
Abstract
Oncostatin M (OSM) is an IL-6/LIF family cytokine that influences mesenchymal progenitor differentiation; however, the mechanisms of this activity have not been fully elucidated. Using uncommitted murine adipose tissue-derived mesenchymal progenitors, we have examined mechanisms of OSM-induced osteogenesis. Murine OSM (mOSM) induced osteogenic differentiation to a greater degree than interleukin (IL)-6 and other members of the gp130 cytokine family, promoting extracellular matrix mineralization as indicated by Alizarin Red S staining. mOSM also increased expression of osteogenesis-associated gene products BMP4, BMP7, Runx-2, and osteocalcin as assessed by immunoblotting and real-time quantitative PCR. Additionally, protein kinase C (PKC) delta activity was upregulated in response to OSM stimulation, and to a greater degree than IL-6. Knockdown of PKCdelta expression by use of RNA interference (RNAi) reduced OSM-mediated osteogenic differentiation and decreased expression of Runx-2. These findings suggest that OSM differentially promotes osteogenesis in non-committed mesenchymal progenitors relative to other gp130 cytokines. This activity correlates with selective activation of PKCdelta in OSM-treated cells, indicating that OSM-induced osteogenesis and upregulation of osteogenic gene products require activity of PKCdelta.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteogenesis is a highly complex biological process, involving temporal–spatial control of multiple gene products coordinated by molecular mechanisms that are not well characterized. Various factors are known to stimulate osteogenesis, notably members of the fibroblast growth factor (FGF) family, insulin-like growth factors (IGFs), as well as members of the transforming growth factor (TGF)-beta-related bone morphogenetic protein (BMP) family (Rickard et al. 1994; Nakamura et al. 2005; Koch et al. 2005). Several other cytokines and growth factors are known to influence osteogenesis, such as members of the interleukin (IL)-6/gp130 cytokine family. Among these, IL-11 and cardiotrophin-1 (CT-1) have been shown to potentiate osteogenesis (Suga et al. 2003; Walker et al. 2008), while leukemia-inhibiting factor (LIF) has been implicated both in inhibiting the differentiation of osteoblast precursors as well as in promoting resorption of bone tissue (Falconi and Aubin 2007). IL-6, depending upon cell type and timing of administration, can either promote or inhibit osteogenic differentiation (Franchimont et al. 2005). Oncostatin M (OSM) has been shown to promote osteoblast proliferation, enhanced osteoblast differentiation (Jay et al. 1996) from precursors including calvarial cells (Malaval et al. 2005), and bone formation in vivo (Walker et al. 2010). In addition, the effects of parathyroid hormone appear to elicit its anabolic effects in osteoblasts through OSM biological activity (Walker et al. 2012). Other studies have indicated that OSM can induce osteogenesis of mesenchymal progenitor cells derived from human adipose tissue (Song et al. 2007). It is increasingly understood that cellular sources of potential bone-forming progenitors are diverse, including bone marrow stroma, adipose tissue, and even within discreet organ-specific tissue reservoirs (Pittenger et al. 1999; De Bari et al. 2001; Sekiya et al. 2002; Zuk et al. 2002). Adipose-derived mesenchymal progenitors have been well described in both mouse and human (Dennis et al. 1999; Rodriguez et al. 2005), and their multipotential characteristics have been shown to closely resemble those of cells isolated from bone marrow, a compartment that is less conveniently accessed, particularly in clinical settings. Therefore, adipose tissue-derived progenitors have an intriguing potential, both as a tool to gain better understanding of mechanisms of developmental or differentiation processes and to identify potential avenues for therapy.
As OSM has been shown to be expressed in local sites of ongoing inflammatory responses (Grenier et al. 2001; Kang et al. 2005) or systemically within patients or animal models of inflammatory disease (Okamoto et al. 1997; Modur et al. 1997; Luzina et al. 2003; O’Hara et al. 2003), the influence of OSM upon altered/improper osteogenic responses and mechanisms through which these occur is of interest. OSM induces substantial responses by various connective tissue cells such as fibroblasts and bone marrow stromal cells (Bamber et al. 1998; Tanaka et al. 2003), and promotes upregulation of gene products associated with connective tissue metabolism, including collagens type I and III, metabolizing enzymes including matrix metalloproteinases, and also MMP inhibitors such as TIMPs 1 and 3 (Duncan et al. 1995; Kerr et al. 1999; Li et al. 2001). While intensive study of the molecular mechanisms underlying OSM-stimulated responses has been ongoing, full understanding of OSM signal transduction and gene regulation is incomplete, notably in the context of its potential role during osteogenic differentiation. This study has examined a potential mechanism of OSM-mediated osteogenesis through its regulation of PKCdelta, a kinase associated with connective tissue responses (Jimenez et al. 2001; Fan et al. 2006; Mishra et al. 2007) and known to confer unique properties in OSM-stimulated connective tissue cells (Chipoy et al. 2004; Smyth et al. 2006).
