
Abstract
The impact of reactive oxygen species and phosphoinositide 3-kinase (PI3K) in differentiating embryonic stem (ES) cells is largely unknown. Here, we show that the silencing of the PI3K catalytic subunit p110α and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 (NOX1) by short hairpin RNA or pharmacological inhibition of NOX and ras-related C3 botulinum toxin substrate 1 (Rac1) abolishes superoxide production by vascular endothelial growth factor (VEGF) in mouse ES cells and in ES-cell-derived fetal liver kinase-1+ (Flk-1+) vascular progenitor cells, whereas the mitochondrial complex I inhibitor rotenone does not have an effect. Silencing p110α or inhibiting Rac1 arrests vasculogenesis at initial stages in embryoid bodies, even under VEGF treatment, as indicated by platelet endothelial cell adhesion molecule-1 (PECAM-1)-positive areas and branching points. In the absence of p110α, tube-like structure formation on matrigel and cell migration of Flk-1+ cells in scratch migration assays are totally impaired. Silencing NOX1 causes a reduction in PECAM-1-positive areas, branching points, cell migration and tube length upon VEGF treatment, despite the expression of vascular differentiation markers. Interestingly, silencing p110α but not NOX1 inhibits the activation of Rac1, Ras homologue gene family member A (RhoA) and Akt leading to the abrogation of VEGF-induced lamellipodia structure formation. Thus, our data demonstrate that the PI3K p110α-Akt/Rac1 and NOX1 signalling pathways play a pivotal role in VEGF-induced vascular differentiation and cell migration. Rac1, RhoA and Akt phosphorylation occur downstream of PI3K and upstream of NOX1 underscoring a role of PI3K p110α in the regulation of cell polarity and migration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The vasculature is the first functional organ to develop during embryonic development for the supply of metabolic substrates (Ribatti 2006). The mechanism of vascular formation has been the subject of intense research for many years (McAuslan and Gole 1980; Tammela et al. 2008), and the mechanisms of vascular stimulation and inhibition remain a major topic of investigation (Muramatsu et al. 2013). Numerous publications have shown that vascular endothelial growth factor (VEGF) plays a pivotal role in the differentiation and proliferation of endothelial cells (vasculogenesis; Carmeliet et al. 1996), as well as blood vessel sprouting and branching (angiogenesis; Risau 1995). In addition, VEGF receptor 2 (Flk-1) is also crucial for the development of the vascular endothelial cell lineage and for the formation of early capillary networks (Millauer et al. 1993). VEGF has been shown to activate several downstream signalling pathways. For example, following VEGF treatment, Flk-1 (Kroll and Waltenberger 1997), phosphoinositide 3-kinase (PI3K; Guo et al. 1995), Akt (also called protein kinase B; Gerber et al. 1998) and extracellular signal-regulated kinase 1/2 (Erk1/2; Colavitti et al. 2002) are activated. As previously demonstrated, PI3K proteins are key regulatory proteins involved in a wide variety of cellular processes, including endothelial proliferation, migration, differentiation and survival (Gerber et al. 1998; Jiang et al. 2000). The PI3K family has been divided into three isoforms (class I, II and III) based on their structural features, lipid substrate specificity and mode of regulation (Fruman et al. 1998). The most important enzymes related to vascular differentiation are class I enzymes, which are further divided into class IA (PI3Kα, β, δ) and class IB (PI3Kγ) enzymes. The class I PI3K isoforms are activated by growth factor receptors and by G-protein-coupled receptors (Wymann et al. 2003). The four subtypes of class I are heterodimers composed of a catalytic subunit with a molecular weight of 110 kDa and a tightly associated regulatory subunit belonging to two distinct families (Wymann and Pirola 1998). The significance of PI3Kα in vascular development has been demonstrated in PI3Kα knockout mice in which impaired migration of endothelial cells and subsequent loss of angiogenic activity have been observed (Graupera et al. 2008).
Increased oxidative stress plays a central role in the pathogenesis of vascular disease (Madamanchi et al. 2005). However, moderate levels of reactive oxygen species (ROS), such as O2 − and hydrogen peroxide (H2O2), are produced in response to the activation of various cell surface receptors and serve as intracellular signals to mediate various biological responses such as cell migration, proliferation, gene expression and angiogenesis (Ushio-Fukai and Urao 2009). Furthermore, many studies have revealed that a major source of ROS is nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), which plays an important role in Flk-1 signalling and angiogenic responses in endothelial cells (Ushio-Fukai 2007; Ushio-Fukai and Urao 2009). Although the importance of ROS for the angiogenesis of ES cells has been reported (Sauer et al. 2005), the upstream signalling events stimulating ROS production by VEGF-PI3Ks are still not well characterized (Chatterjee et al. 2012).
The ability of ES cells to differentiate into endothelial cells and to form vascular structures provides a powerful in vitro model system for studying the molecular mechanisms of the earliest stages of blood vessel formation (Vittet et al. 1996). Vasculogenesis occurs when ES cells are grown within embryoid bodies, which are multicellular spheroidal tissues that have diameters between 0.5 and 1 mm and display gradients in oxygen and nutrients. Moreover, the expression of various endothelial cell markers in ES-cell-derived embryoid bodies has been found to follow the sequence of events occurring in vivo (Kabrun et al. 1997). Interestingly, vascular progenitor cells in 4-day-old embryoid bodies express Flk-1, one of the earliest markers for the endothelial cell lineage (Resch et al. 2012). Furthermore, isolated Flk-1-positive cells from ES cells are able to differentiate in vitro and also in vivo into vascular endothelial cells (Yamashita et al. 2000).
In the present study, we have targeted the catalytic PI3K isoform p110α in ES cells to avoid the side effects of pan-PI3K inhibitors on the immune system, which are largely dependent on p110δ and p110γ (Banham-Hall et al. 2012). In addition, we have unravelled the impact of PI3Kα-NOXs for vasculogenesis/angiogenesis, cell migration and angiogenesis in the ES cell in vitro model and demonstrated that the activation of ras-related C3 botulinum toxin substrate 1 (Rac1) is required to induce NOX1-mediated ROS production. Thus, our study provides an avenue for a better understanding of VEGF effects on blood vessel formation in order to treat pathological blood vessel conditions.
Materials and methods
ES cells and embryoid body culture
The pluripotent ES cell line CGR8 was derived from the inner cell mass of a 3.5-day male pre-implantation mouse embryo (Mus musculus, strain 129). The CGR8 cell line was obtained from the European Collection of Cell Cultures (ECACC, UK) and cultured on gelatine-coated cell culture dishes in Glasgow minimal essential medium (GMEM; Sigma-Aldrich, Taufkirchen, Germany) supplemented with 10 % heat-inactivated (56 °C, 30 min) fetal bovine serum (FBS; Sigma-Aldrich), 2 mM L-glutamine (Biochrom, Berlin, Germany), 45 μM β-mercaptoethanol (Sigma-Aldrich) and 103 U/ml leukaemia inhibitory factor (LIF; Chemicon, Schwalbach, Germany) in a humidified environment containing 5 % CO2 at 37 °C. To generate embryoid bodies, a total of 1 × 107 cells were seeded in spinner flasks (Integra Biosciences, Fernwald, Germany) containing Iscove’s medium supplemented with 16 % FBS (Sigma-Aldrich), 100 μM β-mercaptoethanol (Sigma-Aldrich), 2 mM L-glutamine and 2 mM non-essential amino acids (Biochrom).
Down-regulation of p110α and NOX mediated by short hairpin RNA
pLKO.1-puro (Sigma-Aldrich) derivative plasmids encoding a short hairpin RNA (shRNA) sequence targeting the murine PI3K p110α catalytic subunit or murine NOX1 and NOX2 or a non-targeting control shRNA were separately introduced into undifferentiated CGR8 ES cells by lentiviral particles as previously described (Bekhite et al. 2011). Ecotropic lentiviral particles were generated by using human embryonic kidney (HEK293T)-based packaging cells (Phoenix-Ampho, Invitrogen, Karlsruhe, Germany). Lentiviral particles were added to CGR8 ES cells. A total of three (24 + 36 + 48 h) infection rounds were carried out. On the following day, the cells were selected with 2 μg/ml puromycin (Sigma-Aldrich) for 14 days. Down-regulation of the PI3K p110 catalytic subunit p110α, NOX1 and NOX2 was analysed by reverse transcription plus the polymerase chain reaction (RT-PCR) and western blot (Bartsch et al. 2011; Bekhite et al. 2011).
