
Abstract
Any opacification of the lens can be defined as cataracts, and lens epithelium cells play a crucial role in guaranteeing lens transparency by maintaining its homeostasis. Although several causative genes of congenital cataracts have been reported, the mechanisms underlying lens opacity remain unclear. In this study, a large family with congenital cataracts was collected and genetic analysis revealed a pathological mutation (c.3857 C > T, p.T1287I) in the GBF1 gene; all affected individuals in the family carried this heterozygous mutation, while unaffected family members did not. Functional studies in human lens epithelium cell line revealed that this mutation led to a reduction in GBF1 protein levels. Knockdown of endogenous GBF1 activated XBP1s in the unfolded protein response signal pathway, and enhances autophagy in an mTOR-independent manner. Heterozygous Gbf1 knockout mice also displayed typic cataract phenotype. Together, our study identified GBF1 as a novel causative gene for congenital cataracts. Additionally, we found that GBF1 deficiency activates the unfolded protein response and leads to enhanced autophagy, which may contribute to lens opacity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Any opacification of the lens can be defined as cataracts, which is usually caused by changes in protein constituents or microarchitecture of the lens, or both (Shiels and Hejtmancik 2019). Congenital cataracts are cataracts that occur at birth or develop early in life, constituting the most common cause of visual impairment in children (Beebe 2014; Chan et al. 2012; Zhao et al. 2015). The global prevalence of congenital cataract varies from 2.2 to 13.6 per 10,000 live births(Lenhart and Lambert 2022; Shiels and Hejtmancik 2019). Congenital cataracts can present as either bilateral or unilateral and may occur in isolation or in conjunction with anterior chamber developmental abnormalities such as microcornea and aniridia; additionally, they can be associated with systemic diseases, including metabolic and neurodevelopmental disorders (Maroofian et al. 2023; Shiels and Hejtmancik 2019; Wang et al. 2024). The etiologies of congenital cataracts are diverse, with genetic factors being the primary cause, accounting for 25% of known cases, and most of these variants are inherited as autosomal dominant traits (Li et al. 2020). Currently, more than 50 causative genes of congenital cataracts have been identified; however, congenital cataracts exhibit high heterogeneity, as different variants may result in the same cataract phenotype, while the same variant may also result in different cataract phenotypes; and more than one-third of inherited cataracts have not yet identified the causative genes (Shiels and Hejtmancik 2019, 2021). The mechanisms underlying congenital cataracts remain obscure.
Autophagy is a highly conserved process for cellular degradation and recycling, deficiency in autophagy is associated with various diseases, including cancer, neurodegenerative diseases, and inflammatory disorders (Levine and Kroemer 2019; Parzych and Klionsky 2014). Autophagy is also essential for intracellular quality control and the maintenance of lens transparency; and it involved in the degradation of organelles during the differentiation of lens epithelial cells into lens fibers (Basu et al. 2014; Liu et al. 2022; Morishita and Mizushima 2016). Atg5 and Pik3c3 are two important autophagy proteins and the specific knockout of Atg5 or Pik3c3 in the lens leads to cataracts in mice (Morishita et al. 2013). In humans, six causative genes for non-syndromic congenital cataracts have been reported to function in autophagy: CRYAB (Wignes et al. 2013), FYCO1 (Pankiv et al. 2010), CHMP4B (Sagona et al. 2014), GJA8 (Ping et al. 2021), PIKFYVE (Mei et al. 2022), and RRAGA (Chen et al. 2016). Variants in these genes influence autophagy in different manners and disrupt lens microarchitecture, ultimately leading to cataracts (Shiels and Hejtmancik 2019).
GBF1 (Golgi brefeldin A resistant guanine nucleotide exchange factor 1), locating in the cis-Golgi and endoplasmic reticulum-Golgi intermediate compartment (ERGIC), is a guanine nucleotide exchange factor of the Arf family. Its primary function is to activate ARF1, and it is also involved in COPI vesicle transport (Chia et al. 2021; Kaczmarek et al. 2017; Zou et al. 2021). Previous research has demonstrated that GBF1 is essential for the integrity of the Golgi apparatus (GA); inhibiting the function of GBF1 can cause dissociation and dispersal of the Golgi complex, resulting in the arrest of the secretion of soluble and membrane-anchored proteins, as well as triggering the unfolded protein response (UPR) and potentially causing cell death (Citterio et al. 2008; García-Mata et al. 2003; Ignashkova et al. 2017). GBF1 is also associated with autophagy; in osteoclasts, inhibiting GBF1 leads to Golgi fragmentation, which further activates the endoplasmic reticulum (ER) stress response and enhances autophagy activity in a Beclin1-dependent manner (Wen et al. 2021).
In this study, we identified a novel causative gene for congenital cataracts in a Chinese family, GBF1, which is inherited in an autosomal domain manner. All patients in this family carried the heterozygous mutation (c.3857 C > T, p.T1287I) in the GBF1 gene, while all asymptomatic family members did not. Using the human lens epithelium (HLE) cell line, we found that the p.T1287I mutation reduced GBF1 protein levels. Knockdown of endogenous GBF1 activated the unfolded protein response and enhanced autophagy, as well as increasing XBP1s protein levels and decreasing p-JNK1 protein levels. Heterozygous Gbf1 knockout (Gbf1+/-) mice also exhibited cataract malformation, while their littermate wild-type (Gbf1+/+) mice did not.
Materials and methods
Clinical and phenotypic analysis
The clinical history was obtained and individuals received ophthalmologic examination by ophthalmologists. The diagnosis of congenital cataract was based on lens opacity detected by slit-lamp microscope.
