-AQP4, anti-BrdU/anti-GFAP, anti-BrdU/anti MAP-2 and anti-BrdU/anti-βIV-tubulin. The dilution of the other antibodies was the same as described above.
Relative density of βIV-tubulin+ and GFAP+ cells after 21 days under differentiation assay condition
Six-DIV NEs were differentiated for 21 days and processed for double immunofluorescence for GFAP/βIV-tubulin and DAPI. Specimens were studied under an epifluorescence microscope using the multidimensional acquisition software AxioVision Rel version 4.6 (Zeiss, Aalen, Germany). Four independent experiments were performed. In each one of them, at least four photographs were obtained at a fixed magnification and used to quantitate the relative density of βIV-tubulin+ and GFAP+ cells as compared to the total number of DAPI+ nuclei. The analysis of the data was performed using the Prism GraphPad 6.0 (GraphPad Software, Inc., San Diego, CA).
CSF collection
PN7 nHTx and hyHTx rats were used for CSF collection. Pups were anesthetized with a combination of ketamine (40 mg/kg) and acepromazine (100 mg/kg). CSF was obtained from the cisterna magna (15 μl from each animal, nHTx) or from lateral ventricles (100 to 300 μl) of hyHTx rats. CSF samples from five to seven animals were pooled, centrifuged at 1000g for 30 s; the pellet was discarded and the protein fraction was used for the differentiation experiments.
Effect of CSF from nHTx and hyHTx on neurite outgrowth
NSPCs were collected from the VZ/SVZ nHTx and hyHTx rats and grown into NE after 6 DIV. Then, they were plated on a poly-l-lysine-coated coverslip and cultured in a 24-well culture dish containing basal medium, without growth factors and supplemented with 5% FBS (n = 5), or 5% CSF from nHTx rats (n = 3), or 5% CSF from hyHTx (n = 3). After 7 days in culture, the coverslips carrying the differentiating cells were fixed with 4% paraformaldehyde and processed for double immunofluorescence analysis using anti-βIII-tubulin/anti-GFAP. The specimens were inspected under an epifluorescence microscope using the multidimensional acquisition software AxioVision Rel version 4.6 (Zeiss, Aalen, Germany). In each of the 11 preparations, five pictures were taken at a fixed magnification.
With the support of the SigmaScan Pro.V.5.0, image analysis software (Systat Software Inc., San Jose, CA), data were obtained by measuring the length of neurites of the individual βIII-tubulin+ cells that did not establish contact with other cells. A two-way analysis of variance with a Tukey’s post-test was performed after the determination of the data normality using the D’Agostino-Pearson omnibus test. Data were expressed as average SE with p < 0.0001 for statistical significance. The statistical analysis and normality test of the data was made using Prism GraphPad 6.0 (GraphPad Software, Inc., San Diego, CA).
Transmission electron microscopy
Six-DIV NEs were fixed in 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4 for 2 h, at room temperature. After washing with 0.1 M phosphate buffer, NEs were fixed in 1% osmium tetroxide in 0.1 M phosphate buffer, pH 7.4, for 2 h at 4 °C. Embedding material was a mixture of Epon and Araldite (Mollenhauer 1964). Ultrathin sections were contrasted with uranyl acetate and lead citrate.
Scanning electron microscopy
The coverslips containing the differentiating cells were fixed in 1% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.4 for 2 h, at room temperature. After washing in the same buffer, they were fixed in 1% osmium tetroxide for 2 h at 4 °C. After dehydration in a graded series of ethanol, the cells were air dried for 24 h and ion coated with gold.
Results
According to size distribution, two populations of NE were obtained from NSPCs collected from non-hydrocephalic HTx rats (nHTx).
NSPCs, obtained from the VZ/SVZ of the lateral ventricles of PN1 nHTx rats, were cultured in a medium supplemented with mitogenic factors. After 6 days in culture (6 DIV), the size of most NEs ranged from 40 to 160 μm in diameter, with a small percentage being either smaller than 40 μm or larger than 160 μm. According to a size distribution histogram, two populations were observed: one ranging from 20 to 80 μm and the other from 81 to 240 μm in diameter (Fig. 1a–c). Fusion of 2 or 3 NEs was occasionally seen (Fig. 1b). Each individual NE has a core and a cortex, with different cell density, cell types and proliferation rate (Fig. 1b–h; cf. Guerra et al. 2015a). This zonation of NE allows one to distinguish between a large NE and a large structure resulting from fusion of medium size NE (Fig. 1(b)).

NSPCs collected from the VZ/SVZ of nHTx grow into two populations of NE. The proliferative cells populate the periphery of NE. Neurospheres were studied by phase contrast microscopy (a, b, d–d”), immunofluorescence (e, h, i) and hematoxylin-eosin staining (g, k). a 6DIV NEs. b The core of NE has a high optical density (asterisks). This allows distinguishing large NE from structures resulting from fusion of NE (arrow). c. Frequency distribution of neurosphere size at 6 DIV. d–e Full and broken arrows point to large and small neurospheres growing from 3 to 6 DIV. e NEs after 24 h BrdU labeling. f Quantification in smaller (20–50 μm) and larger (90–140 μm) neurospheres of labeled cells after 3 h and 24 BrdU pulses. D’Agostino-Pearson omnibus test. Data are expressed as average SE. *statistical significance with p < 0.0001. g Six-DIV neurosphere stained with hematoxylin and eosin. h Immunofluorescence for BrdU. After 12 h of exposure to BrdU, most labeled nuclei are at the periphery of the neurosphere. i Detailed view of previous figure showing the decreasing gradient of nucleus BrdU labeling from periphery to core of the NE (1–3). j Densitometric recording of the BrdU-labeled nuclei shown by the large arrow in Fig. 1(i). k After colchicine treatment, the arrested mitoses localized at the periphery of NE. Scale bar, a 200 μm, b 150 μm, d–d” 62 μm, e 66 μm, g–h 35 μm, i 6 μm, k 12.5 μm
Quantification of labeled cells after 3 and 24 h of exposure to BrdU showed that in large NE, 35(± 9 SE) % of nuclei were labeled after 3 h, while after 24 h, about 70(± 10 SE) % of nuclei were labeled. At variance, in small NE, 45(± 15 SE) and 50(± 7 SE) % of nuclei were labeled after 3 and 24 h of BrdU exposure, respectively. This suggests that in small NE, the length of the cell cycle was longer than in the cells of the large NE (Fig. 1d–f). This is supported by the experiment in which individual NEs, identified in phase contrast photographs, were followed twice a day from 3 to 6 DIV; it became clear that small NE grew at a slower rate (Fig. 1d–d”).