Magnetic cell separation and flow cytometry analysis
Four-day-old embryoid bodies were dissociated by incubation with collagenase type II (2 mg/ml, PAA, Coelbe, Germany) at 37 °C for 5–10 min. Magnetic cell sorting (MACS) on MS columns (Miltenyi Biotec, Bergisch-Gladbach, Germany) was applied for cell separation. The separation procedure was carried out by using a phycoerythrin (PE)-conjugated rat anti-mouse Flk-1 antibody (dilution 1:10; BD, Franklin Lakes, USA). Anti-PE MicroBeads (Miltenyi Biotec) were used for labelling. Purity analysis was carried out in a FACSCalibur flow cytometer (Becton Dickinson, Heidelberg, Germany).
Treatment with inhibitors
Embryoid bodies derived from ES cells were incubated in the presence or absence of VEGF-165 (500 pM; Sigma-Aldrich) with the pan-PI3K inhibitor wortmannin (1 μM; Cell Signaling Technology, Frankfurt, Germany) or with 3-(4-morpholinothieno[3,2-d]pyrimidin-2-yl)phenol (compound 15e, 0.5 μM; Alexis Biochemicals, Lörrach, Germany), which is an inhibitor of the catalytic p110α subunit of PI3K (Hayakawa et al. 2006). We also used inhibitors for NOX, i.e. diphenyleneiodonium (DPI, 10 μM; Sigma-Aldrich), 3-benzyl-7-(benzoxazolyl) thio-1,2,3-triazolo(4,5-d)pyrimidine (VAS2870, 50 μM; Vasopharm, Würzburg, Germany), the novel NOX1 inhibitor 2-acetylphenothiazine (2-APT, 0.5 μM), the respiratory chain complex I inhibitor rotenone (2.5 μM) and Rac1 inhibitor (50 μM), all purchased from Calbiochem (Bad Soden, Germany).
Immunofluorescence staining and quantitative analysis
Blood-vessel-like structures in embryoid bodies were identified by immunolabelling platelet endothelial cell adhesion molecule-1 (PECAM-1; CD31) as previously described (Bekhite et al. 2011). Primary antibodies used were: rat monoclonal anti-PECAM-1 (Millipore, Darmstadt, Germany), rabbit polyclonal anti-PECAM-1 (Santa Cruz, Temecula, Calif., USA), rat monoclonal anti-VE-cadherin (BD Biosciences, Heidelberg, Germany) and mouse monoclonal anti-Flk-1 (Abcam, Cambridge, UK). Fluorescence was recorded via a confocal laser scanning microscope (LSM 510, Carl Zeiss, Germany). Quantitative immunofluorescence (IF) was performed as previously described (Sauer et al. 2005). By use of the image analysis facilities of the confocal setup, the antigen-positive vascular areas were identified within the three-dimensional projection of optical sections in embryoid bodies. In order to investigate the changes in actin structures during the migration process, we carried out cytoskeleton staining by phalloidin-Alexa Fluor 488 (Invitrogen, Karlsruhe, Germany). Flk-1+ cells were grown on cover glasses in 24-wells up to semi-confluence and stimulated with VEGF (500 pM) for 30 min. The cells were then fixed with 4 % formaldehyde freshly prepared from paraformaldehyde for 1 h on ice, stained with phalloidin-Alexa Fluor 488 (Invitrogen) for 30 min at room temperature in darkness and analysed by means of LSM 510 (Carl Zeiss).
Measurement of ROS
Intracellular ROS levels were measured by using the fluorescent dye 2’,7’-dichlorodihydrofluorescein diacetate (H2DCF-DA; Invitrogen), which is a nonpolar compound that is converted into a nonfluorescent polar derivative (H2DCF). H2DCF is membrane impermeable and is rapidly oxidized to the highly fluorescent 2’,7’-dichlorofluorescein (DCF) in the presence of intracellular ROS. For the experiments, embryoid bodies were incubated in serum-free medium and 20 μM H2DCF-DA was added. After 20 min, intracellular DCF fluorescence was evaluated via LSM 510 (Carl Zeiss) by using the 488-nm line of an argon ion laser and monitoring fluorescence at > 510 nm.
Measurement of intracellular O2 −
Intracellular generation of O2 − was estimated by using the O2 − indicator dihydroethidium (DHE; Invitrogen), which can be used specifically for measuring intracellular O2 −. Cytosolic DHE exhibits blue fluorescence. However, once this probe is oxidized to ethidium, it intercalates into chromosomal DNA, staining the nucleus with brightly red fluorescence.
Washed ES cells were pre-incubated with 10 μM DHE for 20 min at 37 °C in darkness and then rinsed with E1-buffer (135 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, 1.8 mM CaCl2, pH 7.4) to remove excess dye. For fluorescence excitation, the 488-nm band of the argon ion laser of LSM 510 (Zeiss) was used. Emission was recorded at >650 nm.
RT-PCR and real-time PCR
Total RNA was isolated from embryoid bodies by using the RNeasy Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s recommended procedures, followed by genomic DNA digestion by DNase I (Invitrogen). cDNA was synthesized from 2 μg total RNA by Superscript II reverse transcriptase (Invitrogen) according to manufacturer’s instructions by using random primer. The RT product was diluted 1:10 and PCR was performed by using the primer sets as shown in the supplementary information (Supplementary Material Table S1) and 40 cycles at an annealing temperature of 59 °C. Gel imaging and visualization of bands were performed by GenSnap from SynGene (VWR, Darmstadt, Germany) on a 1 % agarose gel. Real-time PCR was performed by using SYBR-Green (Qiagen, Hilden, Germany) Mastercycler ep realplex (Eppendorf, Hamburg Germany). Relative expression values were obtained by normalizing the CT values of the tested genes to CT values of the internal standard genes by using the ΔΔCT-method.
Western blot assay
ES cells were washed in ice-cold phosphate-buffered saline and subsequently lysed in lysis buffer as described previously (Bekhite et al. 2011). Aliquots of 40 μg protein were separated in 8 or 12 % SDS gels and transferred onto nitrocellulose membrane. The membranes were blocked with 5 % dried fat-free milk in TRIS-buffered saline containing 0.1 % Tween 20 (TBST). Incubation with primary antibodies was performed at 4 °C overnight. Immunoreactive bands were visualised by using horseradish-peroxidase-conjugated secondary antibody and an enhanced chemiluminescence detection kit (Amersham, Freiburg, Germany). Chemiluminescence signals were quantified by using a charge-coupled device (CCD) camera-based chemiluminescence detection system (LAS 3000, Fujifilm, Japan).
The following primary antibodies were used for western blot analyses: rabbit polyclonal anti-D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and anti-phospho-Ras homologue gene family member A (anti-phospho-RhoA; Ser188; Abcam,Cambridge, UK); goat polyclonal anti-mouse NOX1, NOX2, NOX4, p110α, polyclonal rabbit anti-mouse p110β and polyclonal mouse anti-mouse p110δ (Santa Cruz Biotechnology, Heidelberg, Germany); rabbit polyclonal anti-mouse RhoA, Rac1/2/3, phospho-Rac1 (Ser71), Akt and phospho-Akt (Ser473; Cell Signaling Technology, Frankfurt, Germany).
Angiogenesis assay
Matrigel (BD Biosciences, Heidelberg, Germany) was added to 96-well plates on ice and allowed to polymerize for 1 h at 37 °C. The isolated Flk-1+ cells from 4-day-old embryoid bodies incubated in CGR8 medium for 1 day were harvested after trypsin treatment, resuspended in CGR8 medium in the presence or absence of VEGF and plated onto the Matrigel. Matrigel cultures were incubated at 37 °C and photographed after 16 h.
Scratch migration assay
To assess whether p110α and NOX1 played a role in cell migration of Flk-1+ cells, a scratch cell migration assay was performed in 12-well tissue culture plates coated with 0.1 % gelatine for a minimum of 2 h at 37 °C. Upon reaching 80 % confluence, the Flk-1+ cells were scratched by using a pipette tip and cell debris was removed. Cells were cultured for 6 h in order to allow cell migration in the presence or absence of VEGF. Each treatment was performed in triplicate. The mean distance of migrating cells in each well was measured from five randomly chosen fields under light microscopy at 0 h and 6 h (Bhattacharya et al. 2009).