Genetic analysis
Five milliliters of peripheral blood samples were obtained from each family member. Genomic DNA extraction was carried out using the potassium acetate method. Genomic DNA form III:3, III:6, III:9, III:10, IV:2, IV:8, IV:7 underwent whole-exome sequencing. Exome capture was achieved using the SureSelect Human All Exons kit (Agilent Technologies, USA) and then sequenced using a HiSeq2000 sequencer (Illumina, USA) with 150-bp paired-end reads. SNPs were detected from high-quality reads after alignment to the human reference genome (hg19). Variants detection was restricted to the targeted exome regions and flanking regions within 200-bp of each exon. Variants located within intergenic, intronic, and UTR regions, as well as synonymous mutations, and those with a MAF (minor allele frequency) score exceeding 0.5%, were excluded. Pathogenic variants were further confirmed by Sanger sequencing, and the forward primer (5’-cccgctccctgccctaac-3’) and the reverse primer (5’-ttgcttatttgccctcatgctatt-3’) were used for amplifying exon 30 of the GBF1 gene as referenced in Transcript GBF1-201.
Plasmids and siRNAs
Full-length cDNA of the GBF1 gene was digested with NotI-HF (New England Biolabs, USA) and KpnI-HF (New England Biolabs, USA) and subsequently cloned into a p3xFLAG-CMV10 vector. The mutant GBF1 gene was generated using the Mut Express II Fast Mutagenesis Kit V2 (Vazyme, China). All plasmids were validated through enzyme digestion and Sanger sequencing. Control siRNA (siNC) that did not target any human gene, and siRNA specifically targeting human GBF1 (siGBF1) were obtained from Ribobio (Ribobio, China). The target sequence of siGBF1 was 5′-CGAAAUGCCCGAUGGAGCAtt-3’ (Selyunin et al. 2017).
Cell culture and transfection
The human lens epithelial(HLE, SRA 01–04)cell line used in this study is a stable cell line generated from primary cultured human lens epithelial cells transfected with a large T antigen of SV40 and immortalized (Ibaraki et al. 1998; Ping et al. 2021). HLE cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco, USA), and cultured at 37 °C under an atmosphere of 5% CO2. Plasmids and siRNAs were transfected using LipoFectMax (ABP Biosciences, USA) in Opti-MEM Medium (Gibco, USA) according to the manufacturer’s protocol.
Western blot
Forty-eight hours post-transfection, total proteins were extracted from HLE cells. Supernatants were collected by centrifugation at 12,000 g for 30 min, and 5 × loading buffer was added to each sample, followed by heating for 15 min at 95 °C. Total proteins were then separated on 8% or 15% SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE) gels based on their molecular weight and subsequently transferred to polyvinylidene difluoride (PVDF) membranes. Primary antibodies against FLAG, GBF1, p-mTOR, p62, Beclin1, and p-JNK1 were purchased from ABclonal (ABclonal, China); and primary antibodies against Vinculin, LC3, and β-actin were purchased from Proteintech (Proteintech, China). Cycloheximide (MEC, USA) was dissolved in DMSO at a concentration of 50 µg/µL. For cycloheximide chase assay, 24 h after post-transfection, cells were treated with 50 µg cycloheximide for 0, 6 and 12 h, and total proteins were extracted using the methods described above. Primary antibodies against FLAG for cycloheximide chase assay were purchased from Abcam (Abcam, UK). Total proteins from mice lenses were extracted using RIPA lysis buffer. The supernatants were subjected to sonication before centrifugation. Primary antibodies against mouse lens Gbf1 were purchased from Abcam (Abcam, UK).
RNA extracted and real time (RT)-PCR
Forty-eight hours post-transfection, total RNA was extracted using a Trizol kit (Takara, Japan) and reverse transcribed into cDNA using the HiScript II 1st Strand cDNA Synthesis Kit (Vazyme, China). The designed primer sequences for RT-PCR were shown in Supplementary Table 1.
Immunofluorescence
Cells were cultured on glass bottom cell culture dishes (NEST Biotechnology, China) and transfection was initiated 24 h later. Immunofluorescence was started 48 h post-transfection. Cells were rinsed three times with PBS, each for 5 min, and fixed in 4% paraformaldehyde (Biosharp, China) for 15 min at room temperature. Subsequently, cells were rinsed three times with PBS again and permeabilized with 0.5% Triton X-100 (Solarbio, China) in PBS for 20 min. Then, cells were blocked with 1% BSA (Biosharp, China) in TBST for 30 min at room temperature. Cells were incubated with primary antibodies diluted in 5% BSA overnight at 4 °C. After equilibration and reaching room temperature, cells were rinsed three times with PBST, each for 5 min, and incubated with Alexa Fluor-conjugated secondary antibody (Proteintech, China) in 5% BSA for 1 h at room temperature. Subsequently, cells were rinsed three times with PBST again, and stained with DAPI (1 µg/ml) dilute in PBS. Finally, Cells were rinsed three times with PBS again. Observations were performed using a laser scanning confocal microscope (Olympus FV3000, Japan).
Generation of Gbf1 knockout mice
The Gbf1 knockout mouse model was created by Shanghai Model Organisms Center, Inc. Two gRNAs were designed within intron 5 and intron 7 of the Gbf1 gene respectively (gaccaacttcccttaaggctggg and tgtagatagcaatagttctctgg), which led to a 1354-bp deletion comprising exon 6, exon 7, and their flanking intronic regions. Cas9 mRNA and gRNA were obtained by in vitro transcription and subsequently microinjected into the embryos of C57BL/6J mice at the pronuclear stage to obtain F0 mice. F1 mice were obtained through mating between F0 mice and C57BL/6J mice and were subsequently confirmed to carry the 1354-bp deletion by Sanger sequencing. The deletion would cause a frameshift and introduce a premature stop codon at the 143rd amino acid residue.
