
Abstract
Long non-coding RNA (lncRNA) genes represent a large class of transcripts that are widely expressed across species. As most human lncRNAs are non-conserved, we recently employed a unique humanized liver mouse model to study lncRNAs expressed in human livers. We identified a human hepatocyte-specific lncRNA, hLMR1 (human lncRNA metabolic regulator 1), which is induced by feeding and promotes hepatic cholesterol synthesis. Recent genome-wide association studies (GWAS) found that several single-nucleotide polymorphisms (SNPs) from the hLMR1 gene locus are associated with blood lipids and markers of liver damage. These results suggest that dietary and genetic factors may regulate hLMR1 to affect disease progression. In this study, we first screened for nutritional/hormonal factors and found that hLMR1 was robustly induced by insulin/glucose in cultured human hepatocytes, and this induction is dependent on the transcription factor SREBP1. We then tested if GWAS SNPs genetically linked to hLMR1 could regulate hLMR1 expression. We found that DNA sequences flanking rs9653945, a SNP from the last exon of the hLMR1 gene, functions as an enhancer that can be robustly activated by SREBP1c depending on the presence of rs9653945 major allele (G). We further performed CRISPR base editing in human HepG2 cells and found that rs9653945 major (G) to minor (A) allele modification resulted in blunted insulin/glucose-induced expression of hLMR1. Finally, we performed genotyping and gene expression analyses using a published human NAFLD RNA-seq dataset and found that individuals homozygous for rs9653945-G have a higher expression of hLMR1 and risk of NAFLD. Taken together, our data support a model that rs9653945-G predisposes individuals to insulin/glucose-induced hLMR1, contributing to the development of hyperlipidemia and NAFLD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hyperlipidemia is defined as increased blood lipid levels, mainly cholesterol and triglycerides. It is a critical risk factor for heart attack, stroke, and many other cardiometabolic diseases. Currently, hyperlipidemia affects about one-third of the adult population in the U.S. and its management presents a substantial social and financial burden for modern society (Toth et al. 2012). The liver is one of the most important organs in maintaining blood lipid homeostasis. Given its central role in controlling circulating lipids, it is not surprising that chronic liver diseases, including nonalcoholic fatty liver disease (NAFLD), are major risk factors for many cardiovascular disorders. While epidemiological studies support that genetic variants and diets rich in lipids/carbohydrates contribute to NAFLD and hyperlipidemia, how they coordinate to affect hepatic lipid metabolism and promote cardiometabolic diseases in humans has yet to be fully understood.
LncRNAs represent a large class of transcripts that are widely expressed across all mammalian species. Recent lncRNA studies in animal models proved they are crucial nutrient sensors that regulate vital metabolic pathways in a tissue-specific manner (Li et al. 2015; Ruan et al. 2016; Sallam et al. 2018; Yang et al. 2016; Zhao et al. 2014). As most of human lncRNAs are non-conserved across species (Johnsson et al. 2014), we recently used a unique humanized liver mouse model (Hasegawa et al. 2011) to study the regulation and function of human lncRNAs in a physiologically relevant setting (Ruan et al. 2020, 2021). We defined several non-conserved human lncRNA metabolic regulators (hLMRs). One of these human lncRNAs, hLMR1 (Ensemble ID: ENSG00000243818), is exclusively expressed in hepatocytes of the human liver. hLMR1 is upregulated by feeding in the humanized liver mouse model and promotes cholesterol synthesis (Ruan et al. 2021), a process with reported causal effects in both hyperlipidemia and NAFLD (Horn et al. 2022). Consistent with these results, recent GWAS revealed that genetic variants within and around the hLMR1 gene locus are associated with both blood cholesterol levels (Graham et al. 2021), as well as chronic alanine transaminase (Sakaue et al. 2021) (ALT), a biomarker of NAFLD. These experimental and human genetic data suggest that hLMR1 may integrate dietary and genetic signals to promote cholesterol synthesis contributing to the development of hyperlipidemia and NAFLD.
In this study, we found that hLMR1 is robustly induced by insulin/glucose. This induction is controlled by a non-coding genetic variant (rs9653945) associated with the risk of hyperlipidemia and NAFLD. Analyzing a human NAFLD dataset further supports that individuals carrying the risk allele of rs9653945 have elevated expression of hLMR1 and an increased risk of NAFLD. Our study thus provides an example that genetic variants (rs9653945-G) interact with dietary factors (glucose) to regulate human-specific genes (hLMR1) that contribute to the progression of both hyperlipidemia and NAFLD. Our work also supports the potential of blocking hLMR1 as a therapeutic strategy for cardiometabolic diseases.