NEs grow outside inward
After 12 h of exposure to BrdU, labeled cells that had incorporated BrdU during the S phase of the cell cycle were confined to the periphery of large NE (Fig. 1g–j). There was a decreasing gradient of the label from the periphery to the core of NE, suggesting that dividing cells with serial dilutions of BrdU progressively “moved” to the NE core (Fig. 1(i)). Daughter cells with little or no BrdU label formed the core of NE (Fig. 1h).
To investigate the location within the NE of the cells undergoing mitosis, 6-DIV NEs were exposed to different concentrations of colchicine, for 4 h. Colchicine is known to arrest mitosis at metaphase. Cells with arrested mitosis were preferentially located at the periphery of the NE (Fig. 1k).
Four-DIV NEs are formed by proliferative nestin+ cells
After 4 DIV, NE displayed BrdU+/nestin+ cells distributed throughout (Fig. 2a–e). Only occasionally were there a few GFAP+ cells (Fig. 2a’, a”). βIII-tubulin+ cells were not seen (Fig. 2c’). The cells were joined together by N-cadherin+/βcatenin+ adherent junctions (Fig. 2(g, h)); connexin 43+ spots located at the cell periphery suggest the existence of gap junctions (Fig. 2(i)).

Four- and 6-DIV neurospheres from nHTx (PN1) are formed by different cell lineages. The last 24-h in vitro, NEs were exposed to 10 μM of BrdU. Double immunofluorescence of a serial section of anti-BrdU-anti-GFAP (a–a”, b–b”); anti-BrdU-anti-βIII-tub (c–c”, d–d”) and anti-BrdU-anti-nestin (e–e”, f–f”). In 4-DIV neurospheres, approximately 90% of cells are nestin+ and 7% are GFAP+; there were no βIII-tub+ cells. In 6-DIV neurospheres, approximately 65% of the cells are nestin+, 22% are GFAP+ and 20% are βIII-tub+. Cells forming the 4- and 6-DIV neurospheres are joined by adherent and gap junctions (g–l). m, n In block double immunofluorescence of 6-DIV NE using anti-caveolin 1 (green) and anti-nestin (red). a–f” 25 μm; g–l 15 μm; m 50 μm; m’ 2.5 μm; n 7 μm
In 6-DIV neurospheres, NSPCs start to commit into neuron, astrocytes and ependyma cell lineages
The periphery of 6-DIV NE was mainly formed by BrdU+/nestin+ cells (Fig. 2(b)). In block immunofluorescence of 6-DIV NE showed that, in NSC, nestin appeared as a network of filaments distributed throughout the cytoplasm and caveolin 1 (a functional marker of endocytosis) as vesicles distributed in the perikaryon (Fig. 2(m, n)). At the ultrastructural level, NSPCs were characterized by a nucleus with infolded nuclear cisterna, a prominent nucleolus and a cytoplasm poor in organelles and rich in polyribosomes (Fig. 3b, c).

After 6 DIV, NSCs/NPCs from nHTx commit into neuronal, glial and ependymal lineages. Six-DIV neurospheres were studied by phase contrast microscopy (a, a’) and transmission electron microscopy (b–i). a, a’ Phase contrast microscopy photograph of a 6-DIV neurosphere. a’ Detailed magnification of the area framed in (a) showing “cilia-like” structures. b–h Transmission electron microscopy of NE at 6 DIV. b Low magnification of the periphery of NE. c Cells with nuclei with abundant euchromatin and a cytoplasm poor in organelles are observed. d Cell with a well-developed RER, organized like Nissl bodies (neuroblast?). e Cell with numerous intermediate filaments forming bundles (astroblast?). f Two ciliated cells with 9 + 2 cilia (c), basal bodies (BB) and cilia roots (Cr). The cells are joined by adherent (AJ) and gap junctions (GJ). f’ Broken arrow and full arrows point to 9 + 2 and 9 + 0 cilia, respectively. g Higher magnification of the area framed in the previous figure, showing adherent (AJ) and gap junctions (GJ). h, i Multiciliated cells forming adherent (AJ) and gap junctions (GJ) with an undifferentiated cell (NSC/NPC?). Scale bar, a 26 μm; a’ 13 μm; b 5 μm; c 1.7 μm; d 600 nm; e 800 nm; f 800 nm; f’ 400 nm; g 300 nm; h 1.25 μm; i 350 nm
In 6-DIV NE, there were cells displaying neuron (βIII-tubulin) and glial (GFAP) markers; these cells were preferentially located in the core (Figs. 2b, d). The transmission electron microscope revealed the presence of cells undergoing differentiation: (i) cells with a nucleus with abundant euchromatin and packages of RER cisternae in the cytoplasm, resembling Nissl bodies (neuroblast?) (Fig. 3d); and (ii) cells with a nucleus with abundant euchromatin and prominent nucleolus, numerous mitochondria and bundles of intermediate filaments (astroblast?) (Fig. 3e).
The cells were joined together by N-cadherin+/βcatenin+ adherent junctions (Fig. 2(j, k)); connexin 43+ spots located at the cell periphery suggest the existence of gap junctions (Fig. 2l).
Phase contrast microscopy of living 6-DIV NE revealed clusters of cells projecting beating cilia toward the bathing culture medium (Fig. 3a, a’). This finding was confirmed by use of transmission electron microscopy. Clusters of multiciliated cells displaying ultrastructural characteristics of ependymal cells were seen at the periphery of the NE. The cells were rich in polyribosomes, mitochondria and elongated RER cisternae. The cilium complex was formed by the cilia root, the basal corpuscle and the cilium proper; cilia were 9 + 2; a cilium 9 + 0 was also found (Fig. 3f). The multiciliated cells were joined together by adherent junctions and gap junctions (Fig. 3f, g). The multiciliated cells were joined to neighbor undifferentiated cells (NSPCs) by adherent junctions and gap junctions (Fig. 3h, i).
NSPCs of 4- and 6-DIV NE differentiate into neurons, glia and ependyma
Four-DIV NE cultured for 7 days in a medium devoid of growth factors and containing bovine fetal serum was differentiated into (i) isolated βIII-tubulin+ cells displaying one or two slender long neurites and (ii) isolated GFAP+ cells with a few thick, short processes (Fig. 4a, b). When the differentiation assay was prolonged to 21 days, a few multiciliated βIV-tubulin+ were seen in addition to βIII-tubulin+ and GFAP+ cells. Two types of GFAP+ cells were distinguished. One displayed several radiating smooth processes and a small perikaryon (protoplasmatic astrocytes?); the other type had a pear-shaped cytoplasm extending flattened projections filled with bundles of GFAP+ filaments (fibrous astrocytes?) (Fig. 4c, c’). Each cell type formed separate cell clusters (Fig. 4c).