Statistical analysis
Data were expressed as mean values ± SD of three experiments unless otherwise indicated. In each experiment, at least 15 embryoid bodies were analysed. GraphPad InStat-3 software (GraphPad Software, San Diego, Calif., USA) was employed for one-way analysis of variance or Student’s t-test of unpaired data. A value of P < 0.05 was considered significant (*). Data are expressed as the fold or percentage of expression relative to control values.
Results
Flk-1+ cells derived from differentiating ES cells express NOX isoforms and PI3K class IA p110 catalytic subunits
To examine whether members of the NOX family are expressed in vascular progenitor cells, Flk-1+ cells, which represent 9.8 ± 2.7 % of total cells, were isolated from 4-day-old embryoid bodies by MACS. Flow cytometry (FCM) analysis indicated a purity of approximately 96 % Flk-1+ cells (see Supplemental Fig. S1a). Moreover, the relative mRNA expression level of NOX isoforms was examined by using RT-PCR (Fig. 1a, b) and protein synthesis was investigated by western analysis (Fig. 1c-e). Results from the mRNA and protein analysis showed that Flk-1+ cells mainly expressed NOX1 (Fig. 1a, a‘) and synthesized NOX1 protein (Fig. 1c, c‘) and NOX4 mRNA (Fig. 1b, b‘) and NOX4 protein (Fig. 1e, e‘), whereas NOX2 mRNA (Fig. 1a, a‘) and protein (Fig. 1d, d‘) were scarcely present in Flk-1+ cells. In contrast, NOX3 mRNA was absent in Flk-1+ cells (Fig. 1b, b‘). Furthermore, we analysed the mRNA expression of the PI3K class IA p110 catalytic subunits in the Flk-1+ cells. The results showed that PI3K class IA p110 catalytic subunits p110α, β and δ were expressed in Flk-1+ cells in parallel to the first steps of vascular differentiation of ES cells (see Supplemental Fig. S1b, b‘).

Expression of NADPH oxidase (NOX) isoforms in embryoid bodies. Cells of 4-day-old embryoid bodies were sorted by magnetic cell sorting (MACS) and analysed for mRNA expression levels and protein synthesis by using reverse transcription - polymerase chain reaction (RT-PCR) and western blot. mRNA of NOX1 and NOX2 (a, a‘), NOX3 and NOX4 (b, b‘) of 4-day-old sorted Flk-1+, Flk-1− and unsorted cells was amplified by RT-PCR and processed by DNA agarose gel electrophoresis. Polymerase II was used as an internal standard. Specific mRNA level was calculated in relation to polymerase II. D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal standard for western blot analysis of NOX1 (c, c‘), NOX2 (d, d‘) and NOX4 (e, e‘). Graphs under the blots show mean values (± SD) of three independent experiments. *P < 0.05, statistically significant as indicated (n.s not significant)
VEGF induces ROS production mediated by NOXs in vascular progenitor cells
According to the working hypothesis of the present study, VEGF might stimulate vasculogenesis/angiogenesis in ES-cell-derived embryoid bodies by activating ROS production through PI3K. The use of H2DCF as a fluorescent ROS indicator demonstrated that VEGF (500 pM) significantly increased ROS generation in 4-day-old embryoid bodies (Fig. 2a-a‘‘‘‘‘, b). However, no increase in ROS was observed when embryoid bodies were incubated with the NOX inhibitors DPI (10 μM; Fig. 2a‘‘, b) and VAS2870 (50 μM; Fig. 2a‘‘‘) or apocynin (100 μM; Fig. 2a‘‘‘‘‘, b), which inhibits NOX1 and NOX2 activation. In contrast, co-incubation of the embryoid bodies with rotenone (10 μM; Fig. 2a‘‘‘‘, b), a complex I mitochondrial inhibitor, could not abolish the effect of VEGF on ROS generation, thus suggesting a distinct source of ROS from NOX. Furthermore, inhibition of ROS generation by apocynin suggests a contribution of NOX1 and NOX2 to the ROS production of differentiating ES cells (Fig. 2a‘‘‘‘‘, b).

Vascular endothelial growth factor (VEGF)-induced reactive oxygen species (ROS) production in embryoid bodies and O2 − detection in Flk-1+ cells. Five-day-old embryoid bodies were stimulated by VEGF (500 pM) for 30 min. a–a‘‘‘‘‘ DCF (dichlorodihydrofluorescein) fluorescence of embryoid bodies treated with NOX inhibitors diphenyleneiodonium (DPI; 10 μM; a‘‘), VAS2780 (50 μM; a‘‘‘) or apocynin (100 μM; a‘‘‘‘‘) or the respiratory chain complex I inhibitor rotenone (10 μM; a‘‘‘‘) on VEGF-induced ROS generation as indicated. Bars 100 μm. b Mean values (± SD) of ROS generation in three independent experiments. NOX inhibitors DPI, VAS2780 and apocynin completely inhibited ROS generation in embryoid bodies upon VEGF treatment, whereas rotenone did not affect ROS levels. c–c‘‘‘‘ DCF fluorescence of embryoid bodies depleted for p110α (class IA phosphoinositide 3-kinase catalytic subunits p110α), NOX1 and NOX2 or wild-type (pLKO.1) treated with VEGF. Bars 100 μm. d VEGF failed to increase the ROS generation in p110α and NOX1 shRNA gene-inactivated embryoid bodies in comparison with pLKO.1 embryoid bodies. e–e‘‘‘‘‘ O2 − generation of sorted Flk-1+ cells isolated from 4-day-old embryoid bodies in response to VEGF stimulation as analysed by determination of dihydroethidium (DHE) fluorescence. Wild-type cells (pLKO.1) without (e) and with (e‘) VEGF stimulation. Cells depleted for p110α (e‘‘), NOX1 (e‘‘‘), NOX2 (e‘‘‘‘) or wild-type cells (pLKO.1) pre-treated for 30 min with ras-related C3 botulinum toxin substrate 1 (Rac1) inhibitor (50 μM; e‘‘‘‘‘) were used. e–e‘‘‘‘‘ Representative immunofluorescence images showing DHE fluorescence in Flk-1+ ES cells. Bars 100 μm. f Percentage values (± SD) of three independent experiments. *P < 0.05, statistically significant as indicated (sh short hairpin, n.s not significant)
To investigate the involvement of class IA PI3Kα (p110α), NOX1 and NOX2 in VEGF-stimulated ROS generation, we treated 4-day-old shRNA p110α, shRNA NOX1 and shRNA NOX2 embryoid bodies with VEGF (Fig. 2c-c‘‘‘‘, d). In shRNA p110α (Fig. 2c‘‘, d) and shRNA NOX1 (Fig. 2c‘‘‘, d) embryoid bodies, VEGF treatment failed to increase ROS, whereas ROS generation was not affected in shRNA NOX2 embryoid bodies (Fig. 2c‘‘‘‘, d). Thus, our data indicate that p110α and NOX1 but not NOX2 are involved in ROS production during VEGF treatment.
To confirm experiments with the non-specific ROS indicator H2DCF, additional experiments were performed to measure intracellular O2 −. VEGF (500 pM) and DHE (10 μM) were added to medium and, 20 min after stimulation, the cells were examined by microfluorimetry (Fig. 2e-e‘‘‘‘‘). VEGF treatment strongly elevated O2 − generation in plated Flk-1+ cells isolated from 4-day-old embryoid bodies, whereas O2 − remained at the control level when the Flk-1+ cells were isolated from shRNA p110α or shRNA NOX1 embryoid bodies (Fig. 2f). Moreover, to determine whether the ROS production was dependent on Rac1, a Rac1 inhibitor (50 μM) was used. The results demonstrated that VEGF did not increase O2 − production upon pharmacological inhibition of Rac1 (Fig. 2e‘‘‘‘‘, f). Furthermore, we investigated whether NOX2 deficiency in Flk-1+ cells had an impact on O2 − production upon VEGF treatment. For this purpose, we quantified DHE fluorescence in both untreated and VEGF-treated Flk-1+ cells. Knockdown of NOX2 in Flk-1+ cells showed no significant effects on O2 − production upon treatment with VEGF (Fig. 2e‘‘‘‘, f). To exclude cell-line-specific effects, O2 − generation was assessed in Flk-1+ cells isolated from 4-day-old embryoid bodies derived from the ES cell line CCE following inhibition of p110α by Compound 15e (0.5 μM), NOX1 by 2-APT (0.5 μM) and Rac1 activity by Rac-1 inhibitor (50 μM) in the presence or absence of VEGF (Supplemental Fig. S2a-a‘‘‘‘, b). These experiments corroborated our data on CGR8 cells showing that VEGF did not increase O2 − production upon inhibition of p110α or Rac1 and indicating an involvement of class IA PI3Kα-Rac1 and NOX1 in VEGF-mediated O2 − production.