Results
Upregulation of hLMR1 by insulin/glucose
Our previous studies showed that there were 14 isoforms detected in human liver full length RNA-seq analysis (PacBio sequencing). All these detected isoforms have similar gene structure with differences on a few exons. For detecting hLMR1 expression, we designed the primer on the last exon, which is shared by all isoforms. For overexpression of hLMR1, we cloned hLMR1 from a pooled human liver cDNA library, and the cloned isoform covered all the major exons of hLMR1 (Ruan et al. 2021). We found that hLMR1 was suppressed by fasting and induced by refeeding in humanized liver mice (Ruan et al. 2021). In a clinical study, hLMR1 was downregulated by a low-carb diet intervention in obese patients with NAFLD (Mardinoglu et al. 2018) (Supplementary Fig. 1A). These observations indicate that feeding-associated factors like insulin/glucose may regulate the expression of hLMR1. To test this, we treated cultured human HepG2 hepatocytes for eight hours with either 2.5 mM glucose (low glucose), low glucose plus 100 nM insulin, 25 mM glucose (high glucose), or high glucose plus insulin. We found that while insulin and glucose alone showed a mild effect on inducing hLMR1, the combination showed a stronger effect (Supplementary Fig. 1B). We next extended the treatment to 24 h and found that hLMR1 was dramatically upregulated by the combination of insulin and high glucose (Fig. 1A). We noticed that high glucose alone achieved similar induction as high glucose plus insulin in the 24 h treatment conditions, suggesting that glucose concentration is the major factor determining hLMR1 expression. SREBP1c, a known lipogenic transcription factor mediating insulin/glucose-induced gene expression (Wang et al. 2015) was also upregulated in this setting (Fig. 1A). To test if glucose/insulin-induced expression of hLMR1 is dependent on SREBP1c, we knocked down SREBP1 using siRNAs in HepG2 cells treated with insulin/glucose. We found this procedure led to decreased expression of hLMR1 (Fig. 1B). These data support that feeding-associated increase of glucose and insulin upregulates hLMR1 expression through SREBP1.

A Relative expression of hLMR1 in HepG2 cells treated with low (2.5 mM) or high (25 mM) glucose combined with or without 100 nM insulin for 24 h (n = 3 for each group). B Relative mRNA expression in HepG2 cells transfected with siRNA control or siRNA targeting SREBP1 and incubated with high glucose and insulin for 24 h (n = 3 for each group). Data shown as the mean ± SEM. *p < 0.05, **p < 0.01 and ***p < 0.001
rs9653945 is a functional SNP that affects SREBP1c activation
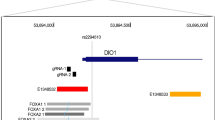
In line with hLMR1’s reported role promoting cholesterol synthesis (Ruan et al. 2021), recent GWAS in general populations and in U.S. veterans found that several SNPs from the hLMR1 gene locus are associated with blood low-density lipoprotein (LDL) cholesterol and ALT levels (Graham et al. 2021; Klarin et al. 2018; Qi and Chatterjee 2018; Sakaue et al. 2021) (Fig. 2A and C). Interestingly, these SNPs are also associated with hepatic expression of hLMR1 based on eQTL data from The Genotype-Tissue Expression project (Consortium 2013) (Fig. 2B). Specifically, for SNPs from this linked region (r2 < 0.8), risk alleles (average allele frequency 65%) that are associated with higher blood lipids/ALT are also linked to higher expression of hLMR1, suggesting these common genetic variants may regulate the expression of hLMR1. Among these SNPs, we were interested in rs9653945 due to its strong association with blood LDL-cholesterol and ALT levels (Fig. 2C), and the hepatic expression of hLMR1 (Fig. 2B) and reported binding events of SREBP1 in the rs9653945 DNA region (Consortium 2012) (Fig. 2D). Furthermore, ChIP analysis in human livers showed that the rs9653945 DNA region is within the peak of enhancer markers, including H3K4me1 and H3K27ac (Roadmap Epigenomics et al. 2015) (Fig. 2D). These observations indicate that the rs9653945 DNA region may interact with SREBP1 in the liver and serve as an enhancer that potentially affects hLMR1 expression. To test this, we prepared a construct that tests the rs9653945 DNA region for enhancer activity upstream of a mini promoter-driven luciferase reporter (Pasquali et al. 2014) (Fig. 2E). We found that SREBP1c robustly activated the reporter carrying the rs9653945 major allele G, while the reporter carrying the rs9653945 minor allele A showed no response to SREBP1c (Fig. 2E). This result suggests that rs9653945 is a causal SNP linking hepatic expression of hLMR1 to blood lipids and ALT.