Slit lamp examination of mice
Ophthalmologists at Tongji Hospital of Tongji Medical College, Huazhong University of Science and Technology, immediately captured photographs after the mice were executed to confirm the phenotype. To eliminate the influence of temperature variations on the lens, mice were acclimated to the same environment as the slit lamp for a minimum of 6 h prior to photography.
Statistical analysis
All experiments were independently performed five times. The data is presented as the mean ± SEM. Statistical analysis was performed by using GraphPad Prism software (Version 7). The RT-PCR results were subjected to two-way test, while the Western Blot results were subjected to Student’s t-test. Data normalization was performed by comparing each data with its corresponding housekeeping proteins, such as β-Actin or Vinculin. Subsequently, the experimental group was compared with the control group to assess the differences. A p-value < 0.05 was deemed statistically significant (Motulsky 2016).
Results
Identification of a mutation in the GBF1 gene in a congenial cataract family
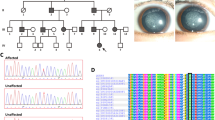
A large Chinese family consisting of 35 members was recruited and 11 members suffered from congenital cataracts (Fig. 1A). The proband was recruited in middle childhood (IV:3) who showed cataracts with microcornea. The youngest sufferer in this family exhibited (IV:8) cataracts with microcornea in infancy. Other sufferers in this family had no ocular abnormalities other than cataracts. Slit-lamp photographs showed severe cataracts of patient II:3, II:5, III:5, and III:10 (Fig. 1B). Detailed information for affected individuals were shown in Supplementary Table 2. To identify the causative gene of this family, DNA samples from five affected individuals (III:3, III:6, III:10, IV:2, IV:8) and two unaffected individuals (III:9, IV:7) were selected for whole-exome sequencing. None of these individuals carried the variants of known pathogenic genes of congenital cataracts, indicating that the cataract phenotype in this family might be due to a mutation in a novel causative gene of cataract.

Identification of the c.3857 C > T variant in the GBF1 gene in a family with congenial cataract. A Pedigree of a congenial cataract family. Affected individuals were depicted with filled circles and unaffected individuals were depicted with empty symbols. The proband was recruited in middle childhood and was indicated by a black arrow. B Slit-lamp photographs showing cataracts of patient II:3, II:5, III:5, and III:10. C DNA sequencing of unaffected individuals and affected individuals. The affected individuals carry the heterozygous variant (c.3857 C > T, p.T1287I) in the GBF1 gene. D The amino acid sequence alignment of the GBF1 protein from different species shows that the T1287 residue is highly conserved
The inheritance pattern of cataract in this family is autosomal dominant. Therefore, several heterozygous mutations present in affected individuals (III:3, III:6, III:10, IV:2, IV:8) but absent in unaffected individuals (III:9, IV:7) were identified from whole-exome sequencing results. These mutations were further validated by Sanger sequencing, and the results showed that only the heterozygous mutation in the GBF1 gene (c.3857 C > T, p.T1287I) co-segregated with the cataract phenotype in the family (Fig. 1C), as all the affected family members carried this heterozygous mutation while all the unaffected family members did not. Genomic DNA from 1000 unaffected individuals that unrelated to this family also subjected to Sanger sequencing and none of them carried the variant in the GBF1 gene. The c.3857 C > T mutation in the GBF1 gene is not reported in the ClinVar database; on the gnomAD database (v4.0.0), the allele frequency of p.T1287I in the GBF1 gene is 0.00006; and according to the dbSNP database, the MAF score of this mutation (rs200015962) in large populations is less than 0.001. Thus, the heterozygous mutation (c.3857 C > T, p.T1287I) in the GBF1 gene is a rare variant and it is not a common single nucleotide polymorphism (SNP). Additionally, the p.Thr1287 residue from different species shows it is highly conserved during evolution (Fig. 1D).
Gbf1 knockout mice exhibit cataract phenotypes
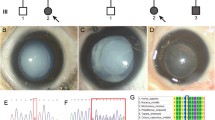
To further confirm the relevance of the GBF1 gene and congenital cataracts, Gbf1 knockout mice were generated through CRISPR/Cas9 gene-editing methods, resulting in a 1354-bp deletion comprising exon 6 and exon 7 with their flanking intronic regions (Fig. 2A). The Gbf1 knockout mice were successfully constructed (Supplementary Fig. 1A-C). Homozygous Gbf1 knockout (Gbf1-/-) mice were found to be embryonic lethal, whereas heterozygous Gbf1 knockout (Gbf1+/-) mice exhibited normal growth and reproduction; these observations align with the data from the MGI (Mouse Genome Informatics) database, which reported that preweaning lethality with complete penetrance in homozygous Gbf1tm1a(KOMP)Wtsi mice (https://www.informatics.jax.org/). Using the OCT (Optical Coherence Tomography) system, both two- and five-month-old Gbf1+/- mice exhibited the cataract phenotype, while littermate Gbf1+/+ mice did not; and the cataract phenotype was notably more pronounced in the five-month-old Gbf1+/- mice (Fig. 2B). Upon slit lamp examination, Gbf1+/- mice showed cataract phenotype, whereas littermate wild-type (Gbf1+/+) mice did not (Supplementary Fig. 1D). While a low penetrance of cataract was observed in one-month-old Gbf1+/- mice (two of the six had exhibited opacification in the lens), nearly all two-month-old Gbf1+/- mice exhibited the cataract phenotype (Fig. 2C). The Gbf1 protein expression levels in lenses of the Gbf1+/- mice were significantly decreased (Supplementary Fig. 1E). RT-PCR results showed a significant increase in Grp78 expression in lenses of the Gbf1+/- mice, indicating the activation of the UPR in mice lens epithelial cells (Supplementary Fig. 1F). Hematoxylin and eosin (HE) staining was used to observe the lenses in both Gbf1+/+ and Gbf1+/-, both groups exhibited normal lens structure and the regular migration of lens epithelial cells from the capsule to the equator (Fig. 2D). However, increased numbers of autophagosomes were observed in lens epithelial cells of Gbf1+/- mice (Fig. 2E). Furthermore, lens epithelial fiber cells of Gbf1+/- mice exhibited abnormal misalignment morphology (Fig. 2F), implying that the microarchitecture of the lenses were disrupted. The presence of cataracts in Gbf1+/- mice further ensured GBF1 as a causative gene for congenital cataracts in humans. Moreover, the enhanced autophagy may be involved in the destruction of the lens microarchitecture, ultimately resulting in lens opacification.