Materials and methods
Isolation and propagation of adipose-derived mesenchymal progenitor cells
Fifteen- to twenty-week-old female C57Bl/6 mice were euthanized and pooled fatty tissue (3 mice per harvest) from the omentum, lining of the small intestine, kidneys and spleen was collected in cold phosphate-buffered saline (PBS). Tissue was finely minced and centrifuged at 500 rpm to separate low density fatty tissue from the dense stromal vascular fraction, followed by repeated (2×) PBS rinses. Heavy fractions were subsequently resuspended in 10 % fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA) -containing Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 0.5 % collagenase and rocked at 37 °C for 1 h. Digested tissue was then filtered through 100-μm strainers, and cells were pelleted at 1000 rpm and resuspended in 10 % FBS/DMEM plus 1 % (v/v) penicillin-streptomycin and 0.5 % fungizone. Viable cultures were passaged for at least 60 population doublings in 60-mm culture dishes at which time an adherent, fibroblast-like cell population was obtained as observed at ×100 magnification in bright field using a CCD-equipped Leica DMRA Image Capture microscope (Leica, Richmond Hill, ON, Canada). We have termed this outgrowing culture C57 adipose-derived mesenchymal progenitor cells (C57Adip MPC). At least 3 separate isolations of cells from the visceral adipose tissue were performed at different times, each one derived from pooled adipose tissue of 3 female C57Bl/6 J mice. Each was followed by extensive culture to rid the explant of non-mesenchymal cell types (the initial culture contains a heterogeneous cell population). During each culture preparation procedure cycle, following a period of 10 or more culture passages, cells were tested for their capacity to be differentiated either to adipocyte-like or osteoblast-like cell populations (see below). Further passaged cells were more homogeneous but maintained the multiple differentiation potential. Subsequently, cultures were either subjected to differentiation experiments (as described below) or frozen in liquid nitrogen for future use.
Materials
Reagents for osteogenic and adipogenic differentiation media (L-ascorbate-2-phosphate, beta-glycerophosphate, dexamethasone, indomethacin, insulin, and methyl-3-butylxanthine) were purchased from Sigma (St. Louis, MO, USA). Recombinant murine oncostatin M (mOSM), interleukin 6 (mIL-6), leukemia inhibitory factor (mLIF) and interleukin 11 (mIL-11) were purchased from R&D Systems (Minneapolis, MN, USA). For flow cytometry, phycoerythrin (PE)-conjugated anti-CD90.2, CD45.2 and CD31, and fluorescein-isothiocyanate (FITC)-conjugated anti-CD44 antibodies were purchased from Becton Dickinson (Franklin Lakes, NJ, USA). PE-conjugated anti-CD105 antibody was purchased from Ebioscience (San Diego, CA, USA). Anti-PKCdelta and anti-PKCepsilon mouse monoclonal antibodies were purchased from Signal Transduction Labs/BD (Mississauga, ON, Canada). Anti-Runx2 and anti-actin goat polyclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-STAT3 rabbit polyclonal antibodies were purchased from Cell Signaling Technology/NEB (Danvers, MA, USA). Purified Histone H1 was purchased from Sigma. Horseradish peroxidase (HRP)-coupled antibody raised against mouse and goat IgG were purchased from Sigma. HRP-anti rabbit IgG was purchased from Santa Cruz. Alizarin Red S and Oil Red O dyes were purchased from Sigma.
Flow cytometric characterization of C57Adip MPC
Subconfluent C57Adip MPC cells grown on 60-mm culture dishes (1 × 106 cells/plate) were washed twice in cold PBS then treated for 5 min at 37 °C with 0.5 ml PBS/500nM EDTA (to reduce adherence)/0.1 % (w/v) bovine serum albumin (Sigma) and gently collected into 6-ml flow cytometry tubes by passage through 40-μm cell strainers. Cells were pelleted at 1000 rpm and resuspended in PBS/500nM EDTA/0.1 % BSA plus 2 μg/μl of FcBlock for 10 min, then 1 μg/μl of respective antibodies were added for 30 min. Surface marker expression was detected by an LSR2 flow cytometer (BD) and collected data were analyzed using FlowJo software (BD).
Induction of osteogenic and adipogenic differentiation
Osteogenesis and adipogenesis differentiation protocols were adapted from Jaiswal et al. (2000). Briefly, cultures of C57Adip MPC were plated in either 24-well or 60-mm dishes (approximately 1 × 105 cells/well or 1 × 106 cells/plate, respectively) and cultured in 10 % FBS/DMEM plus 1 % penicillin-streptomycin/0.5 % fungizone for 2 days after having reached confluence (assessed by visualization on a ×50 magnification inverted microscope). Subsequently, culture media were replaced and cells were grown in either fresh culture medium, or adipogenic medium (containing 10 mM methyl-3-butylxanthine, 2 μg/ml indomethacin, 5 μM insulin and 1 μM dexamethasone) or osteogenic medium (containing 10 mM beta-glycerophosphate, 50 μg/ml ascorbate-2-phosphate and 250 nM dexamethasone). Cells seeded in triplicate were grown for the times indicated as per experimental procedure, with medium being replaced every third day. Initiation of differentiation is considered as day 0 (D0) of the experimental protocol. In cultures in which cytokines were added, they were added (25 ng/ml for all) at the time of differentiation medium addition (D0) and were replaced every third day with medium until the times indicated and subsequently cells were grown in differentiation culture media alone. For experiments in which RNA interference (RNAi) was performed, RNAi oligomers were transfected into cultures 1 day post-seeding (prior to reaching confluence) and media were replaced following overnight incubation until 2 days post-confluence.
Staining and quantitation of osteogenic and adipogenic differentiation
C57Adip MPC cells were cultured in 24-well plates (1 × 105 cells/well) and subjected to differentiation as described above. At the time of staining indicated, cells were washed once with PBS, fixed for 10 min with 10 % phosphate-buffered formalin, and washed again with either PBS (if stained with Alizarin Red S or Oil Red O) or distilled deionized (dd) H20 (if stained with Von Kossa). Stains were prepared as follows: Alizarin Red S was prepared as a 40 mM solution (136 mg of stain in 100 ml H20) and filtered through a 0.45-μm filter prior to use; Oil Red O was prepared as a stock 0.5 % (w/v) solution in 100 % isopropyl alcohol and subsequently diluted 3:2 in dd H20 and filtered through a 0.45-μm filter prior to use; Von Kossa was made as 5 % w/v silver nitrate in dd H20 and protected from light and stored at 4 °C. Cultures stained with Alizarin Red S or Oil Red O were rocked gently at room temperature for 15 min, washed twice with dd H20, and then dried. Cultures stained with Von Kossa were rocked gently at room temperature in darkness for 15 min, exposed for 45 min to direct 100-W incandescent light, washed twice with dd H20, dried and then covered. Microscopic analysis and photography was carried out on a Leica Image Capture microscope equipped with a CCD camera. Images were collected using OpenLab software (Leica) and quantitation of staining was subsequently carried out using Leica QWin software by measuring four photographed, ×100-magnified fields (four photographed fields/well, triplicate wells/experimental condition). For each treatment group that was measured for Alizarin Red S staining (Figs. 2, 5), one representative photographed field used for QWin quantitation is shown.