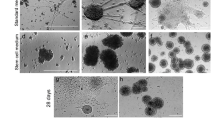
Neurospheres differentiate into neurons and glial cells. Neurospheres of 4 or 6 DIV, with 10 μM BrdU for the last 24 h, were further cultured in a medium devoid of EGF and containing 5% fetal bovine serum (FBS), or cerebrospinal fluid from hydrocephalic and non-hydrocephalic HTx rats. The cells were analyzed by immunofluorescence. (a–c, e–j) and scanning electron microscopy (d–d’). a–c. Four-DIV NEs cultured for 7 days (b) and 21 days (c) in presence of BFS. a Immunofluorescence of 4-DIV NE after 24 h. b Cells of NE differentiated into neurons (βIII-tubulin, green) and astrocytes (GFAP, red). c, c’ Two populations of GFAP+ cells appear after 21 days of differentiation: fibrous astrocytes? (1) and protoplasmatic astrocytes? (2). d, d’ Differentiation assay for 14 days. Scanning electron microscopy showing cells likely corresponding to neurons (d) and (d’) fibrous astrocytes? (1) and protoplasmatic astrocytes? (2). e Immunofluorescence of 6-DIV NE after 24 h BrdU. f, g Double immunofluorescence of 6-DIV neurospheres cultured for 6 (f) and 7 days (g). Differentiating cells are GFAP+ astrocytes (green) and βIII-tubulin neurons (red). h Quantitative analysis of neurite length of βIII-tubulin+ neurons from NE obtained from nHTx and hyHTx rats cultured in the presence of fetal bovine serum (FBS) or CSF from PN7 nHTx and hyHTx rats. Two-way analysis of variance with a Tukey’s post-test. Data are expressed as average SE. Statistical significance between pairs 1:2; 3:4; 5:6; 2:4; 2:6 p < 0.0001. F test 1:2; 2:4; 2:6 variances significantly different. i, j Differentiation of 6-DIV NE in the presence of 5% CSF from non-hydrocephalic (j) and hydrocephalic HTx (k) rats. Differentiating cells are GFAP+ astrocytes (green) and βIII-tubulin neurons (red). Scale bar, a 25 μm; b 20 μm; c 40 μm; c’ 18 μm; d, d’ 10 μm; e 25 μm; f 20 μm; g 37 μm; i–j 20 μm
6-DIV NEs were transferred to a medium devoid of growth factors and containing bovine fetal serum. After 6 h under this condition, the cells forming the NE started to differentiate into βIII-tubulin+ cells displaying one or two processes extending throughout the NE (neuroblasts/neurons) and GFAP+ cells with short radiating processes (glioblasts/astrocytes) (Fig. 4(f)).
After 7 days in the differentiating medium, NE had completely disassembled and some of their cells had differentiated into βIII-tubulin+ neurons, GFAP+ astrocytes (Fig. 4g) and βIV-tubulin+, multiciliated ependymal cells.
In vitro ependymogenesis
NEs were cultured for 6 days and exposed to BrdU during the last 24 h in culture. They were then transferred to a differentiation medium. After 7 days under this condition, a subpopulation of NE cells differentiated into βIV-tubulin+, flattened and ovoid cells. These cells appeared isolated or forming small clusters, surrounded by GFAP+ astrocytes (Fig. 5 b–e). Most βIV-tubulin+ cells displayed bundles of βIV-tubulin+ filaments radiating from a core located near the cell nucleus (Fig. 5a, d). A few βIV-tubulin+ cells displayed a tuft of cilia projecting perpendicularly to the free cell surface (Fig. 5b, c).

A subpopulation of cells of 6-DIV neurospheres differentiate into multiciliated ependymal cells. Six-DIV NEs further cultured for 7 days in the presence of 5% BFS and absence of growth factors were analyzed by double immunofluorescence, using antibodies for βIV-tubulin-N-cadherin (a, c, d, e), βIV-tubulin-GFAP (b). a White full and broken arrows point to N-cadherin, red broken arrow to isolated βIV-tubulin+ filaments and asterisk to bundles of βIV-tubulin+ filaments radiating from a region near the nucleus. b. Double immunofluorescence for βIV-tubulin and GFAP, showing a multiciliated βIV-tubulin+ cell (arrow) mixed with GFAP+ cells. c. Double immunofluorescence for βIV-tubulin and N-cadherin showing a multiciliated cell surrounded by cells forming adherent junctions (arrow). d, e. βIV-tubulin+ cells with different degrees of N-cadherin localization at the plasma membrane domain (broken and full arrows). f, f’ Double immunofluorescence for βIV-tubulin and BrdU. Two of the BrdU+ cells (1, 2) are also βIV-tubulin+. One cell (3) is βIV-tubulin+ but its nucleus is not BrdU-labeled. Scale bar, a 7 μm; b 12 μm; c 10 μm; d 12 μm; e 12 μm; f, f’ 18 μm
Double immunofluorescence for βIV-tubulin and BrdU revealed that βIV-tubulin+, multiciliated ependymal cells had nuclei with varying degree of labeling or no labeling (Fig. 5f, f’). βIV-tubulin+ cells displayed immunoreactive N-cadherin as patches located at the plasma membrane domain at sites where neighbor cells contacted each other (Fig. 5a, d). Some of the βIV-tubulin+ cells showed a continuous N-cadherin+ belt (Fig. 5(e)).
When the differentiation assay was prolonged to 21 days, the number of βIV-tubulin+- and GFAP+-differentiated cells increased significantly (Fig. 6a, d). Considering the total number of cells (DAPI+ nuclei) present in the culture after 21 days of differentiation, multiciliated cells corresponded to 9% and astrocytes to 72% (SE ± 1.7 and 3.5, respectively) (Fig. 6c). GFAP+, AQP4+ astrocytes were densely packed into clusters or a wall-like structure surrounding a cell-free space (Fig. 6a, d). Astrocytes and their processes formed a mesh resembling a cell matrix (Fig. 6b’, d).