PI3K catalytic subunit p110α is essential for vasculogenesis/angiogenesis
Three-dimensional embryoid bodies derived from ES cells (Supplemental Fig. S3a) can differentiate into vessel-like structures formed from cells that are positive for endothelial-specific antigens such as Flk-1 (Supplemental Fig. S3a‘, b), PECAM-1 (Supplemental Fig. S3a‘‘, b‘, c, d) and VE-cadherin (Supplemental Fig. S3c‘, d‘).
To investigate whether class IA PI3Kα, NOX1 and Rac1 are essential for VEGF signalling pathways leading to the vascular differentiation of the ES cells, p110α and NOX1 shRNA embryoid bodies were treated from day 4 to day 10 of cell culture with VEGF (500 pM). This treatment resulted in a significant increase in vascular differentiation in the non-targeting shRNA control cell line (pLKO.1; Fig. 3a, b) as evaluated by quantification of the areas positive for endothelial marker PECAM-1 (Fig. 3c), branching points (Fig. 3d) and FCM analysis of Flk-1+ cells in embryoid bodies (Supplemental Fig. S4a-a‘‘‘‘‘, b). In contrast, stimulation of vascular differentiation upon treatment with VEGF was nearly absent in embryoid bodies co-incubated with the general PI3K inhibitor wortmannin (1 μM; Fig. 3a‘, b‘) and embryoid bodies lacking p110α catalytic subunits (Fig. 3a‘‘, b‘‘). Moreover, incubation of embryoid bodies with Rac1 inhibitor significantly reduced the size of PECAM-1-positive vascular areas (Fig. 3c), nearly abolished the branching points (Fig. 3d) and greatly reduced the number of Flk-1+ cells (Supplemental Fig. S4a, b). Silencing of the p110α catalytic subunit of PI3K resulted in a significant decrease of the PECAM-1-positive vascular area (Fig. 3c) and branching points (Fig. 3d). In the absence of NOX1 (Fig. 3a‘‘‘, b‘‘‘) or pharmacological inhibition of Rac1 (Fig. 3a‘‘‘‘, b‘‘‘‘), a significant reduction of the VEGF-induced PECAM-1-positive vascular area (Fig. 3c) and branching points (Fig. 3d) in the embryoid bodies compared with untreated controls was observed, although a significant enhancement of vascular differentiation still occurred upon VEGF treatment (Fig. 3c, d). These results were confirmed by using embryoid bodies derived from the CCE cell line cultured from day 6 to day 10 with wortmannin (1 μM), the p110α inhibitor compound 15e (0.5 μM) or the NOX1 inhibitor 2-APT (0.5 μM) and Rac1 inhibitor (50 μM) in the absence (Supplemental Fig. S5a-a‘‘‘‘) or presence (Supplemental Fig. S5b-b‘‘‘‘) of VEGF (500 pM). The PECAM-1-positive vascular area was investigated and the analysis showed identical results to those obtained with the CGR8 ES cell line (Supplemental Fig. S5c). However, the much less well developed vascular networks in embryoid bodies derived from the CCE cell line made it difficult to detect branching points (Supplemental Fig. S5b-b‘‘‘‘). Taken together, the results of these experiments demonstrate that the PI3K catalytic subunit p110α, plus Rac1 and NOX1 regulate the vascular differentiation of embryoid bodies.

Class IA PI3K catalytic subunit p110α (p110α) and Rac1 are essential for VEGF-induced vascular differentiation of ES cells. p110α or NOX1 knockdown and wild-type (pLKO.1) ES cells were allowed to differentiate for 10 days. Inhibitors were applied from day 4 to day 10 of cell culture. Cells were stimulated with VEGF (500 pM) or remained unstimulated. Cells were treated with wortmannin (1 μM) or Rac1 inhibitor (50 μM) as indicated. a–a‘‘‘‘, b–b‘‘‘‘ Representative immunofluorescence images for at least three experiments with consistent results showing vascular differentiation in embryoid bodies. Bars 100 μm. c, d Vascular differentiation as indicated by PECAM-1-positive areas (c) and branching points (d). *P < 0.05, statistically significant as indicated (sh short hairpin, n.s not significant)
Level of p110α and NOX1 expression in Flk-1+ cells isolated from shRNA p110α and shRNA NOX1 embryoid bodies
To guarantee a specific and significant inhibition of the NOX1 isoform and the class IA PI3K catalytic subunit p110α, we screened the presence of NOX1 and p110α mRNA by RT-PCR (Fig. 4a, a‘) and of NOX1 and p110α protein synthesis by western blot (Fig. 4b, b‘) in Flk-1+ cells isolated from knockdown embryoid bodies. As shown in Fig. 4, PI3K catalytic subunit p110α (Fig. 4a‘) mRNA and p110α protein (Fig. 4b‘) were significantly decreased in Flk-1+ cells isolated from shRNA p110α embryoid bodies within the time window of vascular differentiation at day 4, whereas no change in the level of NOX1 mRNA expression (Fig. 4a, a‘) and NOX1 protein synthesis (Fig. 4b, b‘) was noted. Conversely, NOX1 mRNA expression (Fig. 4a, a‘) and protein synthesis (Fig. 4b, b‘) were significantly decreased in Flk-1+ cells isolated from 4-day-old shRNA NOX1 embryoid bodies as compared with pLKO.1 cells, whereas no change was detected in the level of p110α mRNA expression and p110α protein. To confirm these findings, real-time PCR for the gene expression of p110α and NOX1 in Flk-1+ cells was performed. The results showed that the mRNA expression level of p110α was significantly decreased to 0.33 ± 0.02 and of NOX1 to 0.34 ± 0.19 in knockdown cells compared with an mRNA expression level of 1.00 ± 0.15 (p110α) and 1.00 ± 0.11 (NOX1) in Flk-1+ wild-type cells (pLKO.1). These data indicated that the silencing of p110α and NOX1 led to more than a 65 % reduction of p110α and NOX1 mRNA expression. In addition, NOX1 expression was not affected by the down-regulation of p110α and the expression of p110α was not influenced by the down-regulation of NOX1.

Level of p110α and NOX1 in Flk-1+ cells isolated from knockdown embryoid bodies. Flk-1-positive cells were sorted by MACS from 4-day-old wild-type (pLKO.1), shRNA p110α or shRNA NOX1 embryoid bodies. a, a‘ mRNA from Flk-1-positive cells was prepared and the expression level of p110α, NOX1 or polymerase II was detected using RT-PCR. Amplification was processed by DNA agarose gel electrophoresis. Representative gels for three repeated experiments are demonstrated. The graph under the blot shows the expression of p110α in relation to that of polymerase II. b, b‘ Western blot analysis of p110α and NOX1. GAPDH was used as an internal standard. Graphs under blots show mean values (± SD). *P < 0.05, statistically significant as indicated (sh short hairpin, n.s not significant)
p110α is essential for formation of tube structures
To further examine the key factors regulating the sprouting of vascular structures, we investigated the formation of tube-like structures of Flk-1+ cells on matrigel; the production of these tubes recapitulates the process of sprouting and tube formation that occurs during angiogenesis. Flk-1+ cells were plated on matrigel-coated 96-multiwell plates. Images were taken after 16 h (Fig. 5a-a‘‘‘‘). VEGF treatment significantly increased the organization of Flk-1+ cells into interconnected tubes (Fig. 5a‘) and enhanced the formation of tube-like structures on matrigel (Fig. 5b, c). In contrast, the pharmacological inhibition of PI3K by wortmannin (1 μM) completely inhibited VEGF-induced tube formation on matrigel (Fig. 5a‘‘). Quantitative analysis showed that both the total length of the tubes and the number of branching points were clearly decreased after knockdown of p110α and NOX1 (Fig. 5b, c). Furthermore, shRNA p110α cells failed to form interconnected tubes, even when stimulated with VEGF (Fig. 5a‘‘‘). Moreover, VEGF lost the ability to increase tube length and branching points in shRNA NOX1 Flk-1+ cells cultured on matrigel (Fig. 5b, c). Similar data were obtained for Flk-1+ cells isolated from CCE ES cells (data not shown). Additionally, we could show that the treatment of human umbilical vein endothelial cells (HUVECs) with specific inhibitors of p110α and NOX1 (Supplemental Fig. S6a-a‘‘‘) led to a significant reduction of VEGF-induced capillary-like tube formation and branching points (Supplemental Fig. S6b, c). Thus, our results suggest that, in various ES cell lines, p110α and NOX1 are directly linked to VEGF-mediated tube formation during angiogenesis.