RNA isolation and quantitative real-time PCR (Taqman)
C57Adip MPC cells were grown to confluence in 60-mm culture dishes in DMEM/10 % FBS. Subsequently, cells were grown in osteogenic medium and cultured in osteogenic medium alone or with 25 ng/ml of the indicated cytokines (replaced with medium for the first two changes or 6 days). At time of harvest, cells were washed twice with cold PBS, and total RNA was extracted from cultures with TRIzol (Invitrogen) according to the manufacturer’s directions. RNA was quantified by spectrophotometric measurement of absorbance at 260 nm. A sample of 1 μg of reverse-transcribed cDNA was analysed for Runx-2, osteocalcin, BMP4, and BMP7 mRNA levels using customized, validated primer probe pairs (Applied Biosystems, Foster City, CA, USA) as described elsewhere (Langdon et al. 2003). Gene expression was quantified using the ΔΔCT method relative to 18S RNA.
Alkaline phosphatase assay
Conditioned media were collected from C57Adip MPC induced to undergo osteogenic differentiation as described above at 9 days post-differentiation and stored at −20 °C. To measure alkaline phosphatase activity, 50 μg p-nitrophenylphosphate (p-NPP) (Sigma) was added to 100-μl samples in triplicate, the reaction was incubated for 1 h at 37 °C protected from light, and stopped with 0.5 M NaOH. Conversion of p-NPP was measured by absorbance (optical density units) at 405 nm.
Immunoblotting
C57Adip MPC were seeded into 60-mm culture dishes and subjected to differentiation as described in “Results”. At the times indicated, cells were washed once with cold PBS and lysed in 25 mM Tris-Cl (pH 7.5)/125 mM NaCl/2 mM EDTA plus 1 % (v/v) Triton X-100 supplemented with aprotinin, PMSF and sodium orthovanadate. Lysates (approximately 300 μl) were collected into 1.5-ml Eppendorf tubes, centrifuged at 10,000 rpm for 10 min at 4 °C, and supernatants were frozen at −70 °C until time of use. For immunoblot experiments, lysates (15 μg/sample as determined by Bradford assay) were resolved on 8 % SDS-PAGE gels and transferred to nitrocellulose membranes. Blots were incubated overnight at 4 °C with primary antibodies directed against the indicated proteins, washed serially in Tris-buffered saline (TBS) supplemented with 0.2 % (v/v) Tween 20, followed by a 1-h, room temperature incubation with suitable horseradish peroxidase (HRP)-conjugated secondary antibodies. Following repeated TBS-Tween 20 washing, blots were treated for 1 min with ECL reagent (Amersham/GE Healthsciences) and subsequently exposed to Kodak X-omat film (Eastman Kodak, Rochester, NY, USA).
Immunoprecipitation and PKCdelta Kinase Activity Assay
Whole cell lysates generated as described above were incubated at 4 °C overnight with 2 μg/ml of anti-PKCdelta monoclonal antibody, followed by 1 h incubation with 15 μg/ml of protein A-agarose. Immune complexes were washed three times in lysis buffer followed by two washes in kinase reaction buffer containing 25 mM Tris (pH 7.5), 0.5 mM EDTA, 5 mM MgCl2 and 0.5 mM DTT. Beads were resuspended in a 20-μl reaction mix containing kinase buffer and 10 μCi of γ32P-ATP, and the assay was initiated by addition of 10 μg of histone H1 per reaction. Samples were incubated for 30 min at 37 °C with occasional mixing, and the reaction was terminated by addition of 10 μl of 4× Laemmli buffer. Samples were resolved on 12 % SDS-PAGE, and gels were dried and exposed to X-omat film for autoradiography. To normalize protein quantities used for kinase activity, cell lysates were also immunoblotted for total STAT3 prior to immunoprecipitation and kinase assay.
Results
Isolation of multipotent mesenchymal progenitors from murine adipose tissue
Isolation and propagation of progenitor cells with the potential to differentiate into various mesenchymal tissues, including adipose and osteoid tissue, have been achieved from sources such as bone marrow and adipose tissue of both human and murine origin (Miyaoka et al. 2006; Song et al. 2007). We have isolated and successfully propagated in long-term culture (over 60 population doublings) cells derived from the omental fatty tissue of female C57Bl/6 mice, and these will be herein referred to as C57Adip mesenchymal progenitor cells (MPC). C57Adip MPC exhibited a nondescript, fibroblast-like morphology in subconfluent culture (Fig. 1a). Flow cytometric analysis to assess surface phenotypic characteristics indicated the cell population was non-hematopoietic (CD45-) and did not arise from endothelial precursors (CD31-) (Fig. 1b, c). Cultures exhibited high expression of both CD90 and CD44, typical ‘core’ mesenchymal markers (Fig. 1d, e). Interestingly, CD105 (endoglin), a TGF-beta receptor superfamily member associated with human mesenchymal progenitors and also murine mesenchymal precursors from bone marrow, was expressed at very low levels (Fig. 1f). Functionally, we assessed and confirmed the differentiation potential of the C57Adip MPC towards an adipogenic lineage using standard adipogenesis-inducing media (see “Materials and Methods”), as assessed by Oil Red O staining (Fig. 1g–i). Relative to the control, mOSM markedly inhibited the differentiation of C57Adip MPC grown in adipogenic medium over as little as 6 days (Fig. 1i).