A subpopulation of cells of 6-DIV neurospheres differentiate into multiciliated ependymal cells. Six-DIV NEs further cultured for 21 days in the presence of 5% FBS were analyzed by double immunofluorescence, using antibodies for βIV-tubulin-aquaporin (a, b, b’), βIV-tubulin-GFAP (d, f-f”) and βIV-tubulin-N-cadherin (e). a Low magnification of a field where βIV-tubulin+ cells (in green, arrows) grow on a matrix of AQP4+ cells (red). b, b’ Detailed magnification of cells from (a) multiciliated βIV-tubulin+ cells grown on a layer of AQP4+ cells. Cells 1, 2 and 3 are shown with the channel for βIV-tubulin (b) and for both channels (b’). Inset. A multiciliated cell is seen overlapping with AQP4+ cells (arrow). c Quantitative analysis of βIV-tubulin+ (9% ± 1.7 SE) and GFAP+ cells (72% ± 3.5 SE) versus the total number of cells. d Low magnification of a field showing the spatial relationship between GFAP+ astrocyte and single or clustered multiciliated βIV-tubulin+ cells. The arrow points to the cluster shown in figures f–f”. e The clustered multiciliated βIV-tubulin+ cells are joined together by N-cadherin-based adherent junctions. f, f’ A cluster of βIV-tubulin+ cells was photographed at different planes of focus (f1, f2) showing that the tips of cilia are in a different plane with respect to that of GFAP+ astrocytes (arrow). f” The same cell clusters shown in f and f’ with only the channel for βIV-tubulin. Scale bar, a 42 μm; b-b’ 12 μm, inset 2.5 μm; d 20 μm; e 10 μm; f–f” 10 μm
Multiciliated, βIV-tubulin+ cells grew intimately associated to the astrocyte matrix. Multiciliated cells were flattened, spherical/ovoid dish-like cells with a major diameter ranging from 30 to 50 μm. The flattened shape of these cells allowed us to look through them and we observed that they were lying on the astrocyte matrix. Individual βIV-tubulin+ cells differed in the number of cilia; some had none, others had a few, while most of them had a tuft of 20–30 cilia projecting from the central part of the cell surface. By going through different planes of focus, it became evident that the cilia projected upward. Cells forming groups were joined together by N-cadherin-based adherent junctions. Video microscopy showed that within each group of multiciliated cells, cilia beat synchronically (see enclosed video) indicating that gap junctions joined these cells.
The CSF of nHTx and hyHTx rats supported the differentiation of NE into neurons and astrocytes
NSPCs obtained from the ventricular/subventricular zone of PN1 nHTx rats were cultured for 6 days to obtain NE. NEs were further cultured for 7 days in medium devoid of growth factors and containing CSF collected either from nHTx or hyHTx HTx rats; they differentiated into βIII-tubulin+ neurons and GFAP+ astrocytes (Fig. 4i, j).
In another experiment, 6-DIV NEs obtained from nHTx and hyHTx rats were cultured for 7 days in the presence of (i) fetal bovine serum, (ii) CSF from nHTx, or (iii) CSF from hyHTx rats. Under these conditions, a quantitative analysis of the neurite length of βIII-tubulin+ neurons was performed. Under the three conditions, NE from hyHTx rats produced neurons with much longer neurites than those of NE from nHTx rats. Furthermore, fetal bovine serum promoted the differentiation of neurons with significantly longer neurites compared with those obtained in the presence of CSF from nHTx or hyHTx rats. Interestingly, no significant differences were seen in the length of neurites of neurons differentiated in the presence of normal versus hydrocephalic CSF (Fig. 4h).
Discussion
NEs formed by NSPCs have been used for transplantation in animal models of brain diseases and brain injuries (Pluchino et al. 2003). A strong body of evidence indicates that in fetal onset hydrocephalus, a junction pathology of NSCs leads to both hydrocephalus and abnormal neurogenesis (Guerra et al. 2015a; McAllister et al. 2017; Rodríguez et al. 2012; Sival et al. 2011). Furthermore, during the perinatal period, the same junction pathology affects the newly differentiated ependymal cells, which further contributes to abnormal CSF flow and hydrocephalus (Páez et al. 2007; Wagner et al. 2003). Since congenital hydrocephalus is, so far, an incurable disease, the grafting of NE formed by normal NSPCs into the CSF of animals with inherited hydrocephalus appears as a promising approach to diminish/repair the outcomes of NSCs pathology. However, there are relevant questions to be answered before proceeding with NE grafting. It seems mandatory to gain insight into the cell biology of NE as they develop in vitro in order to know when to graft and what is actually being transplanted. Similarly, it is important to learn how good a host the normal and the hydrocephalic CSF are for the transplanted NE.
Gaining insight into the cell biology of growing NE
Starting the second day in vitro, the proliferating NSPCs start forming a complex and dynamic structure. Throughout the days in vitro, the clustered NSPCs increase in size and complexity. Thus, after 4 DIV, the cell clusters are exclusively formed by BrdU+, nestin+ NSPCs, joined together by adherent junctions. After two further days in vitro, the cells forming the clusters are a heterogeneous cell population, with a distinct spatial organization. Can both types of cell clusters be regarded as NE? In very few papers out of the long list of publications on NE, a reference to the days in vitro has been made (Ge et al. 2012; Slovinska et al. 2015). There are several papers reporting on the heterogeneous NE-forming cells (Bez et al. 2003; Gil-Perotín et al. 2013; Guerra et al. 2015a; Suslov et al. 2002). Similarly, many papers describe NE as free-floating spherical clusters generated from a single neural stem cell and comprising cells at different stages of maturation in the neuronal and glial lineages (Bez et al. 2003; Gil-Perotín et al. 2013; Guerra et al. 2015a; Marshall et al. 2008; Reynolds and Weiss 1992). Does this mean that the young clusters formed by a homogeneous population of NSPCs are not regarded as NE? This point has been overlooked and is relevant when investigating the cell biology of NE as well as for the design of NE grafting protocols.
NE cells: from multipotency after 4 DIV to commitment after 6 DIV
Under in vitro conditions, to regard a cell as a neural stem cell requires the demonstration of proliferation and extensive renewal and of the capacity to generate a large number of differentiated progeny (Reynolds and Rietze 2005; Reynold and Weiss 1996). In the present investigation, a mixed population of NSPCs from the VZ and SVZ of PN1 nHTx rats was used to obtain NE. Since these primary NEs were not processed for serial passages to test the extensive renewal property that distinguishes neural stem cells from neural progenitor cells, the NE investigated originated from stem and progenitor cells. Although there are certain apparent features that would distinguish NE born from stem cells from those originated by progenitors, such as growth rate or capacity to generate multiciliated ependyma; the acronym NSPCs has been used throughout the text considering the scope and aims of the present investigation.
Virtually, all cells forming 4-DIV NEs were proliferative, uncommitted NSPCs, joined together by adherent junctions. Upon transfer to differentiative conditions, these NEs generated neurons and glial cells. Thus, 4-DIV NEs appear most appropriate for grafting experiments, since uncommitted grafted NSPCs will differentiate in response to clues from the host brain.
When 4-DIV NEs are further cultured for two additional days, they undergo substantial changes. About 40% of their NSPCs have committed into neuronal and glial lineages, with the latter located in the core of the NE. Thus, after 6 DIV, not before, NEs have become what is widely known as complex structures formed by a heterogeneous population of cells in different phases of the cell cycle and in different states of differentiation from NSPCs to neurons and glia (Bez et al. 2003; Marshall et al. 2008; Monni et al. 2011; Gil-Perotín et al. 2013). Consequently, when grafting 6-DIV NEs, it should be kept in mind that they are bags containing NSPC plus cells already committed into neuronal, glial and ependymal lineages.