Angiogenesis assay for Flk-1+ cells after mRNA inactivation of p110α or NOX1. Tube structure was analysed either in the presence or in the absence of VEGF (500 pM) after 16 h of cultivation. The ability to form tubes was evaluated as the total length of tubes and branching points per field. Bars 500 μm. a–a‘‘‘‘ Representative transmitted light images for three independent experiments of tube formation of Flk-1+ cells on matrigel upon treatment with wortmannin (1 μM; a‘‘) or knock down of p110α (a‘‘‘) or NOX1 (a‘‘‘‘) in the absence (a) or presence (a‘) of VEGF (500 pM). VEGF-induced branching points (b) and capillary-like tube formation (c) were significantly inhibited in the presence of the nonspecific PI3K inhibitor wortmannin or after the silencing of p110α and NOX1 in Flk-1+ cells. *P < 0.05, statistically significant as indicated (n.s not significant)
p110α and Rac1 are essential for Flk-1+ cell migration stimulated by VEGF
To address the effect of VEGF on cell migration, which is an essential step for the angiogenesis process, a scratch migration assay was performed. In these experiments, cell migration into the scratch area was assessed 6 h after stimulation of plated Flk-1+ cells with VEGF (500 pM; Fig. 6a-a‘‘‘‘‘). Analysis showed that VEGF treatment significantly increased cell migration (Fig. 6b). To examine whether PI3K was involved in this VEGF-induced migration, we treated the Flk-1+ cells with wortmannin, which totally inhibited the migration of Flk-1+ cells (Fig. 6a‘‘‘, b). To determine whether the silencing of p110α or NOX1 could affect cell migration and hence the ability of the cell to form interconnected network structures, cells depleted for p110α (Fig. 6a‘‘‘‘, b) or NOX1 (Fig. 6a‘‘‘‘‘, b) were used in the scratch assay. Complete inhibition of the migration of the p110α-deficient Flk-1+ cells was observed (Fig. 6a‘‘‘‘, b). Furthermore, the knocking down of NOX1 in Flk-1+ cells caused a significant reduction in cell migration and a decrease in scratch closure compared with the VEGF-treated pLKO.1 control (Fig. 6a‘‘‘‘‘, b). The same results were obtained after treatment of HUVECs (Supplemental Fig. S7a-a‘‘‘‘, b) and Flk-1+ cells isolated from CCE ES cells (data not shown) with p110α- or NOX1-specific inhibitors in the presence or absence of VEGF.

VEGF positively regulates cell migration and cytoskeleton reorganization via PI3K signalling in vitro. a–a‘‘‘‘‘ Representative images of the scratch assay of Flk-1+ cells isolated from 4-day-old embryoid bodies containing Flk-1+ wild-type (pLKO.1), p110α or NOX1 shRNA cells, either alone or in the presence of VEGF (500 pM). Bars 200 μm. b VEGF-treated cells exhibited a highly increased migratory potential, which was totally abolished by the treatment of Flk-1+ cells with wortmannin or the knocking down of the catalytic subunit p110α. c–c‘‘‘‘ Representative images of Flk-1+ cells stained with phalloidin (green) and 4,6-diamidino-2-phenylindole (DAPI; blue) in three independent experiments. Bars 50 μm. Lamellipodia (arrow) were observed at the leading edge of VEGF-stimulated migrating cells. d VEGF-induced actin cytoskeleton reorganization and significant increase in polarized shapes of VEGF-induced Flk-1+ cells. Cell polarization was measured by the formation of lamellipodia in the direction of migration. Wortmannin treatment and the silencing of p110α abrogated VEGF-induced lamellipodia formation and the polarized shapes of Flk-1+ cells. *P < 0.05, statistically significant as indicated (sh short hairpin, n.s not significant)
As shown in representative micrographs of phalloidin-stained Flk-1+ cells (Fig. 6c–c‘‘‘‘), VEGF-treated cells exhibited a significant increase in lamellipodia structures and polarized cell shapes (Fig. 6c‘, d). Wortmannin treatment (Fig. 6c‘‘, d) or knockdown of p110α (Fig. 6c‘‘‘, d) abrogated VEGF-induced lamellipodia structure and polarized cell shape formation compared with the control group. In shRNA NOX1 cells (Fig. 6c‘‘‘‘, d), a significant polarization after VEGF treatment compared with untreated cells was still observed, although this effect was significantly less pronounced as compared with pLKO.1 cells (Fig. 6c‘, d). These data demonstrate that p110α is responsible for the triggering of cell migration stimulated by VEGF and is likely to be involved in ROS generation by NOX1.
VEGF induces Rac1 and RhoA activity in ES cells
A prerequisite for lamellipodia formation is the activation of Rac1 after extracellular stimuli (Disanza et al. 2005). To further elucidate the observed endothelial cell migration during angiogenesis, we performed a western-blot-based Rac1 phosphorylation assay on 5-day-old plated embryoid bodies stimulated with VEGF (500 pM). Phosphorylation of Rac1 increased rapidly within 10 min after the addition of VEGF. Subsequently, Rac1 activation decreased after 30 min but remained on a significantly elevated plateau as compared with 0 min (Supplemental Fig. S8a, b). Moreover, RhoA activation significantly changed from 30 min to 2 h after the addition of VEGF (Supplemental Fig. S8a, b). However, when shRNA p110α embryoid bodies were stimulated with VEGF, Rac1 (Fig. 7a, a‘) and RhoA (Fig. 7b, b‘) activation was absent. In contrast, Rac1 (Fig. 7a, a‘) and RhoA (Fig. 7b, b‘) activation induced by VEGF was significantly increased in shRNA NOX1 embryoid bodies compared with the untreated control group. Pre-treatment of the shRNA NOX1 embryoid bodies with compound 15e (0.5 μM) completely inhibited the increase of Rac1 (Fig. 7c, c‘) and RhoA (Fig. 7d, d‘) activation induced by VEGF. Collectively, these results suggest that the VEGF-induced activation of Rac1 and RhoA in the ES cells is mediated by class-IA-PI3Kα-mediated signalling but is not dependent on NOX1.

VEGF-mediated activation of RhoA (Ras homologue gene family member A) and Rac1 in ES cells depends on PI3Kα. ES cells were stimulated with VEGF (500 pM). Whole cell lysates were blotted and subjected to immunoblotting. Activation of Rac1 (Ser71) and RhoA was analysed by using phospho-specific antibodies. Blots were subsequently re-probed with pan-specific antibodies recognizing Rac1/2/3 (a, c) or RhoA (b, d), respectively. a‘-d‘ Specific activation was quantified as the ratio of phospho-specific to pan-specific signals; mean values (± SD) of three independent experiments. a–d, a‘-d‘ ES cells stably producing shRNA targeting p110α or NOX1 and the pLKO.1 shRNA control were stimulated with VEGF and pre-treated for 30 min with compound 15e (0.5 μM) as indicated. *P < 0.05, statistically significant (sh short hairpin, n.s not significant)
VEGF stimulates Akt activation dependent on p110α and independent of NOX1
VEGF-induced Akt activation showed its maximum at 10 min as demonstrated in Fig. 8. To elucidate whether VEGF-induced Akt activation is p110α-dependent (Fig. 8a, a‘), we blocked p110α by compound 15e (Fig. 8b, b‘), and analysed Akt activation upon VEGF treatment. We found that pre-treatment of the cells with 0.5 μM compound 15e totally abolished the effect of VEGF on Akt activation (Fig. 8b, b‘). To check whether NOX1 is involved in VEGF-induced Akt activation, we blocked NOX1 activity by using 2-APT and examined Akt activity after VEGF treatment (Fig. 8c, c‘). Interestingly, pre-treatment with 0.5 μM 2-APT did not affect VEGF-induced Akt activity. To verify the results obtained with specific inhibitors, we used ES cells shRNA p110α or shRNA RNA NOX1 ES cells. As expected, the down-regulation of p110α resulted in a significant inhibition in Akt phosphorylation upon VEGF treatment (Fig. 8d, d‘). In contrast, a non-significant decrease in Akt phosphorylation was observed after the suppression of NOX1 (Fig. 8d, d‘). Taken together these results suggest that p110α but not NOX1 acts as an upstream effector of Akt activation.