Phenotypic and biochemical characterization of adipose tissue-derived mesenchymal progenitor cells. Adipose tissue-derived mesenchymal progenitors (C57Adip MPC) were isolated from the visceral fat of 15- to 20-week-old C57Bl/6 mice and cultured as described in “Materials and methods”. Subconfluent cell cultures (approx. 1 × 106) were either fixed and observed by bright field microscopy at ×100 magnification (a) or collected from 60-mm dishes with PBS/500nM EDTA/0.1 % bovine serum albumin and subjected to flow cytometric analysis for surface antigens CD45.2 (b), CD31 (c), CD90.2 (d), CD44 (e) and CD105 (f). In (g–i), C57Adip MPC were seeded in 24-well culture dishes (1 × 105cells/well) and subsequently cultured in conditions promoting adipogenic differentiation (see “Materials and Methods”). Following 6 days of incubation in either DMEM/10% FBS (g) or differentiation media supplemented with no OMS (h) or 25 ng/ml mOSM (i), cell cultures were stained with Oil Red O (detection of lipid accumulation), and visualized by microscopy at ×50 magnification (scale bar 20 μm)
Promotion of osteogenesis by Oncostatin M
When treated for 6 days with various gp130 cytokines in osteogenic medium, mineralization was markedly increased as indicated qualitatively by Alizarin Red S staining relative to osteogenic medium treatment alone (Fig. 2a–e). Notably, amongst the cytokine-supplemented groups, IL-6, LIF and IL-11 did not induce an osteogenic response as pronounced as OSM. Staining using Von Kossa (5 % silver nitrate) provided consistent findings (Fig. 2f–j). Randomly selected fields of treated cultures (4 per well, 3 wells per treatment as described in “Materials and Methods”) stained with Alizarin Red S were subsequently quantified for the extent of staining using Leica QWin morphometric software and showed similar trends (Fig. 2k). Intriguingly, OSM was also able to significantly increase osteoid staining relative to the control even in DMEM supplemented with 10 % FBS rather than osteogenic medium, as shown by increased Von Kossa staining (Fig. 2l, m). Identical effects on adipocyte-like and osteoblast-like phenotypes were seen in C57 Adip MPC derived from each of at least 3 separate C57 Adip MPC preparations.

Oncostatin M promotes osteogenesis of C57Adip MPC. C57Adip MPC (1 × 105 cells/well, 24-well dishes) were either left untreated or stimulated with 25 ng/ml gp130 cytokines (as indicated) in osteogenic media for 6 days. Cultures were stained with Alizarin Red S (a–e) or Von Kossa (f–j) and visualized at ×100 magnification (scale bar 20 μm). In (k), levels of Alizarin Red S staining from the samples in (a–e) were quantified in triplicate wells (randomly selected, quadruplicate fields/well) for Alizarin Red S-positive staining using Leica QWin software. *p < 0.01 relative to osteogenesis medium treated alone; #p < 0.01 relative to osteogenesis medium with either mIL6, mLIF or mIL-11. In (l, m), C57Adip MPC were either left untreated or stimulated with 25 ng/ml mOSM in DMEM/10%FBS as indicated for 9 days, after which cells were stained (Von Kossa)
Induction of osteogenic markers by Oncostatin M
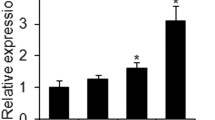
We next assessed whether mOSM selectively induced key factors associated with osteogenesis in differentiation-induced C57Adip MPC cultures. Taqman analysis for the mRNA expression of the osteogenesis master regulator Runx-2 (Fig. 3a) and terminal osteoblast differentiation marker osteocalcin (Fig. 3b) showed that mOSM significantly upregulated expression of these gene transcripts relative to osteogenesis-inducing medium alone. Further, although mRNA expression of Runx-2 and osteocalcin were increased in cells treated with IL-6, LIF or IL-11, the mOSM-induced effects were significantly greater relative to its related cytokines. We also assessed BMP mRNA steady state levels and observed that the addition of 25 ng/ml OSM to osteogenic media resulted in an induction of BMP4 and BMP7 (Fig. 3c, d) mRNA levels 9 days post-differentiation, but not BMP2 (data not shown).

Oncostatin M enhances expression of osteogenic genes in C57Adip MPC. In (a, b) C57Adip MPC were cultured in 60-mm dishes and subjected to osteogenesis induction either in osteogenic medium alone or supplemented with indicated gp130 cytokines (all 25 ng/ml). RNA from day 9 post-differentiation-induced cultures (performed in triplicates) were isolated, reversed transcribed, and measured for Runx-2 (a) or osteocalcin (b) gene expression. *p < 0.01 relative to osteogenic medium alone; #p < 0.01 relative to osteogenic medium with either mIL-6, mLIF or mIL-11. In (c, d), C57Adip MPC RNA was prepared from days 3, 6, and 9 post-differentiation-induced cultures in the absence (Osteo−OSM) or presence (Osteo+OSM) of 25 ng/ml mOSM. RNA collected and measured for BMP4 (c) or BMP7 (d) gene expression. (n = 1 per condition; error bars SD of triplicate measures in qRT-PCR)
PKCdelta contributes to mOSM induction of osteogenesis
Subsequently, we sought to examine whether mOSM-selective signaling intermediates could be identified contributing to its preferential osteogenic effects. Previous studies conducted by our laboratory and others have shown that mOSM and other gp130 cytokines regulate the phosphorylation and activation of various intracellular signaling pathways, including JAK/STAT and MAPK pathways (Chattopadhyay et al. 2007; Schnittker et al. 2013; Wong et al. 2014). Indeed, we were able to show that OSM or IL-6 robustly activated the STAT3 pathway in C57Adip MPC cultures (Fig. 4a). In addition, we have also previously described the capacity of OSM to induce activity of PKCdelta, a calcium-independent or ‘novel’-class PKC isoform in C57Bl/6 lung fibroblasts (Smyth et al. 2006). Therefore, we examined whether mOSM-stimulated C57Adip MPC demonstrated increased PKCdelta activation. Kinase activity assays from C57Adip MPC demonstrated increased PKCdelta activity as indicated by phosphorylation of histone H1 substrate in mOSM-treated cultures as compared to osteogenesis medium treated alone or IL-6-treated cell cultures (Fig. 4b). We next utilized RNAi strategies to selectively ablate PKCdelta. RNAi of PKCdelta could be stably maintained for at least 5 days, and was selective as evidenced by a lack of effect upon the related PKC isoform, PKCepsilon (Fig. 4c). We also assessed 24-h phorbol 12-myristylate 13-acetylate (PMA) pretreated cultures, as PMA transiently depletes cellular levels of multiple PKCs, including both PKCdelta and PKCepsilon.