Heterogeneity of NE
Heterogeneity in the size of NE derived from NSPCs plated individually and cultured under the same conditions has been reported by various authors (Bez et al. 2003; Ge et al. 2012; Guerra et al. 2015a). In the present investigation, the diameter of 6-DIV NEs ranged between 40 and 200 μm, with most of them grouped into two subpopulations of 40–80 μm and 100–160 μm in diameter, respectively. A similar size distribution has been found in 5-DIV NE (Ge et al. 2012). How to explain such a large variation in the size of NE generated by partner clones of NSPCs collected from the VZ/SVZ? Individual follow-up of NE through the 6-DIV and BrdU pulse labeling indicates that small NEs grow at a lower rate and the cycle of their cells is longer, as compared to the large NEs (present research). Are stem cells and progenitors of the neurogenic niche intrinsically diverse, or are they equipotent but respond selectively to different environmental cues? There is evidence supporting both possibilities. Human clonal NE, arranged according to their size, displayed different combinations of transcripts, which contributed to their distinct molecular phenotype. At least four types of clone-forming NSPCs were present within in vitro human stem cells (Suslov et al. 2002). Furthermore, throughout the CNS, there are different neural stem cells also, constituting a variety of clonogenic cells that possess distinct developmental and environmental histories, further contributing to NSPCs heterogeneity (Bonaguidi et al. 2012; Gil-Perotín et al. 2013; Temple 2001; Weiss et al. 1996).
Spatial organization and dynamic behavior of cells forming NE
NEs are not only heterogeneous in size (see above), they also display a diverse population of cells in different states of differentiation from NSPC cells to differentiating neurons and glia (Reynold and Weiss 1992; Bez et al. 2003; Marshall et al. 2008; Monni et al. 2011; Guerra et al. 2015a). After 6-DIV, NEs have a complex tridimensional structure, with a core and a cortex. DNA synthesis (BrdU incorporation) and mitosis (colchicine treatment) occur in nestin+ NSPC cells located in the cortex (present findings; Bez et al. 2003; Guerra et al. 2015a; Monni et al. 2011). The gradient of the BrdU label from cortex to the core suggests that daughter cells originating in the cortex move to the core. Indeed, using ex vivo labeling Wang et al. (2006) showed that cells of the cortex of NE migrate into the core. The core of NE is mainly occupied by cells committed into neuronal and glial lineages, as shown by cell markers and transmission electron microscopy (present findings; Guerra et al. (2015a). An exception to this core location of committed cells is the presence of multiciliated cells in the most superficial layer of the NE cortex (see below). Considering all the findings discussed above, a resemblance between the cortex and core of NE with the ventricular and subventricular zone of the embryo, respectively, may be envisaged.
NE cells are joined together by adherent junctions (Gil-Perotín et al. 2013; Guerra et al. 2015a; Lobo et al. 2003). Although gap junctions have not been previously reported in NE cells, the present ultrastructural and immunocytochemical findings and microinjections of Lucifer Yellow into cells of NE indicate that cells in the NE are coupled through gap junctions (Talaveron et al. 2015).
A subpopulation of NSPCs forming the NE differentiates into ependymal cells
NE cells projecting cilia-like processes have been described (Bez et al. 2003; Monni et al. 2011). In the present investigation, the presence at the periphery of NE of clusters of multiciliated ependymal cells endowed with 9 + 2 cilia has been shown by transmission electron microscopy. These clusters correlate well with those seen under phase contrast of living NE. NEs are motile dynamic structures (Gil-Perotín et al. 2013; Monni et al. 2011); multiciliated ependyma cells may be the propeller of the intrinsic, spontaneous locomotion of NE. The fact that only a subpopulation of NE displays clusters of multiciliated cells further supports the possibility that in the VZ/SVZ of PN1 rats, there is a variety of clonogenic NSPC cells that possess distinct developmental and environmental histories (see above).
The ability of NE to differentiate into neurons, astrocytes and oligodendrocytes when plated on an adhesive substrate, with the mitogens removed, has been widely investigated (see above). However, there is only one publication reporting on the differentiation of NE cells into ependymal cells (Mokrý and Karbanová 2006). NE from NSPCs derived from fetal mouse brain were processed for the differentiation assay for 2 and 3 weeks. The number of multiciliated ependymal cells increased from the 2nd to the 3rd week in culture (Mokrý and Karbanová 2006). In the present work, we confirmed and extended the observations of Mokrý and co-workers (2006). Six-DIV NEs, exposed during the last 24 h to BrdU, were processed for the differentiation assay. After 1 week, a few flat βIV-tubulin+ cells with and without cilia started to establish cell–cell contacts and to develop N-cadherin-mediated junctions; these cells may correspond to ependymoblasts. Some of these cells have a BrdU-labeled nucleus, strongly indicating that multiciliated cells originate from NE cells that had proliferated during the 6th DIV. The dilution of the BrdU label in these cells suggests that βIV-tubulin + cells continue to proliferate after NE disassembling. Similar cells undergoing mitosis have been reported by Mokry and co-workers (2006). This proliferative activity may explain the enormous increase of multiciliated ependymal cells after 3 weeks in culture. The differentiation process also progresses through the 2nd and 3rd week in culture, culminating with patches of ependyma whose cells are joined together by adherent junctions and whose cilia beat synchronously (implying the existence of gap junctions; video enclosed in the Electronic supplementary material).
Under culture differentiation conditions, multiciliated ependymal cells mature much later than neurons, astrocytes and oligodendrocytes (present investigation; Mokrý and Karbanová 2006). This time table of in vitro differentiation implies variable developmental commitments of parental clone-forming cells, mirroring the phenomena of neurogenesis and ependymogenesis occurring during brain development. In rodents, the bulk of neurogenesis occurs from E12 to E18, while ependymogenesis happens from E17 to PN7 (Rodríguez and Guerra 2017). The possibility that ependymal cells are born from subpopulations of NSC with their own time table of differentiation is supported by the evidence that postnatal ependymogenesis does occur in discrete brain regions. In wild-type and hydrocephalic a-SNAP, mutant (hyh) mice postnatal ependymogenesis has been shown to occur in two discrete regions of the brain, the roof of the third ventricle and the dorsal wall of Sylvius aqueduct. The new multiciliated ependymal cells are born from mono-ciliated, nestin+ cells located in the VZ of these two regions (Bátiz et al. 2011). These cells most likely correspond to a subpopulation of NSC that, after birth, remain in these two discrete regions of the VZ, proliferate and differentiate into multiciliated ependyma.
