
Abstract
Systemic Lupus Erythematosus (SLE) is an autoimmune disease with heterogeneous manifestations, including neurological and psychiatric symptoms. Genetic association studies in SLE have been hampered by insufficient sample size and limited power compared to many other diseases. Multiple Sclerosis (MS) is a chronic relapsing autoimmune disease of the central nervous system (CNS) that also manifests neurological and immunological features. Here, we identify a method of leveraging large-scale genome wide association studies (GWAS) in MS to identify novel genetic risk loci in SLE. Statistical genetic comparison methods including linkage disequilibrium score regression (LDSC) and cross-phenotype association analysis (CPASSOC) to identify genetic overlap in disease pathophysiology, traditional 2-sample and novel PPI-based mendelian randomization to identify causal associations and Bayesian colocalization were applied to association studies conducted in MS to facilitate discovery in the smaller, more limited datasets available for SLE. Pathway analysis using SNP-to-gene mapping identified biological networks composed of molecular pathways with causal implications for CNS disease in SLE specifically, as well as pathways likely causal of both pathologies, providing key insights for therapeutic selection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Systemic Lupus Erythematosus (SLE) is a female-dominant autoimmune disease with diverse manifestations across patient populations, including immune dysfunction and systemic inflammation. SLE is characterized by production of various autoantibodies, upregulation of both the innate and adaptive immune systems, and disturbed cytokine balance (Gupta and Kaplan 2021). SLE is associated with the additional risk of developing a range of comorbidities affecting the cardiovascular system, skin, musculoskeletal, kidney and nervous system, among others (Chen and Lee 2022).
Genome wide association studies (GWAS) have emerged as an important tool to investigate complex polygenic autoimmune diseases. As with many analyses relying on population-level statistics, GWAS sample size, defined by the total number of cases and controls, is a key factor in the ability to detect robust disease associations (Uffelmann et al. 2021). Despite the success of candidate genes, Immunochip, and GWAS in mapping SLE susceptibility loci, genetic studies in lupus have been severely limited by the relatively smaller patient populations analyzed that limit the identification of all common variants. It is of note that some SLE susceptibility loci are shared across multiple immune-related conditions, and thus common molecular pathways may contribute to the pathogenesis of SLE and additional inflammatory diseases (Richard-Miceli and Criswell 2012). Leveraging statistical comparison methods between SLE and other larger disease GWAS may provide an avenue to gain information about SLE genetics without relying on meta-analyses or performing de novo studies using an expanded patient cohort, which are time and resource intensive. One candidate for this comparison is Multiple Sclerosis (MS).
MS is a disease of the central nervous system (CNS) characterized by neurodegeneration and autoantibody-driven demyelination (Hauser and Cree 2020). Although rare, there are cases of clinical overlap between MS and SLE as shown by numerous case reports. One study also showed 1–1.2% of SLE patients in their cohort having both MS and SLE, which is a higher prevalence of MS than the general population (estimated at 0.21% in the United States) (Dilokthornsakul et al. 2016). Notably, however, current evidence does not indicate an increased prevalence of SLE in families with a history of MS (Cárdenas-Roldán et al. 2013; Criswell et al. 2005). Both MS and SLE are chronic relapsing autoimmune diseases that have previously been shown to share several genetic risk loci, including SH2B3, IL12A, RPL19P8, CD40, and IRF8 (Richard-Miceli and Criswell 2012). Although MS has classically been viewed as a T-cell mediated disease, one of the most effective treatments is B-cell depleting therapy (Chisari et al. 2022). SLE is also associated with the development of neurological manifestations, termed neuropsychiatric SLE (NPSLE), the most common of which involve mood disorders (up to 80%), cognitive impairment (up to 80%), anxiety (up to 40%), headache (up to 28.3%), and seizure disorders (up to 20%). Neuropsychiatric symptoms can additionally include those more typical of MS, such as demyelinating syndrome (up to 2.7%) (Ota et al. 2022), with some patients developing white matter lesions that are indistinguishable from MS lesions on MRI (Heming et al. 2023). Additionally, up to 60% of NPSLE patients have evidence of oligoclonal bands in their cerebrospinal fluid (CSF), that are also present in up to 95% of MS patients (Graner et al. 2020). MS and SLE patients also both exhibit autoantibodies in common, especially anti-phospholipid antibodies. Despite the rarity of clinical overlap, comparison between the diseases could help deepen understanding of the neuropsychiatric elements of SLE and how nervous system inflammation contributes to the overall disease. This type of comparison additionally may facilitate insight into shared pathologic mechanisms rather than global clinical phenotypes.
Importantly, currently available MS GWAS incorporate a much larger patient population compared to studies performed in SLE. The largest publicly available MS GWAS has 14,498/24091 European cases/controls (International Multiple Sclerosis Genetics Consortium (IMSGC) et al. (2013), whereas the largest publicly available SLE GWAS has 6748/11516 European cases/controls (Langefeld et al. 2017). We posit that statistical genetic comparison methods, such as cross-phenotype association analysis (CPASSOC) to identify overlap in genetic association (Li and Zhu 2017), traditional 2-sample and novel PPI-based mendelian randomization (Kain et al. 2022) along with Bayesian colocalization (COLOC) to identify shared causal SNPs can be applied to the powerful large-scale association studies conducted in MS to facilitate genetic discovery in the smaller, more limited datasets available for SLE. Here, we leverage multiple MS GWAS to identify new risk variants in SLE, thus providing further insight into SLE pathology and establishing a robust methodology to probe smaller GWAS for novel genetic associations.
To investigate the shared genetics of MS and SLE we began by establishing genetic correlation using LDSC and CPASSOC (Bulik-Sullivan et al. 2015). Having established correlative genetic associations between MS and SLE, we investigated causal relationships using traditional MR (Davey Smith and Hemani 2014). We additionally employed a molecular network based MR method to investigate clusters of MS-associated SNPs with causal implications in SLE (Kain et al. 2022). Next, we investigated individual shared causal SNPs in smaller genomic regions using the Bayesian COLOC method (Wallace 2021; Zuber et al. 2022). In all of these analyses, SNPs were mapped to genes to investigate whether they are differentially expressed in SLE patients and to further understand molecular pathways associated with each SNP. Finally, we generated putative molecular pathway associations by aggregating SNP-predicted genes from all of these methods and identified unique causative mechanisms implicated in SLE pathogenesis.
Methods
Data selection
Genome wide association studies (GWAS) were selected primarily based on ancestry of patients, sample size, and public availability. European patient GWAS were selected for this analysis due to the much larger availability of MS data in European patient populations. Additionally, a single ancestry was used because SLE is heterogeneous in different patient populations. The largest publicly available GWAS for European patients were used in this analysis. A trans-ancestral GWAS (GCST011096) was used to validate the claims that CPASSOC and COLOC are identifying SNPs that may be identified in a larger GWAS as significant but were not able to be identified in the other GWAS because of a lack of sufficient sample size. Additionally, data from European patients from GCST007400 was used to validate that these results were identifying relevant loci in European ancestry patients. These GWAS was additionally used to validate that the IVs do not become invalid in a larger GWAS. Differential expression sets were selected from whole-blood SLE datasets.
Linkage disequilibrium score regression (LDSC)
Linkage disequilibrium score regression (LDSC) was used to estimate genome-wide genetic correlations between traits using GWAS summary statistics (Bulik-Sullivan et al. 2015). Pre-processed summary statistics from SLE, MS, and Alzheimer’s GWAS were obtained from the Broad webpage (https://alkesgroup.broadinstitute.org/LDSCORE/all_sumstats/). The SLE dataset used was PASS_Lupus.sumstats.gz; the MS dataset was PASS_Multiple_sclerosis.sumstats.gz; the Alzheimer’s dataset was PASS_Alzheimers_Jansen2019.sumstats.gz. Using the LDSC software provided on github (https:// github.com/bulik/ldsc) and reference data on the Broad webpage (https://alkesgroup.broadinstitute.org/LDSCORE/), including European LD scores 'eur_w_ld_chr' or 'weights_hm3_no_hla' as weights for analyses excluding the HLA region. Using standard parameters, the "ldsc.py" (with the "–rg" flag) script was used to generate genome-wide genetic correlation estimates between SLE and MS or Alzheimer’s disease.
Cross-phenotype association analysis (CPASSOC)
Cross-Phenotype association analysis (CPASSOC) was used as a way to evaluate genetic association between MS and SLE without regards to causality (Zhu et al. 2015). This method provides a way of leveraging a larger GWAS, such as MS, to detect either vertical or horizontal pleiotropic loci with a smaller dataset, such as SLE. The method was implemented using the downloadable CPASSOC R script (http://hal.case.edu/zhu-web/). The method has an option of two test statistics: The Shom method, which is similar to a fixed effect meta-analysis, and the Shet method. The Shet statistic, which is an extension of the Shom statistic, is better able to account for heterogeneity in summary statistics as would likely exist between traits. This method requires a correlation matrix, Z scores for all SNPs in each trait, and sample size of each trait as inputs. The correlation matrix between the MS and SLE GWAS was calculated by LD-pruning all SNPs with r2 = 0.2. SNPs with summary Z score of greater than 1.96 or less than −1.96 were removed to prevent the inflation of the correlation matrix calculation (Li and Zhu 2017). The Shet statistic was then calculated using the Bentham SLE GWAS and GCST001198 MS GWAS summary statistic Z scores as inputs. These SNPs were further filtered to include only SNPs present in the GCST005531, GCST001198 and Bentham GWAS. SNPs reaching genome-wide significance by the Shet method (pCPASSOC < 5 × 10–8) and suggestive significance (p < 1 × 10–3) in both MS and SLE were considered significant.