Oncostatin M induces PKCdelta activity in C57Adip MPC. In (a), C57Adip MPC were cultured in 60-mm dishes and either left untreated or were stimulated with 25 ng/ml mOSM or mIL-6 for 10 min. Whole cell lysates were subjected to kinase activity assay as described in “Materials and Methods”, and the insets show phosphorylated histone autoradiographs, with an immunoblot of input lysate probed for p-tyr-STAT3 or total STAT3 as a control. In (b), histone phosphorylation was quantified and normalized to total PKCdelta expression using ImageJ densitometric software. In (c), AdipMPC were subjected to RNAi as described, and lysed at indicated time points (hours) post-transfection with either (+) PKCdelta targeted RNAi or scrambled RNAi as indicated. Lysates were resolved by SDS-PAGE, and probed for PKCdelta and PKCepsilon, a related novel PKC isoform, by immunoblots
To assess the functional consequences of PKCdelta inhibition, we examined protein lysates for levels of PKCdelta and Runx-2 using immunoblots. Protein lysates collected 3 days after differentiation showed decreased PKCdelta protein signal in OSM-treated cells pre-treated with PKCdelta RNAi relative to the scramble RNAi (Fig. 5a). In parallel, immunoblots indicated that Runx-2 signal was upregulated in scramble RNAi pretreated, mOSM-treated osteogenic-differentiation induced cultures 3 and 9 days post-differentiation relative to osteogenic medium treatment alone (Fig. 5a, b). Runx-2 induction was inhibited by pretreatment with specific PKCdelta RNAi (Fig. 5a, b). The RNAi experiments were performed on C57 Adip MPC derived from at least 2 separate isolations and showed identical results. Induction of osteogenesis as shown using Alizarin Red S staining indicated that PKCdelta RNAi pretreatment reduced mOSM-induced osteoid nodule formation relative to that observed in scramble RNAi control, mOSM-treated cultures (Fig. 5c). Quantitative measurement using QWin morphometric software of multiple imaged fields photographed per treatment indicated PKCdelta RNAi significantly reduced mOSM-stimulated nodule formation (Fig. 5d). Additionally, PKCdelta RNAi treatment reduced day 9 mOSM-induced alkaline phosphatase activity as compared to scramble RNAi treated mOSM-stimulated controls (Fig. 5e).

Oncostatin M-induced osteogenesis of C57Adip MPC is reduced by PKCdelta RNAi. C57AdipMPC cultured in triplicate were subjected to either PKCdelta-targeted RNAi or mock treated with scrambled RNAi oligomers (scr). Subsequently, cells were cultured in osteogenic medium alone or supplemented with mOSM as described in “Materials and Methods” as indicated. Protein lysates were collected at day 3 (a) and day 9 (b) post-differentiation, and resolved by SDS-PAGE. Immunoblotting of lysates were performed for PKCdelta, Runx-2 and actin. In (c), day 9 post-differentiation cultures were stained with Alizarin Red S and microphotographed fields were collected (scale bar 20 μm), while in (d), quantitation of stained area was carried out with QWin software. In (e), supernatants from day 9 post-differentiation cultures were measured for alkaline phosphatase. (*p < 0.01 relative to osteogenic medium alone with scramble RNAi (osteo scr RNAi); #p < 0.01 relative to osteogenic medium + mOSM with targeted PKCdelta RNAi
Discussion
An improved understanding of the molecular mechanisms contributing to osteogenic differentiation will enable the design of strategies either for improving bone maintenance and formation or for intervention in instances of tissue failure or improper deposition. While several factors stimulating osteogenic differentiation are known, their relative contributions and mechanisms through which they respectively induce bone lineage commitment are less well understood. Furthermore, other cytokines and growth factors may play roles that previously were underappreciated. Here, we present findings in a murine model of in vitro osteogenic differentiation indicating that OSM markedly increases osteogenesis of adipose-derived progenitor cells through a mechanism involving PKCdelta. Earlier studies indicated that OSM possessed osteogenic potential (Bellido et al. 1996; Jay et al. 1996); later, it was discovered in human adipose-derived mesenchymal progenitors OSM induced osteogenesis while inhibiting adipogenesis (Song et al. 2007), findings we also observed in murine adipose tissue-derived progenitors here (Figs. 1g–i, 2). Additionally, prior research using transgenic mice overexpressing bovine OSM found that OSM increased bone or osteoid formation (Malik et al. 1995). More recently, in human systems, OSM derived from activated peripheral blood monocytes was shown to induce osteogenesis in mesenchymal stem cells through mechanisms dependent in part on STAT3 (Guihard et al. 2012). It is probable that both PKCdelta and STAT3 contribute to OSM-induced osteogenesis in our mouse system.