Activation of Akt by VEGF is dependent on class 1A PI3Kα but not NOX1. Activation of Akt (Ser473) was analysed by using phospho-specific antibodies. Blots were subsequently reprobed with specific antibodies recognizing Akt. Activation was quantified as the ratio of phospho-specific to pan-specific signals. Graphs under the blots show mean values (± SD) of three independent experiments. a, a‘ Time course of Akt and phospho-Akt (Ser473) upon VEGF treatment. Note that VEGF stimulated Akt phosphorylation after 10 min. The level of total Akt and phospho-Akt (Ser473) in embryoid bodies was determined following 30 min pre-treatment with inhibitor and VEGF for the indicated time. b, b‘ Compound 15e (0.5 μM) inhibited the up-regulation of phospho-Akt (Ser473) induced by VEGF (500 pM). c, c‘ The NOX1 inhibitor, 2-acetylphenothiazine (2-APT), did not affect VEGF-induced Akt activity. d, d‘ Knock down of p110α resulted in significant inhibition of Akt phosphorylation upon VEGF treatment. In contrast, a non-significant decrease of Akt phosphorylation was observed after the silencing of NOX1. *P < 0.05, statistically significant as indicated (n.s not significant)
Discussion
VEGF signalling is known to be crucial for both normal and disease-associated vascular development (Ferrara 2009). Moreover, several studies have demonstrated that VEGF can activate PI3K and stimulate vascular differentiation (Bekhite et al. 2011; Gerber et al. 1998; Jiang and Liu 2009). However, the targeting of vascular development by using wortmannin, a first generation PI3K inhibitor, additionally inhibits PI3K-related kinases such as mTOR (mechanistic target of rapamycin) and PI4-kinase β (Finan and Thomas 2004), which precludes a clear-cut analysis of distinct signalling cascades. Many inhibitors targeting specific PI3K catalytic subunits have been developed to overcome these disadvantages (Marone et al. 2008). Interestingly, the pharmacological inhibition or knockdown of p110α has no effect on ES cell self-renewal but only reduces the proliferation of undifferentiated mouse ES cells (Kingham and Welham 2009). Consistent with this, our previous results have shown that the protein expression of the proliferation marker Ki-67 is significantly decreased in ES cells treated with a pharmacological inhibitor of p110α (Bekhite et al. 2011).
The importance of NOX proteins for ROS production has been linked to the PI3K pathway by several authors (Nakanishi et al. 2014; Seshiah et al. 2002; Xu et al. 2011). However, the precise signalling mechanisms of VEGF on NOX stimulation through PI3K are not yet completely understood (Chatterjee et al. 2012; Kennedy and DeLeo 2008). To elucidate a potential crosslink between PI3K and NOX activation, we have shown here that the VEGF stimulation of intracellular ROS formation is completely inhibited by the NOX inhibitors DPI, VAS2870 and apocynin, but not by rotenone. Previous data have shown that apocynin prevents p47phox and p47phox homologue translocation to the plasma membrane, thereby preventing active and functional NOXs (Banfi et al. 2003; Touyz et al. 2002). ROS formed by VEGF stimulation might be derived from NOX1 and NOX2 (Bedard and Krause 2007). To test the role of NOX1 and NOX2 further and that of p110α in intracellular ROS formation, we have treated shRNA p110α and shRNA NOX1 embryoid bodies with VEGF in this study and observed that VEGF fails to increase the ROS level. In contrast, treatment of shRNA NOX2 embryoid bodies with VEGF does not affect ROS production, indicating that NOX2 is not involved in ROS generation upon VEGF treatment. These data have been confirmed in Flk-1+ cells derived from two different cell lines of ES cells in which VEGF treatment strongly elevates O2 − generation. Furthermore, specific inhibitor or knockdown of p110α or NOX1 in Flk-1+ cells abolishes the effects of VEGF on O2 − production. In contrast, the blocking activity or knockdown of NOX2 in these Flk-1+ cells does not have significant effects on O2 − levels. As is well known, Rac1 plays a critical role in VEGF-induced O2 − formation via NOX activation in various cell types (Abid et al. 2001). Therefore, we have investigated the role of Rac1 in O2 − generation of differentiating ES cells. As expected Rac1 inhibitor abolishes the VEGF-mediated increase in O2 − production, thus indicating an involvement of Rac1 through the activation of NOX1.
In this study, we have shown that the treatment of embryoid bodies with VEGF results in a significant increase in vascular differentiation in pLKO.1 ES cells. This effect is still obvious in NOX1-depleted embryoid bodies but to a significantly lesser degree. Treatment of embryoid bodies with PI3K inhibitors reduces not only the area of PECAM-1-positive cells, but also the tube-like structures in embryoid bodies. Of note, the increase in vascular differentiation upon treatment with VEGF is nearly absent upon the silencing of p110α subunit mRNA expression. Similar findings have been observed in other studies showing that endothelial cell-specific-p110α knock out leads to embryonic lethality at mid-gestation because of severe defects in angiogenic sprouting and vascular remodelling (Graupera et al. 2008). Furthermore, our data have demonstrated that Rac1 inhibitor significantly reduces the size of the PECAM-1-positive area and nearly abolishes branching points in embryoid bodies. To verify these results further, we have repeated this experiment in the mouse ES cell line CCE and obtained comparable data. Thus, vasculogenesis and angiogenesis are apparently controlled by PI3K through p110α subunits and Rac1, independent of the ES cell line employed. These results are consistent with data from others who have demonstrated that PI3K and Rac1 are required for VEGF-dependent angiogenesis in endothelial cells (Zhang et al. 2012). In addition, an essential role has been reported for Rac1 in mediating the effect of VEGF on endothelial cell proliferation and migration in vitro and in inducing angiogenesis in a mouse sponge implant model (Ushio-Fukai et al. 2002).
The defects in vasculogenesis upon interference with p110α and NOX1 might be attributable to the impairment of cell migration. Results from tube-like structure formation and scratch migration assays in Flk-1+ cells indicate that shRNA p110α Flk-1+ cells fail to migrate and to form interconnected tubes, even when stimulated with VEGF. Likewise, the silencing of NOX1 in Flk-1+ cells causes a significant reduction in cell migration and tube length and in branching points upon VEGF treatment. Hence, our findings suggest that both p110α and NOX1 are important for VEGF-induced tube formation. Since VEGF still increases the number of Flk-1+ cells in NOX1 shRNA embryoid bodies, we conclude that NOX1 is not required for the initial stages of vasculogenesis but plays a role in the migration and sprouting of endothelial cells to promote the establishment of capillary-like networks upon VEGF treatment. We further speculate that other NOX isoforms, e.g. NOX4, take over this function in NOX1-depleted cells of embryoid bodies. Similar findings have been observed in other cell lines, such as Flk-1+ cells isolated from the CCE cell line and HUVECs; these results show that p110α and NOX1 inhibition lead to a significant decrease in cell migration and tube length and in branching point numbers upon VEGF treatment. Consistent with our data, others have suggested a role for NOX1 in the stimulation of the branching morphogenesis of sinusoidal endothelial cells (Kobayashi et al. 2004). Mice deficient in NOX1, but not NOX2 or NOX4, exhibit a significant reduction in angiogenesis (Garrido-Urbani et al. 2011).
As is well known, the remodelling of the actin cytoskeleton is essential for cell migration (Lamalice et al. 2007). Our data show that VEGF-treated Flk-1+ cells display polarized shapes attributable to the formation of lamellipodia during cell migration. Pre-treatment with wortmannin or the silencing of p110α in Flk-1+ cells abrogates VEGF-induced lamellipodia structure formation. Moreover, the absence of NOX1 in Flk-1+ cells results in the significant inhibition of polarized cell numbers, although this effect is less pronounced than under conditions of PI3K inhibition. These observations suggest that class IA PI3Kα signalling is required for cytoskeletal changes during cell migration, whereas the silencing of NOX1 in Flk-1+ cells affects lamellipodia structure formation.