Identification of SNP-predicted genes
Expression quantitative trait loci (eQTLs) were identified using GTEx (Lonsdale et al. 2013) version 8 (GTEXportal.org) and mapped to their associated eQTL expression genes (E-Genes). To find SNPs in enhancers and promoters, and their associated transcription factors and downstream target genes (T-Genes), we queried the atlas of Human Active Enhancers to interpret Regulatory variants (HACER, http://bioinfo.vanderbilt.edu/AE/HACER) (Wang et al. 2019). To find SNPs in exons of protein-coding genes (C-Genes) and include proximal genes (P-Genes, within 5 kb), we queried the human Ensembl genome browser’s variant effect predictor (VEP, ensembl.org/info/docs/tools/vep, GRCh38.p12) (Cunningham et al. 2022). We additionally compared these methods of predicting genes to the V2G method of predicting genes (Ghoussaini et al. 2021; Mountjoy et al. 2021).
Network analysis
Protein–protein interaction (PPI) networks of SNP-predicted protein-coding genes were generated by STRING (Szklarczyk et al. 2023) (https://string-db.org, version 11.5), and resulting networks were imported into Cytoscape (Morris et al. 2011) (version 3.9.1) for visualization and partitioned with MCODE via the clusterMaker (Morris et al. 2011) (version 1.2.1) plugin. Metastructures are based on PPI networks. For all metastructures, node gradient shading is proportional to intra-cluster connectivity, cluster size indicates number of genes per cluster and edge weight indicates inter-cluster connections.
Functional gene set analysis
Predicted genes were examined using Biologically Informed Gene Clustering (BIG-C; version 4.4.) (Catalina et al. 2019, 2020; Labonte et al. 2018). BIG-C is a custom functional clustering tool developed to annotate the biological meaning of large lists of genes and has been previously described. I-Scope (Ren et al. 2019) is a custom clustering tool used to identify immune cell types in large gene datasets. The Ingenuity Pathway Analysis (IPA; https://www.qiagenbioinformatics.com) platform and EnrichR (Chen et al. 2013) (https://maayanlab.cloud/Enrichr/) web server provided additional molecular pathway enrichment analysis.
Traditional MR instrumental variable selection
MR methods require that three assumptions be met: (1) the relevance assumption, (2) the exclusion restriction criteria assumption, and (3) the independence assumption. To satisfy the first assumption, SNPs significantly associated with MS (genome-wide significance threshold p-value < 5 × 10–8) were obtained from two different GWAS (International Multiple Sclerosis Genetics Consortium et al. 2011; International Multiple Sclerosis Genetics Consortium (IMSGC) et al. 2013). To satisfy the independence assumption, SNPs weakly associated (p-value < 1 × 10–5) with Rheumatoid Arthritis, Type 1 Diabetes, thyroid disease, cardiovascular disease, and Sjogren’s syndrome were obtained from the Phenoscanner database (www.phenoscanner.medschl.cam.ac.uk) (Kamat et al. 2019; Staley et al. 2016) and were excluded from IVs. HLA-region SNPs were removed as well. To satisfy the independence assumption, SNPs weakly associated with SLE were removed from the IVs. LD clumping was employed using the clump data function (R2 = 0.001, 10,000 kb window, 1000G EA reference population) (Marees et al. 2018) to generate 18 and 31 IVs for GCST001198 and GCST005531 GCST001198 datasets respectively.
IVs were selected for analyses with SLE as the exposure in a similar manner, including SNPs significantly associated with SLE and excluding SNPs weakly associated with Rheumatoid Arthritis, Type 1 Diabetes, thyroid disease, cardiovascular disease, and Sjogren’s syndrome from the Phenoscanner Database. Following LD clumping, analyses included 21 and 22 IVs using the GCST005531 and GCST001198 MS GWAS as the outcomes respectively.
Mendelian randomization (MR)
Mendelian randomization (MR) was used to test for causal relationships between SLE and MS using the MR-Base (https://www.mrbase.org) TwoSampleMR package (Hemani et al. 2018) in R (v0.5.6, https://github.com/MRCIEU/TwoSampleMR). Various sets of MS or SLE-associated genetic variants used as instrumental variables (IVs) and summary statistics for MS or SLE-exposure were manually imported into R and summary statistics were carried out for MR-base compatibility using the ‘format data’ command. All effect sizes and standard errors were obtained from the exposure summary statistics used in each analysis, regardless of the study in which each IV was associated with the exposure. Data from the SLE and all MS-related GWAS studies used in our MR analyses are publicly available and also accessible through the MR-Base software, which was used to obtain the outcome summary statistics via the ‘extract outcome data’ command. The ‘allele harmonization’ command was used to ensure the effect estimates of the exposure and outcome are based on matching alleles, excluding SNPs with completely mismatching alleles from the MR analysis or reversing the effect and non-effect alleles along with the effect estimates when applicable. Because of the allele harmonization step and because some SNPs are absent from the available summary statistics, a small proportion of SNPs used as IVs are absent from the final MR calculations. Steiger filtering was carried out to ensure that all IVs were most strongly acting in the exposure to outcome direction (Hemani et al. 2017). Five individual MR methods were carried out through the TwoSampleMR package, including Simple Mode, Weighted median, MR-RAPS, and MR-PRESSO methods. MR-IVW and MR-Egger heterogeneity test results (Q-value) indicate whether significant heterogeneity was detected, which does not necessarily indicate biased causal estimates especially in GWAS with small sample sizes. Additionally, this statistic may be less informative in this analysis because it assumes that all causal IVs are related to the same parameter; in this analysis it may over-reject the null hypothesis (Burgess et al. 2017). MR-Egger intercepts indicate whether significant directional horizontal pleiotropy was detected, which usually indicates biased causal estimates. A leave-one-out analysis was performed using the ‘mr_leaveoneout’ function. SNPs included as IVs in both the datasets were considered a significant leave-one-out result only if it was significant in both datasets. For single-SNP MR, the ‘MR single-SNP’ function was also carried out using the Wald Ratio method.
PPI-based MR
MS-associated variants from the GCST005531 MS GWAS were linked to their most likely genes, and the genes used to generate PPI-informed gene clusters. The MS-associated SNPs mapping to genes in each of PPI-based clusters were then extracted to “reverse engineer” subsets of SNPs that could be used separately as MS-IVs for MR to independently estimate the causal effects of each PPI-informed SNP-to-Gene module on SLE. SNPs loosely associated with confounders Rheumatoid Arthritis, Type 1 Diabetes, thyroid disease, cardiovascular disease, and Sjogren’s syndrome were removed using the phenoscanner. SNPs loosely associated with SLE were additionally removed using the phenoscanner. MR-IVW estimates were determined for each cluster using the TwoSampleMR package. HLA-region SNPs were conservatively removed from MR analyses. Stringent LD clumping was employed using the clump_data (R2 = 0.001, 10,000 kb window, 1000G EA reference population) function to generate an independent set of MS-IVs. Clusters determined to have a significant effect on SLE by the MR-IVW method were further analyzed using the Simple Mode, Weighted median, MR-RAPS, and MR-PRESSO methods.
COLOC
Colocalization was performed using the Bayesian colocalization method Susie-COLOC (Wallace 2021). This method evaluates the probability of colocalization in any particular region using 4 hypothesis: H0 is that there is no significant association between the region and either trait; H1 is that there is a significant association between the region and the first trait only; H2 is that there is an association between the region and second trait only; H3 is that both traits are associated with the region but with different single causal SNPs; and H4 is that both traits are associated with the region and share the same single causal variant. SLE and MS colocalized SNPs were determined using the coloc R package (v5.1.0.1). SNPs present in both the MS and SLE GWAS were determined and then divided into LD blocks using the lddetect algorithm (Berisa and Pickrell 2016). Beta values, variances, positions, p values, sample sized, and LD matrices were used as the inputs to the coloc functions. The LD matrices were calculated using the ld_matrix function from the ieugwasr package (v0.1.5). Since this LD matrix function is limited to 500 SNPs or less, regions including more than 500 SNPs were broken down further into 500 SNP blocks to be analyzed. Each block of SNPs was then analyzed using first the runsusie function on the MS and SLE SNPs independently and then using the coloc.susie function to analyze colocalization. If the SNPs did not converge by the susie.coloc method, they were analyzed using the coloc.abf function and the SNP with the highest H4 was determined to be the causal SNP. Regions were considered colocalized if they had an H4 > 0.75. Regions containing SNPs reaching H3 > 0.75 and without any significant H4 SNPs were also investigated. These regions could potentially indicate a lack of causal association between MS and SLE. Default prior probabilities were used for all analyses (p1 = 1 × 10–4, p2 = 1 × 10–4, p12 = 5 × 10–6).