Our study indicates that mOSM administration to mesenchymal progenitors enhances a commitment to osteogenic lineage, acting through the upregulation of Runx-2, a critical osteogenic transcription factor (Ducy et al. 1997; Otto et al. 1997), increased expression of BMPs, and increased expression of key structural proteins such as osteocalcin, a terminal marker of osteogenesis. Whether our results indicate an unconventional process of osteogenic differentiation or a process of differentiation unique to adipose-derived cell types remains to be determined. Additionally, although several studies indicate that the degree of osteogenic differentiation may differ between human mesenchymal progenitors derived from bone marrow and alternate reservoirs, we have not assessed the effects of OSM in murine bone marrow in direct comparison to adipose tissue. OSM does mediate PKCdelta activation in lung fibrobasts (Smyth et al. 2006) and in osteosarcoma cells (Chipoy et al. 2004). OSM is indeed expressed in bone marrow; however, further experimentation would be needed to determine if OSM mediates osteoblastogenesis in cells from bone marrow through a PKCdelta mechanism.
Many of the studies of OSM as a regulator of osteogenesis have centred mechanistically upon its induction of Janus kinase/signal transducers and activators of transcription (JAK/STAT) or mitogen-activated protein kinase (MAPK) pathways, in particular OSM induction of STAT3 and extracellular regulated kinase (ERK) 1/2 and ERK 5 (Chipoy et al. 2004; Miyaoka et al. 2006; Song et al. 2007). While these signaling intermediates are clearly induced by OSM, their individual contributions in stimulating osteogenic development have not yet been defined. All gp130 cytokines are capable of inducing STAT3 activation in cells that possess the relevant receptors. ERK signaling has been shown by several studies to be an inconsistent component to osteogenesis, where in early undifferentiated cell types it can promote bone formation, while in later stages of development it may inhibit differentiation (Schindeler and Little 2006). It may be that selection of cell type or the stimulating factor being studied might also influence the importance of ERK signaling to ongoing osteogenic responses. Therefore, we elected to focus upon more OSM-selective signaling intermediates, and in murine systems OSM appears to differentially regulate PKCdelta, a calcium-independent isoform of the PKC family of Ser/Thr kinases (Gschwendt 1999).
Previous work in our laboratory has shown PKCdelta to be required for maximal upregulation of OSM-induced genes such as IL-6, itself a molecule implicated in osteogenesis (Franchimont et al. 2005). Previous work by Chipoy and colleagues (Chipoy et al. 2004) indicated a potential role for PKCdelta in rat osteosarcoma cells and precommitted osteoblast lines, and assessed PKCdelta using rottlerin, an inhibitor-limited specificity of action. Our findings in the murine adipose tissue-derived precursor cell model presented here are consistent with this and show that the stable expression of PKCdelta and consequently its activation through OSM stimulation is a required event to induce maximal OSM-mediated osteoid deposition in vitro. Additionally, while PKCdelta expression at day 9 was restored in the RNAi experiments, Runx-2 protein levels remained at baseline/control levels, indicating that knockdown of PKCdelta levels early in osteogenic differentiation is sufficient to reduce mOSM-dependent effects. PKCdelta has been shown in other connective tissue cell types to affect gene expression of collagens (Fan et al. 2006; Mishra et al. 2007) as well as to alter their migratory capacity (Li et al. 2002; Kruger and Reddy 2003), factors which may be important for mesenchymal progenitor differentiation and homing as well as subsequent bone-forming potential. Additionally, in clinical subjects, PKCdelta has been found to be upregulated selectively in patients with scleroderma, a fibrotic disease with an underlying chronic inflammatory component (Jimenez et al. 2001). As OSM has been isolated in localized tissues from patients suffering chronic conditions such as joints in arthritis (Hui et al. 1997; Cawston et al. 1998) and atherosclerotic plaque in cardiovascular disease (Albasanz-Puig et al. 2011), its potential to modulate connective tissue responses through PKCdelta is a relevant consideration, particularly if it is capable of promoting aberrant responses such as calcification of connective tissue.
The mechanism responsible for OSM induction of PKCdelta in C57Adip MPC, and whether OSM-induced PKCdelta activity influences downstream intermediates and kinases upon osteogenic responses (i.e. STAT proteins or MAPK enzymes including ERK, p38 or JNK), remain to be examined. OSM induces multiple transcription factor families, including activator protein (AP)-1 as well as STATs and Sp1. Recently, it has been shown that zinc finger protein 467 (Zfp467) stimulates adipocyte formation while inhibiting osteoblast commitment in mouse stromal osteoblastic cells (Quach et al. 2011). Based on our findings, we predict that the differential effects of OSM on C57Adip MPC (Figs. 1, 2) cell fate may be through the inhibition of Zfp467. How PKCdelta regulates osteogenic transcription factors including Runx-2, characterized widely as the master regulatory transcription factor promoting osteogenesis, or the osteogenic cytokines BMP4 and BMP7, also remains to be determined. In conclusion, we have demonstrated in a murine adipose-derived mesenchymal cell line that OSM potentiation of osteogenesis and osteogenesis-associated gene product expression and activity are in part dependent upon its activation of PKCdelta. This identifies a novel mechanism in regulation of osteogenic commitment, and provokes interest into further assessment of OSM-mediated effects regulating mesenchymal lineage commitment and contributions to inflammatory disease.