Gbf1 knockout mice exhibited cataract phenotype. A Generation of Gbf1 knockout mouse. Schematic structure of the exon-intron genomic structure of Gbf1 and variant site created with the CRISPR/Cas9 system. Two gRNAs were designed within intron 5 and intron 7 of the Gbf1 gene, which led to a 1354 bp deletion comprising exon 6 and exon 7 and their flanking intronic regions. B OCT scanning results. White dashed ovals indicated opacity observed in the lens. Both two- and five-month-old Gbf1+/− mice exhibited the cataract phenotype, with the latter displaying a more pronounced phenotype. C Incidence of cataract at indicated ages (M = month). D HE staining showed there were no abnormalities in the lenses of Gbf1+/− mice. E Transmission electron microscope (TEM) of Gbf1 knockout mice lenses. The red arrows point to autophagosomes. F TEM of mice lenses showed abnormal morphology of lens epithelial fiber cells in Gbf1+/− mice
Effect of the p.T1287I mutation on GBF1 protein in HLE cells
To access the effect of p.T1287I mutation, Flag-tagged expression vectors of wild-type and mutant-type GBF1 were constructed and transfected into HLE cells. Western blot analysis revealed a reduction in GBF1 protein levels duo to the p.T1287I mutation (Fig. 3A). The reduction in p.T1287I GBF1 protein levels was attributed to the instability of the mutant-type proteins, as cycloheximide chase assay results showed that the mutant-type protein is degraded more rapidly than the wild-type protein (Fig. 3B). Immunofluorescence showed that the mutation did not alter the subcellular localization of GBF1 protein (Fig. 3C). AlphaFold2 was used to evaluate the structure of wild-type and mutant-type GBF1 protein, revealing that the p.T1287I mutation does not appear to affect the conserved α-helix structure of the GBF1 protein (Fig. 3D). This mutation does not localize in the active center, it may influence the interaction of GBF1 with other proteins as it may exert an influence on the loop structure. As the main function of GBF1 is to activate ARF1 and involved in COPI vesicle transport (Chia et al. 2021; Kaczmarek et al. 2017; Zou et al. 2021), the p.T1287I mutation may impact the normal function of Golgi apparatus.

Deficiency of GBF1 results in Golgi apparatus fragmentation and UPR in HLE cells. A Left, western blot analysis of wild-type and mutant-type GBF1 in HLE cells; Right, statistical analysis showed the T1287I variant in GBF1 significantly reduced its protein levels (n = 5; *represents p < 0.05). B Left, western blot analysis of wild-type and mutant-type GBF1 in HLE cells treated with cycloheximide (CHX) for 0, 6, and 12 h. Right, statistical analysis showed the T1287I variant in GBF1 reduced the stability of the mutant-type proteins (n = 3; **represents p < 0.01). C The p.T1287I variant did not affect the localization of GBF1 protein in HLE cells. GBF1 was detected using an antibody against the FLAG tag. Blue, DAPI; Red, FLAG. D The protein structure of wild-type and mutant-type GBF1. Top, alignment of wild-type protein structure (yellow) and mutant-type protein structure (cyan); Bottom, the p.T1287 residue is shown as a red color and the p.I1287 residue is shown as a green color. The p.T1287I variant does not affect the conserved α-helix structure of the GBF1 protein. E The protein expression levels of GBF1 were effectively knockdown by GBF1 siRNA. Left, Western blot analysis of protein levels of GFB1 in HLE cells transfected with siNC and siGBF1; Right, statistical analysis showed protein levels of GBF1 were significantly reduced (n = 5, *represents p < 0.05). F Immunofluorescence of GM130 in HLE cells. In cells transfected with siNC, Golgi apparatus aggregates around the nuclei; and in cells transfected with siGBF1, Golgi apparatus were fragmentation. Blue, DAPI; Green, GM130. G Knockdown of GBF1 could activate UPR. RT-PCR analysis of mRNA expression levels of GBF1, GRP78, XBP1s and ATF4 (n = 5, ***represents p < 0.001, **represents p < 0.01, ns represents not significant). H Left, western blot analysis of protein levels of XBP1s. Right, statistical analysis showed protein levels of XBP1s were significantly increased (n = 5, *represents p < 0.05). I Left, western blot analysis of protein levels of activated ATF6; Right, statistical analysis showed protein levels of activated ATF6 were significantly increased (n = 5, **represents p < 0.01)
Knockdown of GBF1 results in Golgi apparatus fragmentation and UPR in HLE cells
Given that the p.T1287I mutation reduces the GBF1 protein levels, and considering the universal expression of GBF1 across various cell types; thus, to assess the effects of reduced GBF1 protein on HLE cells, we utilized siGBF1 to knockdown the endogenous GBF1 protein (Fig. 3E). Although GBF1 is involved in COPI transport and its deficiency may destroy the integrity of GA, but previous studies have shown that disrupting the function of GBF1 can yield varying effects on GA in different cell lines (Lopes-da-Silva et al. 2019; Selyunin et al. 2017). Therefore, we evaluated the effects of GBF1 deficiency on GA in HLE cell. Immunofluorescence staining for GM130 were performed and found fragmentations of GA in GBF1 deficiency HLE cells, as it became more dispersed throughout the cytosol and exhibited perinuclear punctate structures (Fig. 3F). As GA fragmentation can trigger the UPR, thus, RT-PCR was performed in GBF1 deficiency HLE cells and found a significant increase in GRP78 mRNA levels, indicating the activation of the UPR; moreover, mRNA levels of XBP1s were increased while ATF4 were not altered, suggesting the activation of IRE1α-XBP1s pathway rather than PERK-ATF4 pathway (Fig. 3G). Western blots were performed to further evaluate the UPR signaling pathway and found increased levels of XBP1s protein, providing additional evidence of XBP1s activation (Fig. 3H). In addition, protein levels of 50KDa-ATF6 were also increased, suggesting the activation of ATF6 pathway (Fig. 3I). Together, these results revealed that GBF1 deficiency leads to GA fragmentation and induces UPR, activating the IRE1α-XBP1s pathway and ATF6 pathway in HLE cells.