Results
MS and SLE are genetically correlated and are predicted to share risk loci
To determine whether MS and SLE are genetically correlated, we applied standard LDSC using GWAS summary statistics from MS and SLE. The HLA region was removed to prevent an artificially high correlation estimate. As shown in Fig. 1a, MS and SLE demonstrate a significant genetic correlation (rg = 0.7415, p = 0.0011) indicating that there are shared genetic features between the diseases extending beyond the highly pleiotropic HLA region. In contrast, we did not observe a significant genetic correlation between SLE and Alzheimer’s disease (AD) (rg = −0.0655, p = 0.4304).

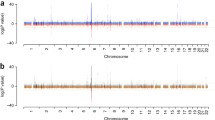
LDSC and CPASSOC detect genetic correlation between MS and SLE and predict shared genetic pathways a Bar plot of LDSC Analysis of MS and AD each correlated with SLE (rg ± standard error). b Manhattan plot of the CPASSOC test statistics with 101 CPASSOC-identified credible SNPs (p.shet < 5 × 10–8) highlighted in green. Grey/black dots indicate SNPs that were not identified by CPASSOC. The blue line shows the cutoff for p < 1 × 10–5, and the red line shows p < 5 × 10–8 on each plot. c Unsupervised MCODE clustering of the 164 putative protein-coding genes. Functional and cell-type enrichments for each cluster were determined using EnrichR and/or BIG-C (black labels) and I-scope (red labels). Bold-type colored labels represent shared functional annotations for matching gene clusters
To investigate individual SNP level genetic association between MS and SLE in greater detail, we used CPASSOC (Zhu et al. 2015). This method can help identify additional SNPs of interest by combining information from multiple datasets to identify variants that are significant in one or more traits (Li and Zhu 2017). Rather than identifying causal SNPs, this method identifies potentially pleiotropic SNPs indicating shared genetics between diseases. We used the SHet test, which is better able to account for heterogeneity between trait summary statistics than the SHom test (Li and Zhu 2017), to identify variants that are significant in both MS and SLE. CPASSOC was implemented using the SLE GWAS from Bentham et al. (GCST003156) and a MS GWAS from Beecham et al. (GCST001198) (Bentham et al. 2015; International Multiple Sclerosis Genetics Consortium (IMSGC) et al. 2013) (Table S1). Using this method, 360 SNPs achieved a genome-wide significant p-value (pshet < 5 × 10–8) with 101 of these SNPs reaching suggestive significance (p < 1 × 10–3) (Liu et al. 2022) in both the MS and SLE GWAS (Fig. 1b, Table S2). Many of these SNPs were identified as significant despite lacking genome-wide significance in the SLE GWAS, supporting the utility of this analysis to identify additional SNPs of interest in lupus. Additionally, the majority of the SNPs became more significant when examined in larger GWAS (GCST011096 and GCST007400) (Langefeld et al. 2017; Wang et al. 2021, p. 38), supporting the utility of this analysis as a discovery tool in GWAS with limited power (Table S2). It is of note that although 43 of the 101 identified SNPs are located in the highly pleiotropic HLA region, 58 SNPs were identified across the rest of the genome, indicating shared genetic associations beyond the HLA region. In order to investigate whether any of these loci were novel, we split the 101 SNPs into genomic regions using the LDetect algorithm. These regions were then compared to loci identified in this SLE GWAS. These regions were then compared to loci identified in this SLE GWAS. CPASSOC identified 20 loci of interest, with 10 of these not having been identified in the Bentham SLE GWAS used for this analysis (Table S2). All of these regions become more significant as GWAS sample size increases, indicating that this analysis is identifying regions that were not identified in smaller GWAS but are likely to be identified as GWAS sample sizes increase.
To identify putative gene(s) influenced by each of the 101 SNPs common to MS and SLE and gain a better understanding of the shared genetic architecture underlying MS and SLE, we mapped SNPs to genes (Kain et al. 2022; Owen et al. 2020, 2023), identifying 164 unique genes for further analysis (Table S3). To determine their functional relevance to SLE, predicted genes were compared to differentially expressed genes (DEGs) derived from multiple SLE whole blood datasets (GSE29536 Banchereau et al. 2012; Obermoser et al. 2013; Park et al. 2013), GSE22098 (Berry et al. 2010), GSE39088 (Ducreux et al. 2016; Lauwerys et al. 2013), GSE45291 (Bienkowska et al. 2014; Petri et al. 2019), GSE49454 (Chiche et al. 2014), GSE61635, GSE88884 (Hoffman et al. 2017; Li et al. 2019). 146 (89.0%) available CPASSOC-predicted genes were significantly differentially expressed in at least one of the selected SLE datasets (Table S4). This indicates that CPASSOC SNP-predicted genetic associations between MS and SLE are functionally relevant in SLE despite their lack of genome-wide significance. To assess molecular networks encoded by the 164 predicted gene set, a protein–protein interaction network was generated in STRINGdb and unsupervised clustering revealed 12 distinct clusters that were functionally enriched in a diverse range of immunological and cellular categories. (Fig. 1c). Functional annotations for General transcription, Transcription factors and DNA repair were shared among clusters 2–4, 6, 8 and 12. In addition, we noted clusters 1 and 11 enriched in a number of type 1 interferon response genes, including TYK2, IRF5 and TNF, heavily implicated in lupus disease development, along with a number of class I and II MHC molecules (Fig. 1c and Table S5). Pathway analysis also revealed Cortisol synthesis and secretion (cluster 4) and parathyroid synthesis and secretion (unclustered), demonstrating the predicted involvement of non-estrogen hormonal regulation in SLE. Cellular enrichment categories included antigen presenting cells (APCs), monocytes, myeloid cells, T, B, and NK cells distributed across the network.
Mendelian randomization shows a unidirectional, positive causal relationship between MS-associated SNPs and SLE
Although CPASSOC detects genetic association between traits, it is not able to establish whether the alleles have a causal relationship. To examine causality, traditional 2-sample MR was used to estimate the association between the effect sizes of instrumental variables (IVs) on SLE and MS. We applied five MR methods, first using MS-associated SNPs as the exposure and SLE as the outcome.
To satisfy the assumptions for IV selection, we carried out two-sample MR analyses using non-HLA SNPs strongly associated (p < 1 × 10–8) with MS, and excluded SNPs weakly associated (p < 1 × 10–5) with SLE or confounders (rheumatoid arthritis, type 1 diabetes, cardiovascular disease, thyroid disease, Sjogren’s syndrome, etc.). This was followed by stringent LD clumping (R2 = 0.001, 10,000 kb window, 1000G EA reference population) to ensure IV independence. Two independent MS GWAS were used as the exposure [GCST005531 (International Multiple Sclerosis Genetics Consortium et al. 2011)], and GCST001198 (International Multiple Sclerosis Genetics Consortium (IMSGC) et al. 2013) and summary statistics from two SLE GWAS [GCST003156 (Bentham et al. 2015), and GCST007400 (Langefeld et al. 2017)] were used as the outcome. Following data harmonization, 19 and 31 IVs from GCST001198 and GCST005531, respectively (Table S6) were tested on the outcome GCST003156 using five different MR methods, some of which account for additional IV-invalidity, pleiotropy, or heterogeneity. None of the IVs overlapped with CPASSOC-predicted SNPs, adding further confidence that the IVs are not violating the MR independence assumption for pleiotropy. Three out of five methods resulted in significant (p < 0.05) positive causal estimates using IVs from GCST001198 on GCST003156, and all five methods resulted in significant causal estimates using IVs from GCST005531 on GCST003156. 32 IVs (Table S6) from GCST005531 were tested on the outcome GCST007400. Three out of five methods were significant for GCST005531 on GCST007400 (Fig. 2a and Figure S1). An additional 13 MR methods were used to further confirm these relationships (Table S7). In contrast, no significant causal estimate was detected using Alzheimer’s disease [GCST001449 (Lambert et al. 2013)] as the exposure (Fig. 2a).

Mendelian Randomization Analysis Demonstrates a Positive-Causal Effect of MS-Associated non-HLA SNPs on SLE a Forest plots of MR causal estimates (beta ± standard error) for MS or AD (exposure) on SLE. b Forest plots of MR causal effects for SLE (beta ± standard error) as the exposure and MS or AD as the outcome. Grey data points indicate insignificant estimates (p > 0.05) and red points indicate significant estimates (p < 0.05). The number of IVs are listed above each graph. MR-EGGER intercept values are included above each graph. c Clustering and pathway analysis of the 110 genes predicted from the 31 IVs derived from GCST005531. Genes predicted by SSMR are highlighted in each cluster. Positive causal SNP-predicted genes are outlined in red, and negative causal SNP-predicted genes are outlined in green. Functional and cell type enrichments for each cluster were determined using BIG-C (black labels) and I-scope (red labels). Bold-type colored labels represent shared functional annotations for matching gene clusters
To eliminate the possibility that the positive causal estimate of MS-associated SNPs on SLE is bidirectional and, therefore, unlikely to represent a true causal relationship, we also carried out two-sample MR analyses using SLE-associated IVs as the exposure and the two MS GWAS as the outcomes (IVs in Table S8). None of the five MR methods were significant using either of the MS or AD GWAS as the outcome (Fig. 2b). This establishes that the relationship is unidirectional with MS-associated SNPs having a positive causal effect on SLE.