A Illustration of the blood lipid-associated SNPs from the hLMR1 gene locus. Red, rs9653945; B Violin plots of hLMR1 QTL data for rs11709868, rs9832727, rs4683438, and rs9653945. Data are adapted from GTEx; C Association of rs9653945-A with decreased circulating LDL-cholesterol, triglyceride, and alanine transaminase levels. Data are adapted from GWAS Catalog (Sollis et al. 2023) (The NHGRI-EBI Catalog of human genome-wide association studies); D Illustration of SREBP1 ChIP-seq in HepG2 cells (https://www.encodeproject.org/experiments/ENCSR000EZP/), H3K4me1 (https://www.encodeproject.org/annotations/ENCSR273DRG/) and H3K27ac (https://www.encodeproject.org/annotations/ENCSR235UQA/) ChIP-seq in human livers in rs9653945 DNA region. Data were visualized in UCSC genome browser. As point by the red arrowhead, the SREBP1 ChIP-seq peak covered the rs9653945 region. rs9653945 was indicated by a dotted line; E Top, illustration of the reporter used; bottom, luciferase assay testing the effects of expressing an active SREBP1c on reporters (n = 3 for each group). Data shown as the mean ± SEM, **p < 0.01 for comparation between Control and SREBP1c
CRISPR base editing of rs9653945 in HepG2 cells affects insulin/glucose-induced hLMR1
To further understand the role of rs9653945 in a native genomic context, we seek to use CRISPR base editing tools to modify the rs9653945 genomic loci in HepG2 cells. In line with our observation that insulin/glucose can robustly activate hLMR1 expression, Taqman genotyping analysis revealed that HepG2 cells are homozygous for the rs9653945 major allele G (Supplementary Fig. 2). As such, we decided to use cytosine base editors (CBEs), which convert C•G base pairs to T•A base pairs (Huang et al. 2021). As no classical NGG PAM motif can be used to design a sgRNA that can include the G allele in the optimized editing window, we decided to use the CBE version that can recognize sgRNAs with an NG PAM motif (Huang et al. 2021). As shown in Fig. 3A, we cloned the designed sgRNA into the sgRNA vector and then co-transfected it with several available CBE-NGs in HepG2 cells. We then purified the genomic DNA and amplified the rs9653945 region for Sanger Sequencing. Among the CBE-NGs, BE4max-NG achieved the best editing efficiency and accuracy (Huang et al. 2021; Thuronyi et al. 2019). As shown in Fig. 3B, around 50% of the G allele was edited to the A allele without unwanted editing. Next, we treated HepG2 cells with high glucose plus insulin, as in Fig. 1A, and found an induced hLMR1 expression in cells with control editing, and significantly compromised induction of hLMR1 in cells with rs9653945 G-to-A editing (Fig. 3C). As a control, the expression of PAQR9, the closest neighbor gene of hLMR1, was mildly upregulated by high glucose plus insulin, but this regulation was not affected by rs9653945 G-to-A editing (Fig. 3D), suggesting the specific regulation of hLMR1 by rs9653945. We further performed a SREBP1 Chromatin immunoprecipitation (ChIP) assay in HepG2 cells with control or rs9653945 base editing in this setting (Fig. 3E). Consistent with the published SREBP1 ChIP-seq data in Fig. 2D, we found SREBP1 showed a strong interaction with the rs9653945 DNA region, and this interaction was reduced by rs9653945 G to A editing. As a negative control, RPL30 showed no enrichment by SREBP1 ChIP. As a positive control, the primer amplifying LDLR (low-density lipoprotein receptor) promoter region was enriched by SREBP1 ChIP (Reed et al. 2008) and this enrichment was not affected by rs9653945 base editing (Fig. 3E). Together with our luciferase reporter assay, these data support that the rs9653945 G allele enhances SREBP1 binding to induce hLMR1 expression.

A Illustration of the design of sgRNA targeting the G allele of rs9653945. B Sanger sequencing of the DNA amplified from Control (no editing) or base editing of rs9653945 HepG2 cells. rs9653945 was pointed out by a red arrow. The G to A switch in edited cells was pointed by a red arrowhead. C hLMR1 and D PAQR9 expression in HepG2 cells with control or rs9653945 G-to-A editing (n = 3 for each group). Data shown as the mean ± SEM. n.s. not significant. *p < 0.05, and ***p < 0.001. E SREBP1 ChIP analyses in HepG2 cells with control or rs9653945 base editing under 25 mM glucose plus 100 nM insulin treatment. Experiments were performed using pooled samples from 24 well plates. The relative enrichment was calculated by normalizing the amount of ChIP DNA to input DNA and comparing with the IgG control (set as 1) as fold enrichment
rs9653945 does not affect the stability of hLMR1
We noticed that rs9653945 localizes on the last exon of hLMR1, which is shared by all hLMR1 isoforms in human livers (Ruan et al. 2021). As such, rs9653945 can potentially affect hLMR1 expression at both the DNA (as demonstrated above) and RNA level (hLMR1-A vs hLMR1-G). To address this, we prepared the constructs that express hLMR1-A or hLMR1-G and transfected them in HepG2 cells and chased their degradation. As shown in Supplementary Fig. 3A, hLMR1-A and hLMR1-G were expressed at a comparable level and showed a similar decay rate with a half-life time of about six hours. The mRNA half-life time of control genes, including GAPDH and SREBP1, was not affected by hLMR1-G as compared with hLMR1-A.