Knockdown of GBF1 leads to autophagy but not apoptosis in HLE cells
As the UPR can induce apoptosis through the PERK-ATF4-CHOP pathway, and given that ATF4 was not activated in GBF1 deficiency HLE cells. Thus, TUNEL staining were performed to confirm if there were apoptosis or not, and the results showed that, knockdown of GBF1 did not induce apoptosis; this observation was consistent with the protein levels of CHOP, which also remained unaltered; in fact, direct cell counting after Trypan blue staining revealed no signs of cell death even 72 h post-transfection; this result was further confirmed by mRNA levels of p53, which also showed no significant alterations in GBF1 knockdown cells (Supplementary Fig. 2). As continuous UPR can also induce autophagy, and lenses of Gbf1+/- mice exhibited an increased number of autophagosomes, we utilized pCMV-GFP-LC3 plasmids to assess autophagy and found aggregations of LC3 in GBF1 knockdown cells (Fig. 4A). Western blots further confirm the induction of autophagy, with increased protein levels of LC3II and decreased protein levels of p62 (Fig. 4B and C), suggesting an enhancement of autophagic flux due to GBF1 deficiency. Collectively, these data demonstrate that knockdown of GBF1 in HLE cells does not induce apoptosis or cell death, but induce autophagy.

Knockdown of GBF1 enhance autophagy in an mTOR independent manner in HLE cells. A Immunofluorescence of LC3 in HLE cells. Left, the GFP-tag LC3 were not gathered in HLE cells transfected with control siRNA; Right, the GFP-LC3 dots were formed in HLE cells transfected with siGBF1. Green, GFP. B Left, western blot analysis of protein levels of LC3II; Right, statistical analysis showed protein levels of LC3II were significantly increased (n = 5, **represents p < 0.01). C Left, western blot analysis of protein levels of p62; Right, statistical analysis showed protein levels of p62 were significantly decreased (n = 5, *represents p < 0.05). D GFP-RFP-LC3 was expressed in HLE cells transfected with siNC and siGBF1 to monitor LC3 puncta. Bottom, Number of GFP, RFP and RFP + GFP- puncta (autolysosomes) was quantified (n = 10 cells from three independent experiments). E Protein levels of p-mTOR and mTOR. The statistical analysis showed protein levels of p-mTOR and mTOR were not altered (n = 5, ns represents not significant). F Left, western blot analysis of protein levels of Beclin1; Right, statistical analysis showed protein levels of Beclin1 were not significantly altered (n = 5, ns represents not significant). G Left, western blot analysis of protein levels of p-PI3K; Right, statistical analysis showed protein levels of p-PI3K were not significantly altered (n = 5, ns represents not significant). H Left, western blot analysis of protein levels of p-JNK1. Right, statistical analysis showed protein levels of p-JNK1were significantly decreased (n = 5, *represents p < 0.05). I Left, western blot analysis of protein levels of ATG5; Right, statistical analysis showed protein levels of ATG5 were significantly increased (n = 5, *represents p < 0.05)
Knockdown of GBF1 leads to autophagy in an mTOR-independent manner in HLE cells
The GFP-RFP-LC3 reporter experiment provided further confirmation of the increased autophagic flux (Fig. 4D). To figure out the underlying mechanism of enhanced autophagy induced by GBF1 deficiency, expression levels of autophagy-related proteins were analyzed. Protein levels of mTOR, p-mTOR, p-PI3K, and Beclin1 were assessed; however, no significant differences were observed (Fig. 4E-G). Thus, in HLE cells, GBF1 involved in autophagy in an mTOR-independent manner. The protein levels of p-JNK1 were further accessed and a significant reduction was observed (Fig. 4H). ATG5 is an essential protein for autophagy elongation, its protein levels were also evaluated, and found a significant increase in GBF1 knockdown cells (Fig. 4I). In conclusion, our findings suggest that in GBF1 knockout HLE cells, increased autophagic flux is regulated by JNK1 and ATG5, rather than p-mTOR and Beclin1.
Discussion
In this study, we identified GBF1 as a novel causative gene of congenital cataracts inherited in an autosomal dominant manner. The mouse model further verified our findings, as Gbf1+/- mice developed cataract phenotypes at an early age, thereby establishing a clear relationship between GBF1 and the cataract phenotype. Using HLE cells, we found that GBF1 deficiency activated the UPR and enhanced autophagy, as well as increasing XBP1s protein levels and decreasing p-JNK1 protein levels.