Importantly, there was no directional pleiotropy detected by the MR-Egger intercept test in any of the estimates (Fig. 2a, b) and the leave-one-out method was used to confirm that no single SNP was driving the causal estimates from MR analysis (Table S9).
Finally, Single-SNP MR (SSMR) was employed to identify individual, non-HLA, MS-associated SNPs with positive or negative estimates on SLE. SSMR identified 10 significant positive causal single SNPs and 1 significant negative causal single SNPs using the GCST005531 as the exposure (Table S10).
We additionally investigated whether another autoimmune disease, rheumatoid arthritis (RA), shows a significant causal effect on SLE (or vice versa). While this analysis did generate some significant results, these results were inconsistent in different datasets and additionally showed evidence of pleiotropy (Figure S2). This analysis acts as an additional negative control, indicating that the same clear-cut relationship does not exist between RA and SLE and MS and SLE.
Pathway analysis of IVs predicts candidate MS pathways in SLE
To understand how the IVs from the MR analysis are expressed in SLE patients, we mapped the MS-associated IVs to 110 candidate causal genes (Table S11). 102 of these genes overlapped with the V2G gene prediction method (Table S12). Overall, 80 (72.7%) SNP-predicted genes were significantly differentially expressed in at least one SLE dataset, indicating that these MS-associated SNPs and molecular pathways are informative in SLE (Table S4).
To elucidate the pathways involved in the SNPs driving the positive causal estimates of MS on SLE, proteins predicted from MS IVs were integrated into PPI based connectivity networks clustered and annotated for functional and cellular categories. This resulted in 11 clusters (Fig. 2c, Table S13), which were enriched in functional categories Transcription factors, Metabolic function, and mRNA processing. A number of clusters were enriched in Gene ontogeny (GO) terms for immune function, including Cytokine-mediated signaling pathway (GO:0019221; cluster 2) and Immunological synapse formation (GO:0001771; cluster 9) (Table S13). Notably, MS-associated pathways were also present, such as the Response to vitamin D (GO:0033280) and Abnormal cholesterol homeostasis (Table S13).
PPI-based MR predicts candidate gene clusters causal of SLE
To obtain a more comprehensive view of the possible impact of MS-derived molecular pathways on SLE, we carried out PPI-based MR, which provides a method to investigate whether individual functional SNP/gene clusters, rather than the whole genome, are potentially causal in SLE (Kain et al. 2022). To accomplish this, we first mapped 1243 MS-associated SNPs with genome-wide significance (p < 5 × 10–8, eliminating confounders) to 548 predicted genes that grouped into 26 distinct clusters based on PPI connectivity mapping (Figure S3). We then identified and collated MS-associated SNPs linked to genes in each of the 26 PPI-based clusters for use as MS IVs to estimate cluster-specific associations with SLE (Table S14). Each cluster underwent stringent LD clumping before MR analysis to identify valid SNPs for use as IVs to estimate cluster-specific associations with SLE. Initial application of MR-IVW to these 26 subsets of MS SNP-derived IVs (Fig. 3a) followed by additional MR methods including simple mode, weighted median, MR-RAPS, and MR-PRESSO (Fig. 3b) yielded 14 significant (p < 0.05) positive causal estimates for SLE. A further 12 methods were also carried out to validate the results on clusters containing more than 2 SNPs (Table S15). Pleiotropy using the MR-Egger intercept test was detected in clusters 10 and 17.

PPI-Based MR Analysis Predicts Individual MS-Associated SNP/gene Clusters Causal in SLE a Forest pots of PPI-based MR results for each cluster using the MR-IVW method (beta ± standard error). b Forest plots of expanded results for significant clusters from part a (beta ± standard error). Clusters 9 and 25 are excluded because they had < 2 IVs. MR-egger intercepts are included (* indicates p < 0.05, and a significant MR-EGGER result). c Cluster metastructures of all significant PPI-based MR clusters based on PPI networks. Functional and cell-type enrichments for each cluster and determined using BIG-C and/or EnrichR (black labels) and I-scope (red labels). Node size is proportional to the number of genes per cluster, node color represents the effect size (beta) and edge weight is proportional to the number of connections between clusters
Genes in significantly causal clusters were analyzed using differential expression to confirm the functional relevance of these genes in SLE (Table S4). MS-derived clusters were then annotated using multiple functional and cellular composition tools. Importantly, pathway analysis indicates a wide range of predicted biological functions known to be involved in MS pathogenesis that have potential causal implications for NPSLE/SLE by MR such as Abnormal excitatory synapse morphology and Abnormal synaptic vesicle clustering (cluster 17) and Acetylcholine neurotransmitter release cycle (cluster 7). (Fig. 3c, Table S16). Although cluster 17 did have evidence of pleiotropy, this pathway indicates a potentially shared, but not causal, pathway between MS and SLE. Shared functional annotations among clusters included transcription factors, immune signaling, MHC class I, chromatin remodeling, intracellular signaling, and cytoskeleton. The largest and most connected genes clusters were clusters 1, 2, 3, and 6, dominated by immune related function, with pathways including signaling by IL, IFN-g signaling, T cell acute lymphoblastic leukemia, TCR signaling, and Cell adhesion molecules. Additional immune pathways were found in smaller clusters, including complement, APCs, monocytes, myeloid cells, NK cells, T cells, and B cells were all predicted to be involved in SLE by these cluster analyses.
COLOC analysis predicted colocalized SNPs/Genes between MS and SLE
Colocalization analysis was used to investigate whether SLE and MS share any causal SNPs in specific genomic regions rather than throughout the whole genome, providing an even more conservative statistical method for evaluating causal relationships between traits than MR (Zuber et al. 2022). The genome was divided into LD blocks using the LDetect algorithm, and each block was individually investigated for colocalization (Berisa and Pickrell 2016) using the Sum of Single Effects (SuSiE) regression framework and COLOC algorithm. Seven regions showed significant colocalization with one or more shared causal SNP satisfying the H4 hypothesis (H4 > 0.75; both traits are associated and share the same single causal SNP) (Table 1). These regions were detected across several chromosomes (3, 5, 6, 7, and 16), providing further support that the genetic associations between MS and SLE are not limited to highly pleiotropic HLA SNPs. Two of these regions are novel SLE loci, as they were not identified in the SLE GWAS used for the analysis. Importantly, several SNPs were colocalized despite not reaching genome-wide significance in either GWAS (Figure S4). All of these SNPs except one additionally become more significant in the larger trans-ancestral SLE GWAS (GCST011096) (Wang et al. 2021, p. 38), indicating the utility of this analysis as a discovery tool for identifying novel loci (Table S17). This illustrates the efficacy of this method to identify SNPs of interest that would otherwise not be detected because of the lack of statistical power in a given GWAS. The significantly colocalized SNPs were mapped to 25 total genes and compared to DEGs in SLE WB datasets to establish their relevance to lupus. Overall, 15 out of 20 detectable genes (75%) were significantly differentially expressed in at least one of the SLE datasets (Table S4). These genes were involved in numerous pathways including IL12 signaling, IFN-G signaling and Monocyte chemotaxis.
In addition to testing the H4 hypothesis, we also investigated the H3 hypothesis that highlights causal implications in both diseases but with distinct lead variants. There were three regions detected by the SuSiE COLOC method with a significant H3 result (H3 > 0.75) (Table S18), indicating that while both regions are associated with the diseases, they are unlikely to be related by a true causal relationship. All three of these regions were located on chromosome 6.
CPASSOC, traditional MR, PPI-based MR, and COLOC pathway meta-analysis predicts both complementary and unique gene associations
To investigate whether the genes from these analyses interact with each other, we conducted a combined pathway meta-analysis with the 553 unique genes predicted with significant results from the multiple approaches employed herein. We observed very little overlap between genes generated from the four analytic methods (MR-IVs, PPI-based MR, CPASSOC and COLOC) with only 1 gene, RMI2, predicted by all four analyses (Fig. 4a). Despite the limited overlap, we investigated whether these predicted genes were involved in similar biological pathways. PPI based connectivity mapping followed by unsupervised clustering revealed 24 distinct clusters, along with a cohort of unclustered genes (Table S19). The majority of clusters were composed of gene combinations derived from all analytic techniques, indicating that each analysis predicts functionally related but unique gene sets (Fig. 4b). Although CPASSOC predicts pleiotropic rather than causal genes, pleiotropic genes were found to cluster with causally predicted genes from traditional MR, PPI-based MR, and COLOC. The clustering pattern of SNP-predicted genes from all analyses adds confidence to the potential causal implications of the pathways from this combined analysis. Furthermore, many of these genes were found to be differentially expressed in SLE datasets (Table S4), highlighting their functional relevance to lupus and the ability of this methodology to identify novel and important components of the genetics of SLE pathophysiology.