PI3K has been found to regulate cell migration through the activation of Rac1 (Williams et al. 2000). In our study, the suppression of p110α by using shRNA significantly inhibits the activation of Rac1, RhoA and Akt in ES cells upon VEGF treatment. VEGF still activates RhoA, Akt and Rac1 in shRNA NOX1 cells but not in shRNA p110α cells, indicating that these factors act upstream of NOX1 and downstream of p110α. This is in agreement with previous studies that have shown that the activation of Rac1 is selectively dependent on the p110α isoform of PI3K (Graupera et al. 2008). Other work has demonstrated that Rac1 can also activate PAK (p21-activated kinase), which can also promote cell migration and tube-like structure formation (Ridley 2001). Since NOX1 is expressed in Flk-1+ cells, and since the shRNA-mediated down-regulation of NOX1 suppresses ROS generation, our findings concur with the observation that VEGF stimulates ROS production via the activation of Rac1-dependent NOX (Ushio-Fukai 2007) or Rac1-regulated NOX1 enzyme (Miyano et al. 2006). Previous research has revealed that ROS are involved in actin cytoskeletal dynamics and cellular motility through their direct interaction with actin or indirectly through an interaction with a number of actin-binding proteins that control barbed-end exposure (gelsolin), actin filament length (cofilin), the association of filaments (filamin, IQGAP) and stress fibre formation (Moldovan et al. 2006).
Taken together, our data demonstrate that the VEGF-VEGFR-PI3K-Akt-Rac1-NOX1 signalling pathway plays a vital role in VEGF-induced blood vessel differentiation. Importantly, these findings provide direct evidence that the activity of p110α in endothelial cells is essential for vascular development and suggest that p110α, NOX1 and their downstream signalling cascade represent promising therapeutic targets for the treatment of numerous human diseases that involve aberrant neovascularization.
References
Abid MR, Tsai JC, Spokes KC, Deshpande SS, Irani K, Aird WC (2001) Vascular endothelial growth factor induces manganese-superoxide dismutase expression in endothelial cells by a Rac1-regulated NADPH oxidase-dependent mechanism. FASEB J 15:2548–2550
Banfi B, Clark RA, Steger K, Krause KH (2003) Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J Biol Chem 278:3510–3513
Banham-Hall E, Clatworthy MR, Okkenhaug K (2012) The therapeutic potential for PI3K inhibitors in autoimmune rheumatic diseases. Open Rheumatol J 6:245–258
Bartsch C, Bekhite MM, Wolheim A, Richter M, Ruhe C, Wissuwa B, Marciniak A, Muller J, Heller R, Figulla HR, Sauer H, Wartenberg M (2011) NADPH oxidase and eNOS control cardiomyogenesis in mouse embryonic stem cells on ascorbic acid treatment. Free Radic Biol Med 51:432–443
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87:245–313
Bekhite MM, Finkensieper A, Binas S, Muller J, Wetzker R, Figulla HR, Sauer H, Wartenberg M (2011) VEGF-mediated PI3K class IA and PKC signaling in cardiomyogenesis and vasculogenesis of mouse embryonic stem cells. J Cell Sci 124:1819–1830
Bhattacharya R, Kwon J, Li X, Wang E, Patra S, Bida JP, Bajzer Z, Claesson-Welsh L, Mukhopadhyay D (2009) Distinct role of PLCbeta3 in VEGF-mediated directional migration and vascular sprouting. J Cell Sci 122:1025–1034
Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380:435–439
Chatterjee S, Browning EA, Hong N, DeBolt K, Sorokina EM, Liu W, Birnbaum MJ, Fisher AB (2012) Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am J Physiol Heart Circ Physiol 302:H105–H114
Colavitti R, Pani G, Bedogni B, Anzevino R, Borrello S, Waltenberger J, Galeotti T (2002) Reactive oxygen species as downstream mediators of angiogenic signaling by vascular endothelial growth factor receptor-2/KDR. J Biol Chem 277:3101–3108
Disanza A, Steffen A, Hertzog M, Frittoli E, Rottner K, Scita G (2005) Actin polymerization machinery: the finish line of signaling networks, the starting point of cellular movement. Cell Mol Life Sci 62:955–970
Ferrara N (2009) Vascular endothelial growth factor. Arterioscler Thromb Vasc Biol 29:789–791
Finan PM, Thomas MJ (2004) PI 3-kinase inhibition: a therapeutic target for respiratory disease. Biochem Soc Trans 32:378–382
Fruman DA, Meyers RE, Cantley LC (1998) Phosphoinositide kinases. Annu Rev Biochem 67:481–507
Garrido-Urbani S, Jemelin S, Deffert C, Carnesecchi S, Basset O, Szyndralewiez C, Heitz F, Page P, Montet X, Michalik L, Arbiser J, Ruegg C, Krause KH, Imhof BA (2011) Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARalpha mediated mechanism. PLoS One 6:e14665
Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N (1998) Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 273:30336–30343
Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, Smith AJ, Ridley AJ, Ruhrberg C, Gerhardt H, Vanhaesebroeck B (2008) Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature 453:662–666
Guo D, Jia Q, Song HY, Warren RS, Donner DB (1995) Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J Biol Chem 270:6729–6733
Hayakawa M, Kaizawa H, Moritomo H, Koizumi T, Ohishi T, Okada M, Ohta M, Tsukamoto S, Parker P, Workman P, Waterfield M (2006) Synthesis and biological evaluation of 4-morpholino-2-phenylquinazolines and related derivatives as novel PI3 kinase p110alpha inhibitors. Bioorg Med Chem 14:6847–6858
Jiang BH, Liu LZ (2009) PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res 102:19–65
Jiang BH, Zheng JZ, Aoki M, Vogt PK (2000) Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc Natl Acad Sci U S A 97:1749–1753
Kabrun N, Buhring HJ, Choi K, Ullrich A, Risau W, Keller G (1997) Flk-1 expression defines a population of early embryonic hematopoietic precursors. Development 124:2039–2048
Kennedy AD, DeLeo FR (2008) PI3K and NADPH oxidase: a class act. Blood 112:4788–4789
Kingham E, Welham M (2009) Distinct roles for isoforms of the catalytic subunit of class-IA PI3K in the regulation of behaviour of murine embryonic stem cells. J Cell Sci 122:2311–2321
Kobayashi S, Nojima Y, Shibuya M, Maru Y (2004) Nox1 regulates apoptosis and potentially stimulates branching morphogenesis in sinusoidal endothelial cells. Exp Cell Res 300:455–462
Kroll J, Waltenberger J (1997) The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J Biol Chem 272:32521–32527
Lamalice L, Le Boeuf F, Huot J (2007) Endothelial cell migration during angiogenesis. Circ Res 100:782–794
Madamanchi NR, Vendrov A, Runge MS (2005) Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol 25:29–38
Marone R, Cmiljanovic V, Giese B, Wymann MP (2008) Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 1784:159–185
McAuslan BR, Gole GA (1980) Cellular and molecular mechanisms in angiogenesis. Trans Ophthalmol Soc U K 100:354–358
Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NP, Risau W, Ullrich A (1993) High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 72:835–846
Miyano K, Ueno N, Takeya R, Sumimoto H (2006) Direct involvement of the small GTPase Rac in activation of the superoxide-producing NADPH oxidase Nox1. J Biol Chem 281:21857–21868
Moldovan L, Mythreye K, Goldschmidt-Clermont PJ, Satterwhite LL (2006) Reactive oxygen species in vascular endothelial cell motility. Roles of NAD(P)H oxidase and Rac1. Cardiovasc Res 71:236–246
Muramatsu F, Kidoya H, Naito H, Sakimoto S, Takakura N (2013)MmicroRNA-125b inhibits tube formation of blood vessels through translational suppression of VE-cadherin. Oncogene 32:414–421
Nakanishi A, Wada Y, Kitagishi Y, Matsuda S (2014) Link between PI3K/AKT/PTEN pathway and NOX proteinin diseases. Aging Dis 5:203–211
Resch T, Pircher A, Kahler CM, Pratschke J, Hilbe W (2012) Endothelial progenitor cells: current issues on characterization and challenging clinical applications. Stem Cell Rev 8:926–939
Ribatti D (2006) Genetic and epigenetic mechanisms in the early development of the vascular system. J Anat 208:139–152
Ridley AJ (2001) Rho GTPases and cell migration. J Cell Sci 114:2713–2722
Risau W (1995) Differentiation of endothelium. FASEB J 9:926–933
Sauer H, Bekhite MM, Hescheler J, Wartenberg M (2005) Redox control of angiogenic factors and CD31-positive vessel-like structures in mouse embryonic stem cells after direct current electrical field stimulation. Exp Cell Res 304:380–390
Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK (2002) Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res 91:406–413
Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R, Freitas C, Duarte A, Isoniemi H, Laakkonen P, Christofori G, Yla-Herttuala S, Shibuya M, Pytowski B, Eichmann A, Betsholtz C, Alitalo K (2008) Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454:656–660
Touyz RM, Chen X, Tabet F, Yao G, He G, Quinn MT, Pagano PJ, Schiffrin EL (2002) Expression of a functionally active gp91phox-containing neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circ Res 90:1205–1213
Ushio-Fukai M (2007) VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal 9:731–739
Ushio-Fukai M, Urao N (2009) Novel role of NADPH oxidase in angiogenesis and stem/progenitor cell function. Antioxid Redox Signal 11:2517–2533
Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW (2002) Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res 91:1160–1167
Vittet D, Prandini MH, Berthier R, Schweitzer A, Martin-Sisteron H, Uzan G, Dejana E (1996) Embryonic stem cells differentiate in vitro to endothelial cells through successive maturation steps. Blood 88:3424–3431
Williams MR, Arthur JS, Balendran A, Kaay J van der, Poli V, Cohen P, Alessi DR (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol 10:439–448
Wymann MP, Pirola L (1998) Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta 1436:127–150
Wymann MP, Zvelebil M, Laffargue M (2003) Phosphoinositide 3-kinase signalling—which way to target? Trends Pharmacol Sci 24:366–376
Xu J, Tian W, Ma X, Guo J, Shi Q, Jin Y, Xi J, Xu Z (2011) The molecular mechanism underlying morphine-induced Akt activation: roles of protein phosphatases and reactive oxygen species. Cell Biochem Biophys 61:303–311
Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, Naito M, Nakao K (2000) Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature 408:92–96
Zhang LJ, Tao BB, Wang MJ, Jin HM, Zhu YC (2012) PI3K p110alpha isoform-dependent Rho GTPase Rac1 activation mediates H2S-promoted endothelial cell migration via actin cytoskeleton reorganization. PLoS One 7:e44590
Acknowledgments
We thank Dr. Martin Förster for his support during FCM and cell sorting procedures. We also thank Dr. Joachim Clement for kindly providing us with the phalloidin-Alexa Fluor 488 dye.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have nothing to declare.