The p.T1287I mutation identified in our study is a loss-of-function mutation, as it has been shown to reduce the protein levels of GBF1. This conclusion is supported by other studies. In zebrafish, the p.L1246R mutation, another variant in the gbf1 gene, was also demonstrated to be a loss-of-function mutation as microinjection of L1246R mRNA into wild-type zebrafish embryos did not result in abnormalities, nor did it rescue the abnormal phenotype of the mutant zebrafish embryos (Chen et al. 2017). Additionally, Bartha et al. have indicated that if a targeted knockout gene is homozygous and embryonic lethal, the heterozygous mice will carry the loss-of-function genotypes (Bartha et al. 2018). Therefore, as homozygous Gbf1 knockout (Gbf1-/-) mice were observed to be embryonic lethal, the Gbf1+/- mice were insufficient for the normal function of the Gbf1 gene.
Both increased autophagy and decreased levels of autophagy have been reported to be associated with congenital cataracts. While variants in ATG5 and PIK3C3, crucial genes in autophagy, have not yet been reported to cause congenital cataracts in humans, loss-of-function mutation of Atg5 and Pik3c3 have been shown to inhibit autophagy and lead to cataracts in mice (Morishita et al. 2013). Variants in the PIKFYV gene and TDRD7 gene have been identified to cause syndromic congenital cataracts in human; and the lenses of pikfyve-/- zebrafish exhibited increased autophagy lysosomes and bafilomycin A1 alleviated the cataract phenotype (Mei et al. 2022); additionally, the Tdrd7−/− mice exhibited increased autophagosomes in the lens with increased LC3II expression levels (Tu et al. 2021). These results were consistent with the observed increase in autophagosomes in the lenses observed in Gbf1+/- mice.
Our research has revealed that in HLE cells, GBF1 deficiency triggers the activation of XBP1s, which may act as an inhibitor of JNK1 to regulate autophagy. A previous study demonstrated that the activation of XBP1s can increase autophagy levels (Margariti et al. 2013). In addition, in A549 cells, JNK has been shown to be a downstream effector of XBP1s, and knockdown of XBP1s leads to reduced phosphorylation of JNK, resulting in decreased cell proliferation and cell viability (Jiang et al. 2022). In fact, JNK1 is a bilateral regulator of autophagy, it can act as a positive regulator that increase autophagy levels (Zhou et al. 2015), and can also act as a negative regulator of autophagy that decrease autophagy levels (Xu et al. 2011). During lens development of the chick, the inactivation of JNK promotes autophagy by inhibiting the activation of mTOR and RPTOR, accelerating the formation of the organelle-free zone (Basu et al. 2014). Our study suggests that JNK1 may act as a negative regulator of autophagy and plays a role in maintaining the microarchitecture of the lens.
In conclusion, we identified a novel causative gene for congenital cataracts, broadening the phenotypic and genetic spectrum of the GBF1 gene, which would assist in precision medicine and facilitate prenatal diagnosis. And our findings illustrate that GBF1 deficiency would increase autophagy autophagic flux, providing valuable insights into the underlying pathogenesis of congenital cataracts.
Data availability
No datasets were generated or analysed during the current study.
References
Bartha I, di Iulio J, Venter JC, Telenti A (2018) Human gene essentiality. Nat Rev Genet 19:51–62. https://doi.org/10.1038/nrg.2017.75
Basu S, Rajakaruna S, Reyes B, Van Bockstaele E, Menko AS (2014) Suppression of MAPK/JNK-MTORC1 signaling leads to premature loss of organelles and nuclei by autophagy during terminal differentiation of lens fiber cells. Autophagy 10:1193–1211. https://doi.org/10.4161/auto.28768
Beebe DC (2014) The physiology and pathobiology of the Lens. In: McManus LM, Mitchell RN (eds) Pathobiology of Human Disease. Academic, San Diego, pp 2072–2083
Chan WH, Biswas S, Ashworth JL, Lloyd IC (2012) Educational paper. Eur J Pediatrics 171:625–630. https://doi.org/10.1007/s00431-012-1700-1
Chen JH, Huang C, Zhang B, Yin S, Liang J, Xu C, Huang Y, Cen LP, Ng TK, Zheng C, Zhang S, Chen H, Pang CP, Zhang M (2016) Mutations of RagA GTPase in mTORC1 pathway are Associated with autosomal Dominant cataracts. PLoS Genet 12:e1006090. https://doi.org/10.1371/journal.pgen.1006090
Chen J, Wu X, Yao L, Yan L, Zhang L, Qiu J, Liu X, Jia S, Meng A (2017) Impairment of Cargo Transportation caused by gbf1 mutation disrupts Vascular Integrity and causes hemorrhage in zebrafish embryos. J Biol Chem 292:2315–2327. https://doi.org/10.1074/jbc.M116.767608
Chia J, Wang SC, Wee S, Gill DJ, Tay F, Kannan S, Verma CS, Gunaratne J, Bard FA (2021) Src activates retrograde membrane traffic through phosphorylation of GBF1. Elife 10. https://doi.org/10.7554/eLife.68678