Genes predicted from all analyses integrate into related pathways a Venn diagram showing overlap between genes predicted from all four analyses (IVs, PPI-based MR, COLOC, and CPASSOC). b Individual cluster composition is displayed by percentage. c Cluster metastructures using 553 genes predicted by all analyses. Functional and cell type enrichments were determined using BIG-C and/or EnrichR (black labels) and I-scope (red labels). Node size is proportional to the number of genes per cluster, node color represents clusters with shared functionality designated in bold. Edge weight is proportional to the number of connections between clusters
Of the 24 derived gene clusters derived from the combined genes emanating from each approach, the largest, most connected clusters (clusters 1, 2, 3 and 11) were enriched in immune-related functional categories including Immune signaling, Immune cell surface and Class I and II MHC (Fig. 4c and Table S20). Additional immune cell related pathways such as interferon gamma signaling, Th17 cell differentiation, cytokine signaling in the immune system, and activation of C3 and C5 were also detected. Notably, other clusters were enriched in lipid metabolism and steroid biosynthesis along with mitochondrial function. A number of neural pathways were identified in cluster 8 and include Acetylcholine neurotransmitter release cycle, nerve growth factor pathway, and abnormal excitatory synapse morphology (Fig. 4c and Table S20). Gene expression (transcription), spliceosome, triacylglyceride biosynthesis, and negative regulation of cell–cell adhesion were detected in various other clusters.
Discussion
Small sample sizes of available SLE GWAS limit their capabilities to identify common genetic variants associated with the disease. We used several statistical approaches to characterize genetic correlation and causation between MS and SLE GWAS to identify shared genetic variants. This study identifies a method of leveraging the larger MS GWAS to identify variants of interest in the SLE GWAS. These findings suggest genetic correlation and pleiotropy between MS and SLE as shown by LDSC and CPASSOC. MR established a unidirectional, positive causal association between MS and SLE. Moreover, PPI-based MR expanded this positive causal estimation to include 14 positive causal gene clusters. COLOC predicted a number of genomic regions with similar patterns in MS and SLE. Each analysis was able to identify novel SNPs and/or genes of interest that did not reach genome-wide significance in the SLE GWAS but showed a significant association between the SLE and MS GWAS. Interestingly, SNP-predicted genes from each analysis clustered together based on protein–protein interactions to identify functional pathways predicted by all analyses. These methods provide a way to expand networks of relevant SLE SNPs by comparing to larger GWAS. Pathways predicted by SNP-predicted genes included a number of immune-mediated pathways as well as neural pathways related to synapse morphology. A number of other pathways related to steroid synthesis, transcription, cell adhesion, and more were also identified.
The use of CPASSOC allows identification of individual pleiotropic SNPs between diseases, lending additional support to the genetic correlation evidence from LDSC. This method allows for the identification of pleiotropy between MS and SLE beyond general genetic overlap of genome-wide significant SNPs. In this context, CPASSOC can leverage a large GWAS such as MS to identify pleiotropic SNPs that may not reach significance in a smaller GWAS, such as carried out in SLE, because of a lack of statistical power. Although important to remove in causal analyses, pleiotropic SNPs are able to provide information about shared pathways involved in diseases even if they are not mediated by the same factors. Identification of pleiotropic loci between diseases could allow expansion of therapies into multiple diseases and allow identification of potential shared side-effects of these therapies (Barrio-Hernandez et al. 2023). CPASSOC additionally provides a way to identify shared SNPs of interest without respect to causality.
Understanding causal relationships between traits in addition to correlation can provide additional information as to how the diseases are related. Causal estimates indicate that the same genetic changes are occurring in both diseases rather than occurring because of different causes or intermediaries. The result showing a significant, unidirectional, positive causal relationship of MS on SLE provides novel insight that the genetics of MS are acting in a similar manner in SLE. Additionally, the SNPs used as IVs can be used to determine candidate causal genes that may be driving the significant causal association between MS and SLE. These candidate causal genes may be playing a role in SLE through similar mechanisms as they act in MS. Expansion of traditional MR into PPI-based MR provides a method to determine causal pathways between MS and SLE. This method also highlights individual functional gene clusters and pathways contributing most to a causal effect of MS on SLE (Kain et al. 2022). CPASSOC and COLOC additionally identify SNPs that may be pruned in the MR IV-selection process but are of interest.
We additionally confirmed that this causal relationship between MS and SLE through several controls. There was no causal estimate indicated between Alzheimer’s disease and SLE. The same consistent causal relationship is not found in other autoimmune diseases. We investigated whether RA showed a causal relationship with SLE, considering that RA and SLE also share genetic and clinical features (Farh et al. 2015; Lu et al. 2021). Although there were some significant results from these analyses, they were inconsistent across datasets and additionally showed evidence of bidirectionality and pleiotropy. One potential explanation for the lack of a causal relationship is the extensive shared features between RA and SLE. This may result in over-pruning of variants in the MR IV selection process, as any variants associated with both diseases must be removed to avoid pleiotropy. Additionally, MS provides the advantage of an organ-specific comparison rather than a global clinical comparison to SLE. Both MS and SLE are complex diseases that might have both shared and disparate genetic elements. Our results suggest that there are shared genetic elements that could influence the common molecular features of these otherwise complex traits. The finding of unidirectional, positive causal variants between MS and SLE that were consistent across datasets and showed clear unidirectionality and lack of pleiotropy strongly indicates a shared molecular causality of some pathogenic features of the two autoimmune diseases.
COLOC is a method of determining shared causal SNPs between diseases without regards to pleiotropy. It also provides a more conservative causal estimation than MR despite having fewer assumptions. Similarly to CPASSOC, COLOC does not require causal SNPs to reach genome-wide significance in the traits of interest, allowing the identification of SNPs of interest in GWAS with limited power. All four of these analytic methods provide an avenue to probe the SLE GWAS for robust associations that are not otherwise able to be identified. Importantly, the SNPs identified by both CPASSOC and COLOC became more significant in the trans-ancestral validation GWAS. While many SNPs still did not reach genome-wide significance even in this larger GWAS, the decreased p-value with increased sample size suggests that these SNPs may reach genome-wide significance in a GWAS with large enough sample size. This validation supports the utility of these analyses as a discovery tool to uncover novel disease loci in GWAS with a limited sample size.
Having identified these SNPs of interest, we linked SNPs to genes using amino acid changes in encoded proteins, proximity, expression quantitative trait loci (eQTL) predictions and regulatory elements as previously reported (Kain et al. 2022; Owen et al. 2020, 2023) in order to interrogate potential associated genes as comprehensively as possible. Interestingly, many of the genes predicted by CPASSOC, traditional MR, significant PPI-based MR clusters, and COLOC clustered together into functionally similar networks. These methods together expand the number of genes that can be identified as relevant in SLE beyond any one analysis. The efficacy of this kind of analysis to predict functionally relevant genes is further confirmed in the significant DE results for most genes in all analyses. These results highlight how MS genetics can be used to identify both known and novel pleiotropic and causal loci in SLE.
All four of these methods of identifying associations map to genes with functional relevance in SLE as shown by DE analysis. These pathways identify a spectrum of biological processes that are shared between MS and SLE. The main category of pathways of interest are immune pathways. Although SLE has traditionally been viewed as a type 1 IFN mediated disease, this analysis also identified IFN-g, a type 2 interferon, as a shared disease feature. Complement pathways were also identified across analyses. Monocytes, T cells, B cells, APCs, and NK cells were all implicated in the pathway analyses, indicating that this association between diseases is not limited to an individual immune cell type. Interestingly, however, Th17 cells were specifically implicated in the pathway meta-analysis. This cell type has been shown to be associated with both MS and NPSLE (Canatar and Ayaz 2022; Okamoto et al. 2010). Gamma-delta T cells were also identified as a relevant cell population, which has been shown to be associated with SLE, and have functions similar to Th1, Th2, Th17, and Treg cells (Wu et al. 2016).
Another potential area of interest is neurological pathways. Given the presence of CNS disease in some SLE patients (Kampylafka et al. 2013; Ota et al. 2022), identification of potential neurological pathways or risk factors in SLE could help expand potential treatment options and further understanding of the biological underpinnings of this disease manifestation. Additionally, the lack of available large-scale data on NPSLE increases the need for these trans-trait methods to further understand specific disease manifestations. MS provides an informative way to bring out these nervous system pathways, as some of the CNS-mediated MS disease manifestations may be found in SLE. This analysis identified pathways, including Acetylcholine release cycle and abnormal excitatory synapse morphology, from candidate causal genes. One potential reason for the identification of these pathways could be the role of anti-NMDAR antibodies in NPSLE. These antibodies interfere with excitatory glutamate-releasing synaptic terminals (Faust et al. 2010). However, further study is required to determine the cause of this shared dysfunctional excitatory synapse pathway. To date, NPSLE has been characterized as having a wide range of causes resulting in different symptoms. These causes include autoantibodies, inflammation, vasculopathy, and neural cell damage (Manca 2022; Wang et al. 2022; Wen et al. 2016). Both autoimmune and ischemic pathways have been identified as leading to NPSLE, leading to stroke/cognitive dysfunction and seizures/psychosis, respectively (Sarwar et al. 2021). This analysis primarily identifies potential autoimmune-mediated pathways underlying NPSLE. However, Vitamin D biosynthesis was identified as a shared pathway between MS and SLE in this analysis, and has been shown to be involved in MS and other autoimmune diseases, including SLE (Johnson and Thacher 2023). Low vitamin D has also been associated with atherosclerosis as well as other cardiovascular disease manifestations, and identified as a potential mediator of NPSLE pathogenesis (Kassi et al. 2013; Rizzoni et al. 2019; Sarwar et al. 2021; Zhou et al. 2022). However, this pathway may also be pointing to vitamin D in autoimmunity, as vitamin D may regulate the function of monocytes, dendritic cells, T-cells, and B-cells downstream of vitamin D receptors expressed on these cell types (Chen et al. 2007; Sintzel et al. 2017). Vitamin D has also been shown to upregulate neuron calcium uptake, upregulate microglial activity, and activate astrocytes (Miclea et al. 2020). Further research is needed to understand the interplay of these pathways. Interestingly, cell–cell adhesion was implicated across a number of analyses. This pathway has a number of potential implications in NPSLE, including blood–brain barrier (BBB) disruption.