References
Albasanz-Puig A, Murray J, Preusch M et al (2011) Oncostatin M is expressed in atherosclerotic lesions: a role for Oncostatin M in the pathogenesis of atherosclerosis. Atherosclerosis 216:292–298. doi:10.1016/j.atherosclerosis.2011.02.003
Bamber B, Reife RA, Haugen HS, Clegg CH (1998) Oncostatin M stimulates excessive extracellular matrix accumulation in a transgenic mouse model of connective tissue disease. J Mol Med (Berl) 76:61–69
Bellido T, Stahl N, Farruggella TJ et al (1996) Detection of receptors for interleukin-6, interleukin-11, leukemia inhibitory factor, oncostatin M, and ciliary neurotrophic factor in bone marrow stromal/osteoblastic cells. J Clin Invest 97:431–437. doi:10.1172/JCI118432
Cawston TE, Curry VA, Summers CA et al (1998) The role of oncostatin M in animal and human connective tissue collagen turnover and its localization within the rheumatoid joint. Arthritis Rheum 41:1760–1771. doi:10.1002/1529-0131(199810)41:10<1760::AID-ART8>3.0.CO;2-M
Chattopadhyay S, Tracy E, Liang P et al (2007) Interleukin-31 and oncostatin-M mediate distinct signaling reactions and response patterns in lung epithelial cells. J Biol Chem 282:3014–3026. doi:10.1074/jbc.M609655200
Chipoy C, Berreur M, Couillaud S et al (2004) Downregulation of osteoblast markers and induction of the glial fibrillary acidic protein by oncostatin M in osteosarcoma cells require PKCdelta and STAT3. J Bone Miner Res 19:1850–1861. doi:10.1359/JBMR.040817
De Bari C, Dell’Accio F, Tylzanowski P, Luyten FP (2001) Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum 44:1928–1942. doi:10.1002/1529-0131(200108)44:8<1928::AID-ART331>3.0.CO;2-P
Dennis JE, Merriam A, Awadallah A et al (1999) A quadripotential mesenchymal progenitor cell isolated from the marrow of an adult mouse. J Bone Miner Res 14:700–709. doi:10.1359/jbmr.1999.14.5.700
Ducy P, Zhang R, Geoffroy V et al (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89:747–754
Duncan MR, Hasan A, Berman B (1995) Oncostatin M stimulates collagen and glycosaminoglycan production by cultured normal dermal fibroblasts: insensitivity of sclerodermal and keloidal fibroblasts. J Invest Dermatol 104:128–133
Falconi D, Aubin JE (2007) LIF inhibits osteoblast differentiation at least in part by regulation of HAS2 and its product hyaluronan. J Bone Miner Res 22:1289–1300. doi:10.1359/jbmr.070417
Fan J, Guan S, Cheng C-F et al (2006) PKCdelta clustering at the leading edge and mediating growth factor-enhanced, but not ecm-initiated, dermal fibroblast migration. J Invest Dermatol 126:1233–1243. doi:10.1038/sj.jid.5700149
Franchimont N, Wertz S, Malaise M (2005) Interleukin-6: An osteotropic factor influencing bone formation? Bone 37:601–606. doi:10.1016/j.bone.2005.06.002
Grenier A, Combaux D, Chastre J et al (2001) Oncostatin M production by blood and alveolar neutrophils during acute lung injury. Lab Invest 81:133–141
Gschwendt M (1999) Protein kinase C delta. Eur J Biochem 259:555–564
Guihard P, Danger Y, Brounais B et al (2012) Induction of osteogenesis in mesenchymal stem cells by activated monocytes/macrophages depends on oncostatin M signaling. Stem Cells 30:762–772. doi:10.1002/stem.1040
Hui W, Bell M, Carroll G (1997) Detection of oncostatin M in synovial fluid from patients with rheumatoid arthritis. Ann Rheum Dis 56:184–187
Jaiswal RK, Jaiswal N, Bruder SP et al (2000) Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. J Biol Chem 275:9645–9652
Jay PR, Centrella M, Lorenzo J et al (1996) Oncostatin-M: a new bone active cytokine that activates osteoblasts and inhibits bone resorption. Endocrinology 137:1151–1158. doi:10.1210/endo.137.4.8625883
Jimenez SA, Gaidarova S, Saitta B et al (2001) Role of protein kinase C-delta in the regulation of collagen gene expression in scleroderma fibroblasts. J Clin Invest 108:1395–1403. doi:10.1172/JCI12347
Kang HJ, Kang JS, Lee SH et al (2005) Upregulation of oncostatin m in allergic rhinitis. Laryngoscope 115:2213–2216. doi:10.1097/01.mlg.0000187819.89889.4a
Kerr C, Langdon C, Graham F et al (1999) Adenovirus vector expressing mouse oncostatin M induces acute-phase proteins and TIMP-1 expression in vivo in mice. J Interferon Cytokine Res 19:1195–1205. doi:10.1089/107999099313145
Koch H, Jadlowiec JA, Campbell PG (2005) Insulin-like growth factor-I induces early osteoblast gene expression in human mesenchymal stem cells. Stem Cells Dev 14:621–631. doi:10.1089/scd.2005.14.621
Kruger JS, Reddy KB (2003) Distinct mechanisms mediate the initial and sustained phases of cell migration in epidermal growth factor receptor-overexpressing cells. Mol Cancer Res 1:801–809
Langdon C, Kerr C, Tong L, Richards CD (2003) Oncostatin M regulates eotaxin expression in fibroblasts and eosinophilic inflammation in C57BL/6 mice. J Immunol 170:548–555
Li WQ, Dehnade F, Zafarullah M (2001) Oncostatin M-induced matrix metalloproteinase and tissue inhibitor of metalloproteinase-3 genes expression in chondrocytes requires Janus kinase/STAT signaling pathway. J Immunol 166:3491–3498
Li W, Nadelman C, Gratch NS et al (2002) An important role for protein kinase C-delta in human keratinocyte migration on dermal collagen. Exp Cell Res 273:219–228. doi:10.1006/excr.2001.5422
Luzina IG, Atamas SP, Wise R et al (2003) Occurrence of an activated, profibrotic pattern of gene expression in lung CD8+ T cells from scleroderma patients. Arthritis Rheum 48:2262–2274. doi:10.1002/art.11080