The capacity of NSPCs to form ependyma in vitro opens new possibilities for investigating cell and molecular mechanisms involved in ependymogenesis and for stimulating the use of NSPC grafting to replace ependymal cells lost to disruption of the ventricular zone in fetal onset hydrocephalus (Rodríguez and Guerra 2017).
Hydrocephalic CSF as a supportive medium to host NE
CSF is a heterogeneous and highly dynamic compartment that changes its molecular composition while moving through the various ventricular and subarachnoidal compartments. Signal molecules transported from blood to CSF or secreted into CSF by circumventricular organs and CSF-contacting neurons use the CSF to reach their targets in the brain (including the prenatal and postnatal neurogenic niche). This allows cross talk between brain regions located beyond the blood-brain barrier, thus keeping the brain milieu private (Bueno and García-Fernández 2016; Gato et al. 2014; Guerra et al. 2015b; Lehtinen et al. 2011; Rodríguez 1976). CSF proteomics has shown a wealth of over 200 proteins (Parada et al. 2006; Zappaterra et al. 2007). A long series of peptides and neurotransmitters are also present in CSF (Guerra et al. 2015b). Many of these compounds play a role in neuronal and glial proliferation and differentiation (Buddensiek et al. 2010; Zhu et al. 2015). During development, the multiple signal molecules of fetal CSF play a fundamental role in promoting survival, proliferation, and differentiation of NSPCs (Gato et al. 2005; Lehtinen et al. 2011).
How would NSPCs behave when grafted into CSF with its wealth of hundreds of signal molecules? Would the CSF be a friendly or unfriendly host? Would grafted NSPCs recognize the CSF as their former particular milieu, a sort of “coming back home?” Furthermore, there are important changes in the CSF proteome of hydrocephalic mutant rats as compared with that of non-hydrocephalic littermates (Vío et al. 2008; Ortloff et al. 2013). How would a hydrocephalic CSF host normal NSPCs? These are relevant questions apparently overlooked in previous investigations of NSPCs grafting into the CSF (Bai et al. 2003; Ohta et al. 2004; Pluchino et al. 2003; Wu et al. 2002). The present investigation has partially addressed these questions. The CSF of both nHTx and hyHTx rats disassembled 6-DIV NE and triggered the differentiation of their NSPCs into neurons, glia and ependyma suggesting that under both conditions, the CSF would be a friendly medium to host normal NE. However, conclusive evidence would result from the actual grafting of NE into the CSF of normal and hydrocephalic animals (experiment in progress). There is one paper reporting that epidermal neural crest stem cells cultured in the presence of CSF from normal adult rats differentiate into neurons and glia (Pandamooz et al. 2013). This finding and the fact that CSF is a circulatory system in close contact with different parts of the central nervous system, led these authors to conclude that CSF is a practical route to deliver stem cells.
The present findings contribute to the rationale of NE grafting into the brain, the CSF in particular. They should stimulate new attempts to use this approach for the treatment of certain brain pathologies such as congenital hydrocephalus.
Abbreviations
- AJ:
-
adherent junctions
- AQP4:
-
aquaporin 4
- BrdU:
-
bromodeoxyuridine
- CSF:
-
cerebrospinal fluid
- DIV:
-
days in vitro
- EGF:
-
epidermal growth factor
- FBS:
-
fetal bovine serum
- FGF:
-
fibroblast growth factor
- GFAP:
-
glial fibrillary acidic protein
- GJ:
-
gap junctions
- hyHTx:
-
hydrocephalic Texas rat
- NSPCs:
-
neural stem and progenitor cells
- NE:
-
neurospheres
- nHTX:
-
non-hydrocephalic Texas rat
- PN:
-
postnatal
- SVZ:
-
subventricular zone
- VZ:
-
ventricular zone
References
Ahn YS, Chang YS, Sung DK, Sung SI, Yoo HS, Lee HJ, Oh WI, Park WS (2013) Mesenchymal stem cells prevent hydrocephalus after severe intraventricular hemorrhage. Stroke. https://doi.org/10.1161/STROKEAHA.112.679092
Armstrong RJ, Svendsen N (2000) Neural stem cells: from cell biology to cell replacement. Cell Transplant 9:139–152
Bai H, Suzuki Y, Noda T, Wu S, Kataoka K, Kitada K, Ohta M, Chou H, Ide C (2003) Dissemination and proliferation of neural stem cells on injection into the fourth ventricle of the rat: a transplantation. J Neurosci Methods 124:181–187
Bátiz LF, Jimenez A, Guerra M, Rodríguez-Perez LM, Toledo CD, Vio K, Páez P, Pérez- Fígares JM, Rodrígue EM (2011) New ependymal cells are born postnatally in two discrete regions of the mouse brain and support ventricular enlargement in hydrocephalus. Acta Neuropathol 121:721–735. https://doi.org/10.1007/s00401-011-0799-x
Bez A, Corsini E, Curti D, Biggiogera M, Colombo A, Nicosia R, Pagano S, Parati A (2003) Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res 993:18–29. https://doi.org/10.1016/j.brainres.2003.08.061
Bonaguidi MA, Song J, Ming G, Song H (2012) A unifying hypothesis on mammalian neural stem cell properties in the adult hippocampus. Curr Opin Neurobiol 22:754–761. https://doi.org/10.1016/j.conb.2012.03.013
Buddensiek J, Dressel A, Kowalski M, Runge U, Schroeder H, Hermann A, Kirsch M, Storch A, Sabolek M (2010) Cerebrospinal fluid promotes survival and astroglial differentiation of adult human neural progenitor cells but inhibits proliferation and neuronal differentiation. BMC Neurosci 11:48. https://doi.org/10.1186/1471-2202-11-48
Bueno D, Garcia-Fernández J (2016) Evolutionary development of embryonic cerebrospinal fluid composition and regulation: an open research field with implications for brain development and function. Fluids Barriers CNS 13:5. https://doi.org/10.1186/s12987-016-0029-y
Del Bigio MR (2001) Pathophysiologic consequences of hydrocephalus. Neurosurg Clin N Am 12:639–649
Del Bigio MR (2010) Neuropathology and structural changes in hydrocephalus. Devel Dis Res Rev 16:16–22. https://doi.org/10.1002/ddrr.94