It is of interest that these genetic associations are present despite a lack of widespread overlap between MS and SLE in patient populations. Diagnosis of both MS and SLE concurrently is rare, although possible (Fanouriakis et al. 2014). However, the diseases do share some clinical features that could result from the presence of a causal estimate, including the presence of autoantibodies, B cell involvement and oligoclonal bands in the CSF. This points to the efficacy of this kind of analysis, and of MR specifically, even without the presence of an obvious causal relationship between disease diagnosis to act as a discovery tool. MR has traditionally been used between factors with a clearer clinical relationship, but robust MR analyses, as shown in this study, can be used as a discovery tool and performed between diseases without an obvious clinical association.
The novel approach to genetic analysis reported here provides an avenue to assess common causative alleles between SLE and other diseases further by leveraging related disease GWAS. This could be especially important in analyzing even smaller SLE GWAS with patients of different ancestries. Given the higher prevalence of SLE in people of non-European ancestry (Demkova et al. 2023; Lewis and Jawad 2017), further understanding SLE in these populations is crucial. These methods could additionally be applied to determine therapies that may be helpful in multiple diseases based on shared pleiotropic or causal loci.
There are several limitations to this study. First, this analysis was conducted using SLE and MS GWAS with largely European-ancestry patients. Given the heterogeneity of the disease in different patient populations (Allen et al. 2021; Owen et al. 2022), further analysis in other ancestries is required. The GWAS used as a validation tool is a trans-ancestral GWAS, limiting its ability to confirm population-specific effects in SLE. MS is also most prevalent in European populations, as opposed to SLE which is most prevalent in non-European populations (Langer-Gould et al. 2022; Walton et al. 2020). Analyses in different patient populations would be of interest as a future direction. Additionally, all DE was conducted in whole blood datasets, which may not reflect the differential expression of these genes in the CNS or other tissues. Moreover, all genes in the pathway analysis are limited to protein-coding genes and interactions included in STRINGDb. Furthermore, the ability to identify pathways of interest is also limited by the biases of the pathway-prediction platforms used. Finally, the COLOC analysis specifically was limited in that the LD matrix calculation methods were limited to SNPs with a minor allele frequency of greater than 0.01, so some more rare variants may not have been detected by this analysis.
In summary, we have identified a method of identifying additional robust SNP and gene associations within a GWAS of limited sample size. These results have provided information about shared molecular pathways in MS and SLE, as well as potential insight into CNS disease in SLE and novel treatment options.
Data availability
This study did not generate any new datasets or code.
References
Allen ME, Rus V, Szeto GL (2021) Leveraging heterogeneity in systemic lupus erythematosus for new therapies. Trends Mol Med 27(2):152–171. https://doi.org/10.1016/j.molmed.2020.09.009
Banchereau R, Jordan-Villegas A, Ardura M, Mejias A, Baldwin N, Xu H, Saye E, Rossello-Urgell J, Nguyen P, Blankenship D, Creech CB, Pascual V, Banchereau J, Chaussabel D, Ramilo O (2012) Host immune transcriptional profiles reflect the variability in clinical disease manifestations in patients with Staphylococcus aureus infections. PLoS ONE 7(4):e34390. https://doi.org/10.1371/journal.pone.0034390
Barrio-Hernandez I, Schwartzentruber J, Shrivastava A, Del-Toro N, Gonzalez A, Zhang Q, Mountjoy E, Suveges D, Ochoa D, Ghoussaini M (2023) Network expansion of genetic associations defines a pleiotropy map of human cell biology. Nat Genet 55(3):3
Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, Martín J, Fairfax BP, Knight JC, Chen L, Replogle J, Syvänen A-C, Rönnblom L, Graham RR, Wither JE, Rioux JD, Alarcón-Riquelme ME, Vyse TJ (2015) Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet 47(12):1457–1464. https://doi.org/10.1038/ng.3434
Berisa T, Pickrell JK (2016) Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics 32(2):283–285. https://doi.org/10.1093/bioinformatics/btv546
Berry MPR, Graham CM, McNab FW, Xu Z, Bloch SAA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J (2010) An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466(7309):973–977
Bienkowska J, Allaire N, Thai A, Goyal J, Plavina T, Nirula A, Weaver M, Newman C, Petri M, Beckman E, Browning JL (2014) Lymphotoxin-LIGHT pathway regulates the interferon signature in rheumatoid arthritis. PLoS ONE 9(11):e112545. https://doi.org/10.1371/journal.pone.0112545
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, Duncan L, Perry JRB, Patterson N, Robinson EB, Daly MJ, Price AL, Neale BM (2015) An atlas of genetic correlations across human diseases and traits. Nat Genet 47(11):1236–1241
Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG (2017) Sensitivity analyses for robust causal inference from mendelian randomization analyses with multiple genetic variants. Epidemiology 28(1):30–42
Canatar C, Ayaz F (2022) Sensitivity analyses for robust causal inference from mendelian randomization analyses with multiple genetic variants. Eng Applic 1(2):2
Cárdenas-Roldán J, Rojas-Villarraga A, Anaya J-M (2013) How do autoimmune diseases cluster in families? A systematic review and meta-analysis. BMC Med 11(1):73. https://doi.org/10.1186/1741-7015-11-73
Catalina MD, Bachali P, Geraci NS, Grammer AC, Lipsky PE (2019) Gene expression analysis delineates the potential roles of multiple interferons in systemic lupus erythematosus. Commun Biol 2(1):1
Catalina MD, Owen KA, Labonte AC, Grammer AC, Lipsky PE (2020) The pathogenesis of systemic lupus erythematosus: harnessing big data to understand the molecular basis of lupus. J Autoimmun 110:102359. https://doi.org/10.1016/j.jaut.2019.102359
Chen JH, Lee CTC (2022) Explore comorbidities associated with systemic lupus erythematosus: a total population based case control study. QJM 115(1):17–23
Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE (2007) Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol 179(3):1634–1647
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR (2013) Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform 14(1):128. https://doi.org/10.1186/1471-2105-14-128
Chiche L, Jourde-Chiche N, Whalen E, Presnell S, Gersuk V, Dang K, Anguiano E, Quinn C, Burtey S, Berland Y, Kaplanski G, Harle J-R, Pascual V, Chaussabel D (2014) Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthr Rheumatol 66(1583):1595. https://doi.org/10.1002/art.38628
Chisari CG, Sgarlata E, Arena S, Toscano S, Luca M, Patti F (2022) Rituximab for the treatment of multiple sclerosis: A review. J Neurol 269(1):159–183. https://doi.org/10.1007/s00415-020-10362-z
Criswell LA, Pfeiffer KA, Lum RF, Gonzales B, Novitzke J, Kern M, Moser KL, Begovich AB, Carlton VEH, Li W, Lee AT, Ortmann W, Behrens TW, Gregersen PK (2005) Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet 76(4):561–571
Cunningham F, Allen JE, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Austine-Orimoloye O, Azov AG, Barnes I, Bennett R, Berry A, Bhai J, Bignell A, Billis K, Boddu S, Brooks L, Charkhchi M, Cummins C (2022) Ensembl 2022. Nucleic Acids Res 50(988):995
Davey Smith G, Hemani G (2014) Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet 23(R1):R89–R98. https://doi.org/10.1093/hmg/ddu328
Demkova K, Morris DL, Vyse TJ (2023) Genetics of SLE: Does this explain susceptibility and severity across racial groups? Rheumatology 62(15):21
Dilokthornsakul P, Valuck RJ, Nair KV, Corboy JR, Allen RR, Campbell JD (2016) Multiple sclerosis prevalence in the United States commercially insured population. Neurology 86(11):1014–1021. https://doi.org/10.1212/WNL.0000000000002469