Citterio C, Vichi A, Pacheco-Rodriguez G, Aponte AM, Moss J, Vaughan M (2008) Unfolded protein response and cell death after depletion of brefeldin A-inhibited guanine nucleotide-exchange protein GBF1. Proc Natl Acad Sci U S A 105:2877–2882. https://doi.org/10.1073/pnas.0712224105
García-Mata R, Szul T, Alvarez C, Sztul E (2003) ADP-ribosylation factor/COPI-dependent events at the endoplasmic reticulum-golgi interface are regulated by the guanine nucleotide exchange factor GBF1. Mol Biol Cell 14:2250–2261. https://doi.org/10.1091/mbc.e02-11-0730
Ibaraki N, Chen SC, Lin LR, Okamoto H, Pipas JM, Reddy VN (1998) Human lens epithelial cell line. Exp Eye Res 67:577–585. https://doi.org/10.1006/exer.1998.0551
Ignashkova TI, Gendarme M, Peschk K, Eggenweiler HM, Lindemann RK, Reiling JH (2017) Cell survival and protein secretion associated with Golgi integrity in response to Golgi stress-inducing agents. Traffic 18:530–544. https://doi.org/10.1111/tra.12493
Jiang H, Jiang Q, He Y, Li X, Xu Y, Liu X (2022) XBP1s promotes the development of lung adenocarcinoma via the p–JNK MAPK pathway. Int J Mol Med 49. https://doi.org/10.3892/ijmm.2022.5089
Kaczmarek B, Verbavatz JM, Jackson CL (2017) GBF1 and Arf1 function in vesicular trafficking, lipid homoeostasis and organelle dynamics. Biol Cell 109:391–399. https://doi.org/10.1111/boc.201700042
Lenhart PD, Lambert SR (2022) Current management of infantile cataracts. Surv Ophthalmol 67:1476–1505. https://doi.org/10.1016/j.survophthal.2022.03.005
Levine B, Kroemer G (2019) Biological functions of Autophagy genes: a Disease Perspective. Cell 176:11–42. https://doi.org/10.1016/j.cell.2018.09.048
Li J, Chen X, Yan Y, Yao K (2020) Molecular genetics of congenital cataracts. Exp Eye Res 191:107872. https://doi.org/10.1016/j.exer.2019.107872
Liu Z, Huang S, Zheng Y, Zhou T, Hu L, Xiong L, Li DW, Liu Y (2022) The lens epithelium as a major determinant in the development, maintenance, and regeneration of the crystalline lens. Prog Retin Eye Res: 101112. https://doi.org/10.1016/j.preteyeres.2022.101112
Lopes-da-Silva M, McCormack JJ, Burden JJ, Harrison-Lavoie KJ, Ferraro F, Cutler DF (2019) A GBF1-Dependent mechanism for environmentally responsive regulation of ER-Golgi transport. Dev Cell 49:786–801e6. https://doi.org/10.1016/j.devcel.2019.04.006
Margariti A, Li H, Chen T, Martin D, Vizcay-Barrena G, Alam S, Karamariti E, Xiao Q, Zampetaki A, Zhang Z, Wang W, Jiang Z, Gao C, Ma B, Chen YG, Cockerill G, Hu Y, Xu Q, Zeng L (2013) XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J Biol Chem 288:859–872. https://doi.org/10.1074/jbc.M112.412783
Maroofian R, Kaiyrzhanov R, Cali E, Zamani M, Zaki MS, Ferla M, Tortora D, Sadeghian S, Saadi SM, Abdullah U, Karimiani EG, Efthymiou S, Yeşil G, Alavi S, Al Shamsi AM, Tajsharghi H, Abdel-Hamid MS, Saadi NW, Al Mutairi F, Alabdi L, Beetz C, Ali Z, Toosi MB, Rudnik-Schöneborn S, Babaei M, Isohanni P, Muhammad J, Khan S, Al Shalan M, Hickey SE, Marom D, Elhanan E, Kurian MA, Marafi D, Saberi A, Hamid M, Spaull R, Meng L, Lalani S, Maqbool S, Rahman F, Seeger J, Palculict TB, Lau T, Murphy D, Mencacci NE, Steindl K, Begemann A, Rauch A, Akbas S, Aslanger AD, Salpietro V, Yousaf H, Ben-Shachar S, Ejeskär K, Al Aqeel AI, High FA, Armstrong-Javors AE, Zahraei SM, Seifi T, Zeighami J, Shariati G, Sedaghat A, Asl SN, Shahrooei M, Zifarelli G, Burglen L, Ravelli C, Zschocke J, Schatz UA, Ghavideldarestani M, Kamel WA, Van Esch H, Hackenberg A, Taylor JC, Al-Gazali L, Bauer P, Gleeson JJ, Alkuraya FS, Lupski JR, Galehdari H, Azizimalamiri R, Chung WK, Baig SM, Houlden H, Severino M (2023) Biallelic MED27 variants lead to variable ponto-cerebello-lental degeneration with movement disorders. Brain 146:5031–5043. https://doi.org/10.1093/brain/awad257
Mei S, Wu Y, Wang Y, Cui Y, Zhang M, Zhang T, Huang X, Yu S, Yu T, Zhao J (2022) Disruption of PIKFYVE causes congenital cataract in human and zebrafish. Elife 11. https://doi.org/10.7554/eLife.71256
Morishita H, Mizushima N (2016) Autophagy in the lens. Exp Eye Res 144:22–28. https://doi.org/10.1016/j.exer.2015.08.019
Morishita H, Eguchi S, Kimura H, Sasaki J, Sakamaki Y, Robinson ML, Sasaki T, Mizushima N (2013) Deletion of autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem 288:11436–11447. https://doi.org/10.1074/jbc.M112.437103
Motulsky HJ (2016) GraphPad Statistics Guide. http://www.graphpad.com/guides/prism/10/statistics/index.htm
Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjørkøy G, Johansen T (2010) FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol 188:253–269. https://doi.org/10.1083/jcb.200907015
Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20:460–473. https://doi.org/10.1089/ars.2013.5371
Ping X, Liang J, Shi K, Bao J, Wu J, Yu X, Tang X, Zou J, Shentu X (2021) Rapamycin relieves the cataract caused by ablation of Gja8b through stimulating autophagy in zebrafish. Autophagy 17:3323–3337. https://doi.org/10.1080/15548627.2021.1872188