Fuchs E, Segre JA (2000) Stem cells: a new lease on life. Cell 100:143–155. https://doi.org/10.1016/S0092-8674(00)81691-8
Gato A, Alonso MI, Martin C, Carnicero E, Moro JA, De la Mano A, Fernandez JM, Lamus F, Desmond ME (2014) Embryonic cerebrospinal fluid in brain development: neural progenitor control. Croat Med J 55:299–305. https://doi.org/10.3325/cmj
Gato A, Moro JA, Alonso MI, Bueno D, De La Mano A, Martín C (2005) Embryonic cerebrospinal fluid regulates neuroepithelial survival, proliferation, and neurogenesis in chick embryos. Anat Rec A Discov Mol Cell Evol Biol 284:475–484. https://doi.org/10.1002/ar.a.20185
Ge D, Song K, Guan S, Dai M, Ma X, Liu T, Cui Z (2012) Effect of the neurosphere size on the viability and metabolism of neural stem/progenitor cells. African J Biotech 11:3976–3985. https://doi.org/10.5897/AJB11.3324
Gil-Perotín S, Duran-Moreno M, Cebrián-Silla A, Ramírez M, García-Belda P, García-Verdugo JM (2013) Adult neural stem cells from the subventricular zone: a review of the neurosphere assay. Anat Rec 296:1435–1452. https://doi.org/10.1002/ar.22746
Guerra M, Henzi R, Ortloff A, Lichtin N, Vío K, Jiménez A, Dominguez-Pinos MD, González C, Jara MC, Hinostroza F, Rodríguez S, Jara M, Ortega E, Guerra F, Sival DA, den Dunnen W, Pérez-Figares JM, McAllister JP, Johanson CE, Rodríguez EM (2015a) A cell junction pathology of neural stem cells is associated with ventricular zone disruption, hydrocephalus, and abnormal neurogenesis. J Neuropathol Exp Neurol 74:653–671. https://doi.org/10.1097/NEN.0000000000000203
Guerra MM, González C, Caprile T, Jara M, Vío K, Muño RI, Rodríguez S, Rodríguez EM (2015b) Understanding how the subcommissural organ and other periventricular secretory structures contribute via the cerebrospinal fluid to neurogenesis. Front Cell Neurosc 9:480. https://doi.org/10.3389/fncel.2015.00480
Jones HC, Bucknall RM (1987) Changes in cerebrospinal fluid pressure and outflow from the lateral ventricles during development of congenital hydrocephalus in the H-Tx rat. Exper Neurol 98:573–583. https://doi.org/10.1016/0014-4886(87)90266-4
Kim DE, Schellingerhout D, Ishii K, Shah K, Weissleder R (2004) Imaging of stem cell recruitment to ischemic infarcts in a murine model. Stroke 35:952–957. https://doi.org/10.1161/01.STR.0000120308.21946.5D
Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hil A, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, D’Ercole AJ, Wong ET, LaMantia AS, Walsh CA (2011) The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron 69:893–905. https://doi.org/10.1016/j.neuron.2011.01.023
Lobo M, Alonso F, Redondo C, López-Toledano M, Caso E, Herranz A, Paíno C, Reimers D, Bazán E (2003) Cellular characterization of epidermal growth factor-expanded free-floating neurospheres. J Histoch Cytoch 51:89–103. https://doi.org/10.1177/002215540305100111
Marshall GP, Ross HH, Suslov O, Zheng T, Steindler D, Laywell E (2008) Production of neurospheres from CNS tissue. Meth Mol Biol 438:135–150 doi:https://doi.org/10.1007/978-1-59745-133-812
McAllister JP (2012) Pathophysiology of congenital and neonatal hydrocephalus. Sem Fet Neonat Med 17:285–294. https://doi.org/10.1016/j.siny.2012.06.004
McAllister JP, Guerra MM, Castaneyra Ruiz L, Jimenez A, Dominguez-Pinos MD, Sival D, den Dunnen W, Morales D, Schmidt R, Rodríguez EM, Limbrick D (2017) Ventricular zone disruption in human neonates with intraventricular hemorrhage. J Neurop Exper Neurol 76:358–375. https://doi.org/10.1093/jnen/nlx017
Mokrý J, Karbanová J (2006) Foetal mouse neural stem cells give rise to ependymal cells in vitro. Fol Biol (Praha) 52:149–155
Mollenhauer HH (1964) Plastic embedding mixtures for use in electron microscopy. Stain Technol 39:111–114
Monni E, Congiu T, Massa D, Nat R, Diana A (2011) Human neurospheres: from stained sections to three-dimensional assembly. Transl Neurosci 2:43–48. https://doi.org/10.2478/s13380-011-0007-4
Neuhuber B, Barshinger AL, Paul C, Shumsky JS, Mitsui T, Fischer I (2008) Stem cell delivery by lumbar puncture as a therapeutic alternative to direct injection into injured spinal cord. J Neurosurg Spine 9:390–399. https://doi.org/10.3171/SPI.2008.9.10.390
Ohta M, Yoshihisa S, Toru N, Kazuya K, Hirotomi C, Namiko I, Masaaki K, Matsumoto N, Dezawa M, Suzuki S, Ide C (2004) Implantation of neural stem cells via cerebrospinal fluid into the injured root. Neuroreport 15:1249–1253. https://doi.org/10.1097/01.wnr.0000129998.72184.e1
Ortega E, Muñoz RI, Luza N, Guerra F, Guerra M, Vio K, Henzi R, Jaque RS, McAllister JP, Rodrigue EM (2016) The value of early and comprehensive diagnoses in a human fetus with hydrocephalus and progressive obliteration of the aqueduct of sylvius: case report. BBMC Neurology 16:45. https://doi.org/10.1186/s12883-016-0566-7
Ortloff AR Vío K, Guerra M, Jaramillo K, Kaehne T, Jones H, McAllister JP, Rodríguez EM (2013) Role of the subcommissural organ in the pathogenesis of congenital hydrocephalus in the HTx rat. Cell Tiss Res 352:707–725. https://doi.org/10.1007/s00441-013-1615-9
Páez P, Bátiz LF, Roales-Buján R, Rodríguez-Pérez LM, Rodríguez S, Jiménez AJ, Rodríguez EM, Pérez-Fígare JM (2007) Patterned neuropathologic events occurring in hyh congenital hydrocephalic mutant mice. J Neuropathol Exper Neurol 66:1082–1092. https://doi.org/10.1097/nen.0b013e31815c1952
Pandamooz S, Mohammad N, Alinezhad F, Zarghami A, Pourghasem M (2013) The influence of cerebrospinal fluid on epidermal neural crest stem cells may pave the path for cell-based therapy. Stem Cell Res Ther 4(4):84. https://doi.org/10.1186/scrt235
Parada C, Gato A, Aparicio M, Bueno D (2006) Proteome analysis of chick embryonic cerebrospinal fluid. Proteomics 6:312–320. https://doi.org/10.1002/pmic.200500085
Pluchino S, Quattrini A, Brambilla E, Gritti E, Salani G, Dina G, Galli R, Del Carro U, Amadio S, Bergami A, Furlan R, Comi G, Vescovi AL, Martino G (2003) Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature 422:688–694. https://doi.org/10.1038/nature01552