Ducreux J, Houssiau FA, Vandepapelière P, Jorgensen C, Lazaro E, Spertini F, Colaone F, Roucairol C, Laborie M, Croughs T, Grouard-Vogel G, Lauwerys BR (2016) Interferon α kinoid induces neutralizing anti-interferon α antibodies that decrease the expression of interferon-induced and B cell activation associated transcripts: analysis of extended follow-up data from the interferon α kinoid phase I/II study. Rheumatology (oxford) 55(10):1901–1905. https://doi.org/10.1093/rheumatology/kew262
Fanouriakis A, Mastorodemos V, Pamfil C, Papadaki E, Sidiropoulos P, Plaitakis A, Amoiridis G, Bertsias G, Boumpas DT (2014) Coexistence of systemic lupus erythematosus and multiple sclerosis: prevalence, clinical characteristics, and natural history. Semin Arthritis Rheum 43(6):751–758. https://doi.org/10.1016/j.semarthrit.2013.11.007
Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJH, Shishkin AA, Hatan M, Carrasco-Alfonso MJ, Mayer D, Luckey CJ, Patsopoulos NA, De Jager PL, Kuchroo VK, Epstein CB, Daly MJ (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 513:7539. https://doi.org/10.1038/nature13835
Faust TW, Chang EH, Kowal C, Berlin R, Gazaryan IG, Bertini E, Zhang J, Sanchez-Guerrero J, Fragoso-Loyo HE, Volpe BT, Diamond B, Huerta PT (2010) Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc Natl Acad Sci USA 107(43):18569–18574. https://doi.org/10.1073/pnas.1006980107
Ghoussaini M, Mountjoy E, Carmona M, Peat G, Schmidt EM, Hercules A, Fumis L, Miranda A, Carvalho-Silva D, Buniello A, Burdett T, Hayhurst J, Baker J, Ferrer J, Gonzalez-Uriarte A, Jupp S, Karim MA, Koscielny G, Machlitt-Northen S (2021) Open targets genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res 49(1311):1320
Graner M, Pointon T, Manton S, Green M, Dennison K, Davis M, Braiotta G, Craft J, Edwards T, Polonsky B, Fringuello A, Vollmer T, Yu X (2020) Oligoclonal IgG antibodies in multiple sclerosis target patient-specific peptides. PLoS ONE 15(2):e0228883. https://doi.org/10.1371/journal.pone.0228883
Gupta S, Kaplan MJ (2021) Bite of the wolf: Innate immune responses propagate autoimmunity in lupus. J Clin Investig 131(3):e144918. https://doi.org/10.1172/JCI144918
Hauser SL, Cree BAC (2020) Treatment of multiple sclerosis: a review. Am J Med 133(12):1380-1390.e2. https://doi.org/10.1016/j.amjmed.2020.05.049
Hemani G, Tilling K, Davey Smith G (2017) Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 13(11):e1007081. https://doi.org/10.1371/journal.pgen.1007081
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, Tan VY, Yarmolinsky J, Shihab HA, Timpson NJ, Evans DM, Relton C, Martin RM, Davey Smith G, Gaunt TR (2018) The MR-Base platform supports systematic causal inference across the human phenome. Elife 7:e34408
Heming M, Müller-Miny L, Rolfes L, Schulte-Mecklenbeck A, Brix TJ, Varghese J, Pawlitzki M, Pavenstädt H, Kriegel MA, Gross CC, Wiendl H (2023) Supporting the differential diagnosis of connective tissue diseases with neurological involvement by blood and cerebrospinal fluid flow cytometry. J Neuroinflammation 20(1):46. https://doi.org/10.1186/s12974-023-02733-w
Hoffman RW, Merrill JT, Alarcón-Riquelme MME, Petri M, Dow ER, Nantz E, Nisenbaum LK, Schroeder KM, Komocsar WJ, Perumal NB, Linnik MD, Airey DC, Liu Y, Rocha GV (2017) Gene expression and pharmacodynamic changes in 1,760 systemic lupus erythematosus patients from two phase III trials of BAFF blockade with tabalumab. Arthr Rheumatol 69(643):654
International Multiple Sclerosis Genetics Consortium (IMSGC), Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, Shah TS, Spencer C, Booth D, Goris A, Oturai A, Saarela J, Fontaine B, Hemmer B, Martin C, Zipp F, D’Alfonso S, Martinelli-Boneschi F, McCauley JL (2013) Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 45(11):1353–1360. https://doi.org/10.1038/ng.2770
International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium, Sawcer S, Hellenthal G, Pirinen M, Spencer CCA, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Compston A (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476(7359):214
Johnson CR, Thacher TD (2023) Vitamin D: Immune function, inflammation, infections and auto-immunity. Paediatr Int Child Health. https://doi.org/10.1080/20469047.2023.2171759
Kain J, Owen KA, Marion MC, Langefeld CD, Grammer AC, Lipsky PE (2022) Mendelian randomization and pathway analysis demonstrate shared genetic associations between lupus and coronary artery disease. Cell Rep Med 3(11):100805. https://doi.org/10.1016/j.xcrm.2022.100805
Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J, Butterworth AS, Staley JR (2019) PhenoScanner V2: An expanded tool for searching human genotype-phenotype associations. Bioinformatics 35(22):4851–4853. https://doi.org/10.1093/bioinformatics/btz469
Kampylafka EI, Alexopoulos H, Kosmidis ML, Panagiotakos DB, Vlachoyiannopoulos PG, Dalakas MC, Moutsopoulos HM, Tzioufas AG (2013) Incidence and prevalence of major central nervous system involvement in systemic lupus erythematosus: a 3-year prospective study of 370 patients. PLoS ONE 8(2):e55843. https://doi.org/10.1371/journal.pone.0055843
Kassi E, Adamopoulos C, Basdra EK, Papavassiliou AG (2013) Role of vitamin D in atherosclerosis. Circulation 128(23):2517–2531. https://doi.org/10.1161/CIRCULATIONAHA.113.002654
Labonte AC, Kegerreis B, Geraci NS, Bachali P, Madamanchi S, Robl R, Catalina MD, Lipsky PE, Grammer AC (2018) Identification of alterations in macrophage activation associated with disease activity in systemic lupus erythematosus. PLoS ONE 13(12):e0208132. https://doi.org/10.1371/journal.pone.0208132
Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, Jun G, DeStefano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thornton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45(12):12
Langefeld CD, Ainsworth HC, Graham DSC, Kelly JA, Comeau ME, Marion MC, Howard TD, Ramos PS, Croker JA, Morris DL, Sandling JK, Almlöf JC, Acevedo-Vásquez EM, Alarcón GS, Babini AM, Baca V, Bengtsson AA, Berbotto GA, Bijl M (2017) Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun 8:16021. https://doi.org/10.1038/ncomms16021
Langer-Gould AM, Gonzales EG, Smith JB, Li BH, Nelson LM (2022) Racial and ethnic disparities in multiple sclerosis prevalence. Neurology 98(18):e1818–e1827. https://doi.org/10.1212/WNL.0000000000200151
Lauwerys BR, Hachulla E, Spertini F, Lazaro E, Jorgensen C, Mariette X, Haelterman E, Grouard-Vogel G, Fanget B, Dhellin O, Vandepapelière P, Houssiau FA (2013) Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon α-kinoid. Arthr Rheum 65(2):447–456. https://doi.org/10.1002/art.37785
Lewis MJ, Jawad AS (2017) The effect of ethnicity and genetic ancestry on the epidemiology clinical features and outcome of systemic lupus erythematosus. Rheumatology 56(67):77
Li X, Zhu X (2017) Cross-phenotype association analysis using summary statistics from GWAS. Methods Mol Biol 1666:455–467
Li Y, Higgs RE, Hoffman RW, Dow ER, Liu X, Petri M, Wallace DJ, Dörner T, Eastwood BJ, Miller BB, Liu Y (2019) A Bayesian gene network reveals insight into the JAK-STAT pathway in systemic lupus erythematosus. PLoS ONE 14(12):e0225651. https://doi.org/10.1371/journal.pone.0225651
Liu Q, Tang B, Zhu Z, Kraft P, Deng Q, Stener-Victorin E, Jiang X (2022) A genome-wide cross-trait analysis identifies shared loci and causal relationships of type 2 diabetes and glycaemic traits with polycystic ovary syndrome. Diabetologia 65(9):1483–1494. https://doi.org/10.1007/s00125-022-05746-x
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H (2013) The genotype-tissue expression (GTEx) project. Nat Genet 45(6):6. https://doi.org/10.1038/ng.2653
Lu H, Zhang J, Jiang Z, Zhang M, Wang T, Zhao H, Zeng P (2021) Detection of genetic overlap between rheumatoid arthritis and systemic lupus erythematosus using GWAS summary statistics. Front Genet 12:656545. https://doi.org/10.3389/fgene.2021.656545
Manca E (2022) Autoantibodies in neuropsychiatric systemic lupus erythematosus (NPSLE): can they be used as biomarkers for the differential diagnosis of this disease? Clin Rev Allergy Immunol 63(2):194–209. https://doi.org/10.1007/s12016-021-08865-2