Additional information
This work was supported by the Excellence Cluster Cardio-Pulmonary System (ECCPS) of the German Research Foundation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Purity of Flk-1+ cells and PI3K catalytic subunit expression in vascular progenitor isolated from embryoid bodies. Flk-1+ cells were sorted by MACS from 4-day-old embryoid bodies. a Flk-1+ cells were labelled by using phycoerythrin (PE)-conjugated rat anti-mouse Flk-1 antibody and processed by FCM. b, b‘ Mean values (± SD) of three independent experiments for mRNA expression of the class IA PI3K catalytic subunits (p110α, β, δ). *P < 0.05, statistically significant as indicated. (GIF 2501 kb)
Fig. S2
VEGF-induced O2 − production is dependent on PI3K-Rac1 activation in Flk-1+ cells derived from the ES cell line CCE. O2 − generation of sorted Flk-1+ cells isolated from 4-day-old embryoid bodies in response to VEGF stimulation was analysed by determination of DHE fluorescence. a-a‘‘‘‘ Representative DHE fluorescence images of Flk-1+ cells incubated with compound 15e (0.5 μM) (a‘‘), a specific p110α inhibitor, and the NOX1 inhibitor 2-APT (0.5 μM; a‘‘‘) and Rac1 inhibitor (50 μM; a‘‘‘‘) in the absence (a) or presence (a‘) of VEGF (500 pM). b Percentage values (± SD) of three independent experiments. *P < 0.05, statistically significant as indicated (n.s not significant). (GIF 5698 kb)
Fig. S3
Vessel-like structures in embryoid bodies derived from ES cells. a Representative image of embryoid body stained with endothelial markers Flk-1 (green) and PECAM-1 (red). To visualize the shape of embryoid bodies, transmitted light images were recorded (blue). a Overlay image. a‘ Flk-1. a‘‘ PECAM-1. b Flk-1. b‘ PECAM-1. The cell nuclei in b, b‘ were labelled with DAPI (blue). c, d Representative images of embryoid bodies stained with endothelial markers PECAM-1 (green) and VE-cadherin (red). c PECAM-1, c‘ VE-cadherin, c‘‘ Overlay image. d PECAM-1. d‘ VE-cadherin. d‘‘ Overlay image. (GIF 16111 kb)
Fig. S4
Number of Flk-1+ cells in p110α or NOX1 knockdown and wild-type (pLKO.1) ES cells. Cells were differentiated for 10 days and treated with wortmannin (1 μM) or Rac1 inhibitor (50 μM) as indicated. a–a‘‘‘‘‘, b FCM analysis for three independent experiments; Flk-1+ cell numbers in embryoid bodies and after application of inhibitors from day 4 to day 10 of cell culture either in the absence or presence of VEGF (500 pM). *P < 0.05, statistically significant as indicated (n.s not significant). (GIF 2979 kb)
Fig. S5
Effects of pan-PI3K inhibitor (wortmannin) and specific inhibitor of p110α (compound 15e), NOX1 (2-APT) and Rac1 (Rac1 inhibitor) on vascular differentiation of the ES cell line CCE. ES cells were differentiated for 10 days. Wortmannin (1 μM), compound 15e (0.5 μM), the NOX1 inhibitor 2-APT (0,5 μM) or Rac1 inhibitor (50 μM) was applied from day 4 to day 10 of cell culture and the cells were stimulated with VEGF (500 pM) as indicated. a–a‘‘‘‘, b-b‘‘‘‘ Representative immunofluorescence images for at least three experiments showing vascular differentiation in embryoid bodies under conditions as indicated. Bars 100 μm. c Percentage values (± SD) of PECAM-1-positive areas for three independent experiments. *P < 0.05, statistically significant as indicated (n.s not significant). (GIF 8254 kb)
Fig. S6
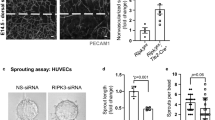
Tube formation of human umbilical vein endothelial cells (HUVECs) on matrigel treated with p110α (compound 15e) or NOX1 inhibitor (2-APT). Tube structures were analysed either in the presence or in the absence of VEGF (500 pM) after 16 h of cultivation. The ability to form tubes was expressed as total length of tubes and branching points per field. Bars 200 μm. a–a‘‘‘ Representative transmitted light images for three independent experiments of tube formation of HUVECs on matrigel upon treatment with either compound 15e (0.5 μM) or 2-APT (0,5 μM) in the presence or absence of VEGF (500 pM). VEGF-induced tube-like formation (b) and branching points (c) were significantly inhibited in the presence of the specific p110α or NOX1 inhibitor in HUVECs. *P < 0.05, statistically significant as indicated (n.s not significant). (GIF 3862 kb)
Fig. S7
VEGF positively regulates cell migration of HUVECs in vitro. a–a‘‘‘ Representative images of the scratch assay in HUVECs treated with either compound 15e (0.5 μM) or the NOX1 inhibitor 2-APT (0.5 μM) in the presence or absence of VEGF (500 pM). b VEGF-treated cells exhibited a highly enhanced migratory potential, which was significantly inhibited by treatment of HUVECs with compound 15e or 2-APT. *P < 0.05, statistically significant as indicated (n.s not significant). (GIF 2923 kb)
Fig. S8
Kinetics of Rac1 and RhoA activation in ES cells. Cells were subjected to immunoblotting at the indicated time points after stimulation with VEGF. Activation of Rac1 (Ser71) and RhoA was analysed by using phospho-specific antibodies. a Blots were subsequently reprobed with pan-specific antibodies recognizing Rac1/2/3 or RhoA, respectively. b Specific activation was quantified as the ratio of phospho-specific to pan-specific signals. Graphs under the blots show mean values (± SD) of three independent experiments. *P < 0.05, statistically significant as indicated (n.s not significant). (GIF 2538 kb)
Table S1
(DOC 38 kb)
ESM 1
(DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Bekhite, M.M., Müller, V., Tröger, S.H. et al. Involvement of phosphoinositide 3-kinase class IA (PI3K 110α) and NADPH oxidase 1 (NOX1) in regulation of vascular differentiation induced by vascular endothelial growth factor (VEGF) in mouse embryonic stem cells. Cell Tissue Res 364, 159–174 (2016). https://doi.org/10.1007/s00441-015-2303-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-015-2303-8