Sagona AP, Nezis IP, Stenmark H (2014) Association of CHMP4B and autophagy with micronuclei: implications for cataract formation. Biomed Res Int 2014:974393. https://doi.org/10.1155/2014/974393
Selyunin AS, Iles LR, Bartholomeusz G, Mukhopadhyay S (2017) Genome-wide siRNA screen identifies UNC50 as a regulator of Shiga toxin 2 trafficking. J Cell Biol 216:3249–3262. https://doi.org/10.1083/jcb.201704015
Shiels A, Hejtmancik JF (2019) Biology of inherited cataracts and opportunities for treatment. Annu Rev Vis Sci 5:123–149. https://doi.org/10.1146/annurev-vision-091517-034346
Shiels A, Hejtmancik JF (2021) Inherited cataracts: genetic mechanisms and pathways new and old. Exp Eye Res 209:108662. https://doi.org/10.1016/j.exer.2021.108662
Tu C, Li H, Liu X, Wang Y, Li W, Meng L, Wang W, Li Y, Li D, Du J, Lu G, Lin G, Tan YQ (2021) TDRD7 participates in lens development and spermiogenesis by mediating autophagosome maturation. Autophagy 17:3848–3864. https://doi.org/10.1080/15548627.2021.1894058
Wang Q, Wang D, Qin T, Zhang X, Lin X, Chen J, Chen W, Zhao L, Huang W, Lin Z, Li J, Dongye M, Wu X, Wang X, Li X, Lin Y, Tan H, Liu Y, Lin H, Chen W (2024) Early diagnosis of syndromic congenital cataracts in a large cohort of congenital cataracts. Am J Ophthalmol 263:206–213. https://doi.org/10.1016/j.ajo.2023.10.022
Wen C, Zhou Y, Xu Y, Tan H, Pang C, Liu H, Liu K, Wei L, Luo H, Qin T, He C, Liu C, Zhou C (2021) The Regulatory Role of GBF1 on Osteoclast activation through EIF2a mediated ER stress and novel marker FAM129A induction. Front Cell Dev Biol 9:706768. https://doi.org/10.3389/fcell.2021.706768
Wignes JA, Goldman JW, Weihl CC, Bartley MG, Andley UP (2013) p62 expression and autophagy in αB-crystallin R120G mutant knock-in mouse model of hereditary cataract. Exp Eye Res 115:263–273. https://doi.org/10.1016/j.exer.2013.06.026
Xu P, Das M, Reilly J, Davis RJ (2011) JNK regulates FoxO-dependent autophagy in neurons. Genes Dev 25:310–322. https://doi.org/10.1101/gad.1984311
Zhao L, Chen XJ, Zhu J, Xi YB, Yang X, Hu LD, Ouyang H, Patel SH, Jin X, Lin D, Wu F, Flagg K, Cai H, Li G, Cao G, Lin Y, Chen D, Wen C, Chung C, Wang Y, Qiu A, Yeh E, Wang W, Hu X, Grob S, Abagyan R, Su Z, Tjondro HC, Zhao XJ, Luo H, Hou R, Jefferson J, Perry P, Gao W, Kozak I, Granet D, Li Y, Sun X, Wang J, Zhang L, Liu Y, Yan YB, Zhang K (2015) Lanosterol reverses protein aggregation in cataracts. Nature 523:607–611. https://doi.org/10.1038/nature14650
Zhou YY, Li Y, Jiang WQ, Zhou LF (2015) MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep 35. https://doi.org/10.1042/bsr20140141
Zou YJ, Shan MM, Pan ZN, Pan MH, Li XH, Sun SC (2021) Loss of Arf Guanine Nucleotide Exchange factor GBF1 activity disturbs Organelle Dynamics in Mouse oocytes. Microsc Microanal 27:400–408. https://doi.org/10.1017/s1431927620024885
Acknowledgements
We would like to thank Prof. Jiahuai Han from Xiamen University and Prof. Rongjia Zhou from Wuhan University for their kindly donating of plasmids. And we would also like to thank Prof. Qing Wang from Huazhong University of Science and Technology for his kindly donating of HLE cells.
Funding
This work was supported by National Key R&D Program of China (2023YFC2706302), the National Natural Science Foundation of China (81000079, 81170165, and 81870959), the Program for HUST Academic Frontier Youth Team (2016QYTD02), and the Fundamental Research Funds for the Central Universities (HUST: 2019JYCXJJ035).
Author information
Authors and Affiliations
Contributions
WMJ, CMZ, YLL, JG, CY, DZZ, XPZ, and YYT performed experiments. CMZ and GGL took clinical care of the patients and provided advice. WMJ, CMZ, YLL, and JG. analyzed and interpreted data. JG, SW, ZC, and GGL performed slit-lamp microscope examination and confirmed the cataract phenotype of mice. WMJ wrote the manuscript. XQZ led and supervised the work and designed the experiments. All authors edited the paper with input from the other authors, and approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the ethics committee on human subject research at Huazhong University of Science and Technology (2019S1160) and all animal procedures and experiments were approved by the Animal Care Committee of Huazhong University of Science and Technology (S672). All these studies were conformed with the Helsinki Declaration of 1975 (as revised in 2008) concerning Human and Animal Rights.
Consent to participate and to publish
Written informed consent was obtained from all patients or their caregivers for genetic testing and the publication of article.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jia, W., Zhang, C., Luo, Y. et al. GBF1 deficiency causes cataracts in human and mouse. Hum. Genet. (2024). https://doi.org/10.1007/s00439-024-02697-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00439-024-02697-8