Marees AT, de Kluiver H, Stringer S, Vorspan F, Curis E, Marie-Claire C, Derks EM (2018) A tutorial on conducting genome-wide association studies: quality control and statistical analysis. Int J Methods Psychiatr Res 27(2):e1608. https://doi.org/10.1002/mpr.1608
Miclea A, Bagnoud M, Chan A, Hoepner R (2020) A brief review of the effects of vitamin D on multiple sclerosis. Front Immunol 11:781. https://doi.org/10.3389/fimmu.2020.00781
Morris JH, Apeltsin L, Newman AM, Baumbach J, Wittkop T, Su G, Bader GD, Ferrin TE (2011) clusterMaker: a multi-algorithm clustering plugin for cytoscape. BMC Bioinform 12(1):436. https://doi.org/10.1186/1471-2105-12-436
Mountjoy E, Schmidt EM, Carmona M, Schwartzentruber J, Peat G, Miranda A, Fumis L, Hayhurst J, Buniello A, Karim MA, Wright D, Hercules A, Papa E, Fauman EB, Barrett JC, Todd JA, Ochoa D, Dunham I, Ghoussaini M (2021) An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat Genet 53(11):1527–1533. https://doi.org/10.1038/s41588-021-00945-5
Obermoser G, Presnell S, Domico K, Xu H, Wang Y, Anguiano E, Thompson-Snipes L, Ranganathan R, Zeitner B, Bjork A, Anderson D, Speake C (2013) Systems scale interactive exploration reveals quantitative and qualitative differences in response to influenza and pneumococcal vaccines. Immunity 38(4):831–844
Okamoto H, Kobayashi A, Yamanaka H (2010) Cytokines and chemokines in neuropsychiatric syndromes of systemic lupus erythematosus. J Biomed Biotechnol 2010:268436. https://doi.org/10.1155/2010/268436
Ota Y, Srinivasan A, Capizzano AA, Bapuraj JR, Kim J, Kurokawa R, Baba A, Moritani T (2022) Central Nervous system systemic lupus erythematosus: pathophysiologic, clinical, and imaging features. Radiographics 42(1):212–232. https://doi.org/10.1148/rg.210045
Owen KA, Price A, Ainsworth H, Aidukaitis BN, Bachali P, Catalina MD, Dittman JM, Howard TD, Kingsmore KM, Labonte AC, Marion MC, Robl RD, Zimmerman KD, Langefeld CD, Grammer AC, Lipsky PE (2020) Analysis of trans-ancestral SLE risk loci identifies unique biologic networks and drug targets in African and European ancestries. Am J Hum Genet 107(5):864–881. https://doi.org/10.1016/j.ajhg.2020.09.007
Owen KA, Grammer AC, Lipsky PE (2022) Deconvoluting the heterogeneity of SLE: the contribution of ancestry. J Allergy Clin Immunol 149(1):12–23. https://doi.org/10.1016/j.jaci.2021.11.005
Owen KA, Bell KA, Price A, Bachali P, Ainsworth H, Marion MC, Howard TD, Langefeld CD, Shen N (2023) Molecular pathways identified from single nucleotide polymorphisms demonstrate mechanistic differences in systemic lupus erythematosus patients of Asian and European ancestry. Sci Rep 13(1):1
Park J, Munagala I, Xu H, Blankenship D, Maffucci P, Chaussabel D, Banchereau J, Pascual V, Cunningham-Rundles C (2013) Interferon signature in the blood in inflammatory common variable immune deficiency. PLoS ONE 8(9):e74893. https://doi.org/10.1371/journal.pone.0074893
Petri M, Fu W, Ranger A, Allaire N, Cullen P, Magder LS, Zhang Y (2019) Association between changes in gene signatures expression and disease activity among patients with systemic lupus erythematosus. BMC Med Genomics 12(1):4. https://doi.org/10.1186/s12920-018-0468-1
Ren J, Catalina MD, Eden K, Liao X, Read KA, Luo X, McMillan RP, Hulver MW, Jarpe M, Bachali P, Grammer AC, Lipsky PE, Reilly CM (2019) Selective histone deacetylase 6 inhibition normalizes B cell activation and germinal center formation in a model of systemic lupus erythematosus. Front Immunol 10:2512. https://doi.org/10.3389/fimmu.2019.02512
Richard-Miceli C, Criswell LA (2012) Emerging patterns of genetic overlap across autoimmune disorders. Genome Med 4(1):6. https://doi.org/10.1186/gm305
Rizzoni D, Rizzoni M, Nardin M (2019) Vitamin D and ischaemic heart disease: a casual or A causal association? High Blood Press Cardiovasc Prev 26(2):151–155. https://doi.org/10.1007/s40292-019-00302-y
Sarwar S, Mohamed AS, Rogers S, Sarmast ST, Kataria S, Mohamed KH, Khalid MZ, Saeeduddin MO (2021) neuropsychiatric systemic lupus erythematosus: a 2021 update on diagnosis management, and current challenges. Cureus 13(9):17969
Sintzel MB, Rametta M, Reder AT (2017) Vitamin D and multiple sclerosis: a comprehensive review. Neurol Therapy 7(1):59–85. https://doi.org/10.1007/s40120-017-0086-4
Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, Paul DS, Freitag D, Burgess S, Danesh J, Young R, Butterworth AS (2016) PhenoScanner: a database of human genotype-phenotype associations. Bioinformatics 32(20):3207–3209. https://doi.org/10.1093/bioinformatics/btw373
Szklarczyk D, Kirsch R, Koutrouli M, Nastou K, Mehryary F, Hachilif R, Gable AL, Fang T, Doncheva NT, Pyysalo S, Bork P, Jensen LJ, von Mering C (2023) The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res 51(D1):D638–D646. https://doi.org/10.1093/nar/gkac1000
Uffelmann E, Huang QQ, Munung NS et al (2021) Genome-wide association studies. Nat Rev Methods Primers 1(1):1
Wallace C (2021) A more accurate method for colocalisation analysis allowing for multiple causal variants. PLoS Genet 17(9):e1009440. https://doi.org/10.1371/journal.pgen.1009440
Walton C, King R, Rechtman L, Kaye W, Leray E, Marrie RA, Robertson N, La Rocca N, Uitdehaag B (2020) Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Multiple Scler 26(14):1816–1821
Wang J, Dai X, Berry LD, Cogan JD, Liu Q, Shyr Y (2019) HACER: an atlas of human active enhancers to interpret regulatory variants. Nucleic Acids Res 47(D1):D106–D112. https://doi.org/10.1093/nar/gky864
Wang YF, Zhang Y, Lin Z, Zhang H, Wang TY, Cao Y, Morris DL, Sheng Y, Yin X, Zhong SL (2021) Identification of 38 novel loci for systemic lupus erythematosus and genetic heterogeneity between ancestral groups. Nat Commun 12(1):1
Wang X, Feng D, Ke Y, Gu L, Lv C, Zhang M, Wang Q, Wang Y (2022) Brain-reactive antibodies are potential biomarkers for evaluating therapeutic efficacy in NPSLE patients. Neuropsychiatr Dis Treat 18:1329–1340. https://doi.org/10.2147/NDT.S359698
Wen J, Stock AD, Chalmers SA, Putterman C (2016) The role of B cells and autoantibodies in neuropsychiatric lupus. Autoimmun Rev 15(9):890–895. https://doi.org/10.1016/j.autrev.2016.07.009
Wu M, Yang J, Li X, Chen J (2016) The role of γδ T Cells in systemic lupus erythematosus. J Immunol Res 2016:2932531. https://doi.org/10.1155/2016/2932531
Zhou A, Selvanayagam JB, Hyppönen E (2022) Non-linear Mendelian randomization analyses support a role for vitamin D deficiency in cardiovascular disease risk. Eur Heart J 43(18):1731–1739. https://doi.org/10.1093/eurheartj/ehab809
Zhu X, Feng T, Tayo BO, Liang J, Young JH, Franceschini N, Smith JA, Yanek LR (2015) Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am J Hum Genet 96(1):21–36
Zuber V, Grinberg NF, Gill D, Manipur I, Slob EAW, Patel A, Wallace C, Burgess S (2022) Combining evidence from Mendelian randomization and colocalization: review and comparison of approaches. Am J Human Genet 109(5):767–782. https://doi.org/10.1016/j.ajhg.2022.04.001
Acknowledgements
The work presented in this manuscript was funded by a grant awarded to P.E.L. and A.C.G. of the RILITE Research Institute by the John and Marcia Goldman Foundation. The funder provided support in the form of salaries for authors (S.K, K.A.O. and D.S.). The authors gratefully acknowledge additional support from the Lupus Research Alliance (LRA).
Funding
John and Marcia Goldman Foundation, Lupus Research Alliance
Author information
Authors and Affiliations
Contributions
Conceptualization: P.E.L., A.C.G., and K.A.O; methodology, P.E.L; software, formal analysis, and investigation: S.K.; writing-original draft: S.K., K.A.O., and P.E.L.; writing-review and editing: K.A.O, P.E.L; funding acquisition: P.E.L and A.C.G.; Supervision: P.E.L. and A.C.G.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kerns, S., Owen, K.A., Schwalbe, D. et al. Examination of the shared genetic architecture between multiple sclerosis and systemic lupus erythematosus facilitates discovery of novel lupus risk loci. Hum. Genet. 143, 703–719 (2024). https://doi.org/10.1007/s00439-024-02672-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-024-02672-3