Malaval L, Liu F, Vernallis AB, Aubin JE (2005) GP130/OSMR is the only LIF/IL-6 family receptor complex to promote osteoblast differentiation of calvaria progenitors. J Cell Physiol 204:585–593. doi:10.1002/jcp.20312
Malik N, Haugen HS, Modrell B et al (1995) Developmental abnormalities in mice transgenic for bovine oncostatin M. Mol Cell Biol 15:2349–2358
Mishra R, Zhu L, Eckert RL, Simonson MS (2007) TGF-beta-regulated collagen type I accumulation: role of Src-based signals. Am J Physiol Cell Physiol 292:C1361–C1369. doi:10.1152/ajpcell.00370.2006
Miyaoka Y, Tanaka M, Naiki T, Miyajima A (2006) Oncostatin M inhibits adipogenesis through the RAS/ERK and STAT5 signaling pathways. J Biol Chem 281:37913–37920. doi:10.1074/jbc.M606089200
Modur V, Feldhaus MJ, Weyrich AS et al (1997) Oncostatin M is a proinflammatory mediator. In vivo effects correlate with endothelial cell expression of inflammatory cytokines and adhesion molecules. J Clin Invest 100:158–168. doi:10.1172/JCI119508
Nakamura Y, Tensho K, Nakaya H et al (2005) Low dose fibroblast growth factor-2 (FGF-2) enhances bone morphogenetic protein-2 (BMP-2)-induced ectopic bone formation in mice. Bone 36:399–407. doi:10.1016/j.bone.2004.11.010
O’Hara KA, Kedda M-A, Thompson PJ, Knight DA (2003) Oncostatin M: an interleukin-6-like cytokine relevant to airway remodelling and the pathogenesis of asthma. Clin Exp Allergy 33:1026–1032
Okamoto H, Yamamura M, Morita Y et al (1997) The synovial expression and serum levels of interleukin-6, interleukin-11, leukemia inhibitory factor, and oncostatin M in rheumatoid arthritis. Arthritis Rheum 40:1096–1105. doi:10.1002/1529-0131(199706)40:6<1096::AID-ART13>3.0.CO;2-D
Otto F, Thornell AP, Crompton T et al (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89:765–771
Pittenger MF, Mackay AM, Beck SC et al (1999) Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147
Quach JM, Walker EC, Allan E et al (2011) Zinc finger protein 467 is a novel regulator of osteoblast and adipocyte commitment. J Biol Chem 286:4186–4198. doi:10.1074/jbc.M110.178251
Rickard DJ, Sullivan TA, Shenker BJ et al (1994) Induction of rapid osteoblast differentiation in rat bone marrow stromal cell cultures by dexamethasone and BMP-2. Dev Biol 161:218–228. doi:10.1006/dbio.1994.1022
Rodriguez A-M, Pisani D, Dechesne CA et al (2005) Transplantation of a multipotent cell population from human adipose tissue induces dystrophin expression in the immunocompetent mdx mouse. J Exp Med 201:1397–1405. doi:10.1084/jem.20042224
Schindeler A, Little DG (2006) Ras-MAPK signaling in osteogenic differentiation: friend or foe? J Bone Miner Res 21:1331–1338. doi:10.1359/jbmr.060603
Schnittker D, Kwofie K, Ashkar A et al (2013) Oncostatin M and TLR-4 ligand synergize to induce MCP-1, IL-6, and VEGF in human aortic adventitial fibroblasts and smooth muscle cells. Mediators Inflamm 2013:317503. doi:10.1155/2013/317503
Sekiya I, Larson BL, Smith JR et al (2002) Expansion of human adult stem cells from bone marrow stroma: conditions that maximize the yields of early progenitors and evaluate their quality. Stem Cells 20:530–541. doi:10.1634/stemcells.20-6-530
Smyth DC, Kerr C, Richards CD (2006) Oncostatin M-induced IL-6 expression in murine fibroblasts requires the activation of protein kinase Cdelta. J Immunol 177:8740–8747
Song HY, Jeon ES, Kim JI et al (2007) Oncostatin M promotes osteogenesis and suppresses adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells. J Cell Biochem 101:1238–1251. doi:10.1002/jcb.21245
Suga K, Saitoh M, Kokubo S et al (2003) Interleukin-11 acts synergistically with bone morphogenetic protein-2 to accelerate bone formation in a rat ectopic model. J Interferon Cytokine Res 23:203–207. doi:10.1089/107999003765027401
Tanaka M, Hirabayashi Y, Sekiguchi T et al (2003) Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood 102:3154–3162. doi:10.1182/blood-2003-02-0367
Walker EC, McGregor NE, Poulton IJ et al (2008) Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J Bone Miner Res 23:2025–2032. doi:10.1359/jbmr.080706
Walker EC, McGregor NE, Poulton IJ et al (2010) Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest 120:582–592. doi:10.1172/JCI40568
Walker EC, Poulton IJ, McGregor NE et al (2012) Sustained RANKL response to parathyroid hormone in oncostatin M receptor-deficient osteoblasts converts anabolic treatment to a catabolic effect in vivo. J Bone Miner Res 27:902–912. doi:10.1002/jbmr.1506
Wong S, Botelho FM, Rodrigues RM, Richards CD (2014) Oncostatin M overexpression induces matrix deposition, STAT3 activation, and SMAD1 Dysregulation in lungs of fibrosis-resistant BALB/c mice. Lab Invest. doi:10.1038/labinvest.2014.81
Zuk PA, Zhu M, Ashjian P et al (2002) Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13:4279–4295. doi:10.1091/mbc.E02-02-0105
Acknowledgments
The authors want to thank Christine Demers, Carrie M. Langdon, and Rebecca Rodrigues (McMaster University) for excellent technical assistance, and Steven Wong (McMaster University) for review of the manuscript. This work was supported by CIHR grant # 102565.
Conflict of Interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Smyth, D.C., Takenaka, S., Yeung, C. et al. Oncostatin M regulates osteogenic differentiation of murine adipose-derived mesenchymal progenitor cells through a PKCdelta-dependent mechanism. Cell Tissue Res 360, 309–319 (2015). https://doi.org/10.1007/s00441-014-2099-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-014-2099-y