Reynolds BA, Weiss S (1992) Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science (New York, NY) 255:1707–1710. https://doi.org/10.1126/science.1553558
Reynolds BA, Weiss (1996) Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol 175(1):1–13. https://doi.org/10.1006/dbio.1996.0090
Reynolds BA, Rietze R (2005) Neural stem cells and neurospheres. Re-evaluating the relationship. Nature Meth 2:333–336. https://doi.org/10.1038/nmeth58
Rietze RL, Valcanis H, Brooker GF, Thomas T, Voss K, Bartlett PF (2001) Purification of a pluripotent neural stem cell from the adult mouse brain. Nature 412:736–739. https://doi.org/10.1038/35089085
Rodriguez EM (1976) The cerebrospinal fluid as a pathway in neuroendocrine integration. J Endocrinol 71:407–443. https://doi.org/10.1677/JOE.0.0710407
Rodriguez EM, Guerra MM (2017) Neural stem cells and foetal-onset hydrocephalus. Pediatr Neurosurg 52:446–461. https://doi.org/10.1159/000453074
Rodríguez EM, Guerra MM, Vío K, González C, Ortloff A, Bátiz LF, Rodríguez S, Jara MC, Munoz RI, Ortega E, Jaque J, Guerra F, Sival DA, den Dunnen WFA, Jimenez A, Dominguez-Pinos MD, Pérez-Figares JM, McAllister JP, Johanson C (2012) A cell junction pathology of neural stem cells leads to abnormal neurogenesis and hydrocephalus. Biol Res 45:231–241. https://doi.org/10.4067/S0716-97602012000300005
Satake K, Lou J, Lenke LG (2004) Migration of mesenchymal stem cells through cerebrospinal fluid into injured spinal cord tissue. Spine 29:1971–1979. https://doi.org/10.1097/01.brs.0000138273.02820.0a
Seaberg RM, van der Kooy D (2002) Adult rodent neurogenic regions: the ventricular subependyma contains neural stem cells, but the dentate gyrus contains restricted progenitors. Journal Neurosc 22:1784–93 doi:22/5/1784 [pii] ET - 2002/03/07
Sival DA, Guerra MM, den Dunnen WFA, Bátiz LF, Alvial G, Castañeyra-Perdomo A, Rodríguez EM (2011) Neuroependymal denudation is in progress in full-term human foetal spina bifida aperta. Brain Pathol 21:163–179. https://doi.org/10.1111/j.1750-3639.2010.00432.x
Slovinska L, Szekiova E, Blasko J, Devaux S, Salzet M, Cizkova D (2015) Comparison of dynamic behavior and maturation of neural multipotent cells derived from different spinal cord developmental stages: an in vitro study. Acta Neurobiol Exp 75:107–114
Suslov ON, Kukekov VG, Ignatova TN, Steindler DA (2002) Neural stem cell heterogeneity demonstrated by molecular phenotyping of clonal neurospheres. Proc Nat Acad Sci 99:14506–14511. https://doi.org/10.1073/pnas.212525299
Talaverón R, Fernández P, Escamilla R, Pastor AM, Matarredona ER, Sáez JC (2015) Neural progenitor cells isolated from the subventricular zone present hemichannel activity and form functional gap junctions with glial cells. Front Cell Neurosci 9:411–422. https://doi.org/10.3389/fncel.2015.00411
Temple S (2001) The development of neural stem cells. Nature 414:112–117
Vio K, Rodríguez S, Yulis CR, Oliver C, Rodriguez EM (2008) The subcommissural organ of the rat secretes Reissner’s fiber glycoproteins and CSF-soluble proteins reaching the internal and external CSF compartments. Cerebr Fluid Res 5:1–14. https://doi.org/10.1186/1743-8454-5-3
Wagner C, Batiz LF, Rodriguez S, Jiménez AJ, Páez P, Tomé M, Pérez-Fígares JM, Rodríguez EM (2003) Cellular mechanisms involved in the stenosis and obliteration of the cerebral aqueduct of hyh mutant mice developing congenital hydrocephalus. J Neuropathol Exp Neurol 62:1019–1040
Wang TY, Sen A, Behie LA, Kallos MS (2006) Dynamic behavior of cells within neurospheres in expanding populations of neural precursors. Brain Res 1107:82–96. https://doi.org/10.1016/j.brainres.2006.05.110
Weiss S, Reynolds BA, Vescovi A, Morshead C, Craig CG, van der Kooy D (1996) Is there a neural stem cell in the mammalian forebrain? Trends Neurosc 19:387–393. https://doi.org/10.1016/S0166-2236(96)10035-7
Williams MA, McAllister JP, Walker ML (2007) Priorities for hydrocephalus research: report from a National Institutes of Health-sponsored workshop. J Neurosurg 107:345–357
Wu S, Suzuki Y, Noda Y, Bai H, Kitada M, Kataoka K, Nishimura Y, Ide C (2002) Immunohistochemical and electron microscopic study of invasion and differentiation in spinal cord lesion of neural stem cells grafted through cerebrospinal fluid in rat. J Neurosc Res 69:940–945. https://doi.org/10.1002/jnr.10346
Zappaterra MD, Lisgo SN, Lindsay S, Gygi SP, Walsh CA, Ballif BA (2007) A comparative proteomic analysis of human and rat embryonic cerebrospinal fluid. J Prot Res 6:3537–3548. https://doi.org/10.1021/Pr07047w2
Zhu M, Feng Y, Dangelmajer F, Guerrero-Cazares H, Chaichana KL, Smith CL, Levchenko A, Lei T, Quinones-Hinojosa A (2015) Human cerebrospinal fluid regulates proliferation and migration of stem cells through insulin-like growth factor-1. Stem Cells Dev 24:160–171. https://doi.org/10.1089/scd.2014.0076
Acknowledgements
The authors wish to acknowledge the valuable technical support of Mrs Clara Jara, and Confocal and Electron Microscopy Core Facilities of Universidad Austral de Chile. Monoclonal antibodies against nestin and BrdU were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biological Sciences, Iowa City, Iowa.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by Fondecyt 1111018 to EMR; Hydrocephalus Association Established Investigator Award No. 51002705 to PM, EMR, CJ; Doctoral CONICYT Fellowship (Chile) to RH.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Henzi, R., Guerra, M., Vío, K. et al. Neurospheres from neural stem/neural progenitor cells (NSPCs) of non-hydrocephalic HTx rats produce neurons, astrocytes and multiciliated ependyma: the cerebrospinal fluid of normal and hydrocephalic rats supports such a differentiation. Cell Tissue Res 373, 421–438 (2018). https://doi.org/10.1007/s00441-018-2828-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-018-2828-8