rs9653945 associates with the expression of hLMR1 and the progression of NAFLD
Together with the published eQTL (Consortium 2013) (Fig. 2B) and GWAS data (Graham et al. 2021; Klarin et al. 2018; Sakaue et al. 2021) (Fig. 2C), our experimental analyses established the causality of rs9653945, which potentially links hLMR1 expression with blood cholesterol and ALT levels. To further explore the pathophysiological significance of the rs9653945-hLMR1 axis, we next asked if we could directly capture these associations in a single dataset of metabolic diseases. To address this, we re-analyzed a published RNA-seq dataset of human NAFLD, which includes health controls, obese individuals, NAFLD and nonalcoholic steatohepatitis (NASH) patients (Suppli et al. 2019). As rs9653945 is on the largest and shared exon of all hLMR1 isoforms, we were able to determine the genotyping information of rs9653945 and match this information with the expression of hLMR1 or status of disease (see “Methods”). As shown in Fig. 4A, in this disease dataset, the average expression of hLMR1 is higher in the rs9653945 (G/G) group than in the rs9653945 (A/A) group. Compared with health/obese individuals, NAFLD and NASH groups show a trend of higher frequency of rs9653945-G allele (Fig. 4B and Supplementary Table 1).

A DESeq2-normalized counts of hLMR1 for each genotype of rs9653945. Each dot represents a normalized count of hLMR1 in each sample. The colored lines indicate the mean of hLMR1 expression in each genotype of the groups. B The proportion of rs9653945 genotypes (%) in Healthy/Obese, or NAFLD/NASH group
Discussion
hLMR1 mediated linkage among insulin resistance, hyperlipidemia and NAFLD
Cardiometabolic diseases, including hyperlipidemia, NAFLD and type 2 diabetes, are highly correlated, but the underlying genetic and dietary factors contributing to this correlation are unclear. This study provides evidence that insulin resistance associated hyperglycemia, can robustly induce hepatic expression of hLMR1 (Fig. 1A), a human-specific lncRNA that promotes cholesterol synthesis and affects circulating cholesterol levels (Ruan et al. 2021). As increased cholesterol deposit in the liver is relevant to the pathogenesis of NAFLD, our studies may partially explain the tight connection among insulin resistance, hyperlipidemia and NAFLD. We provide evidence that this regulatory cascade is dependent on the presence of the major allele of rs9653945, a common genetic variant that is associated with blood lipids as well as the risk of NAFLD (Fig 2B and C). As both the genetic variant and hLMR1 are non-conserved, our work thus defined a human-specific metabolic regulation and emphasize the importance of using a human-like system to understand metabolic regulation in health and diseases.
Rs9653945 DNA region as an exonic enhancer
It is known that cardiometabolic diseases are caused by an interplay between genetic and dietary factors, but how they coordinate with each other to affect disease progress is not clear. In our study, we noticed that at the basal condition in either the reporter assay (vector control) or the treatment experiment (low glucose group), the activity of the reporter or the expression of hLMR1 showed no differences between the setting of rs9653945 major allele G and minor allele A. However, when an active SREBP1c was introduced in the reporter assay or a high insulin plus glucose treatment was applied, the effects of G/A alleles started to differentiate, with the G allele showing a higher activity interacting with SREBP1 to induce hLMR1 expression (Fig. 3E and C), suggesting that the rs9653945 DNA region may serve as an exonic enhancer. Exonic enhancers are DNA elements from exon regions that also function as enhancers of the gene in which they reside or (a) nearby gene(s) (Ahituv 2016). Exonic enhancers can be activated by the binding of transcription factors and cofactors, which subsequently leads to the activation of their target promoters, either through looping interactions between the enhancer and the promoter or via other mechanisms such as tracking or chromatin modifications. Our data suggest that DNA region flanking rs9653945 serves as an enhancer to activate hLMR1 expression possibly through looping interactions with hLMR1 promoter.
CRISPR base editing of rs9653945
The traditional strategies to experimentally define causal SNPs heavily rely on reporter assay. While it is straightforward and powerful, cloning-based reporter assay may not be able to capture long-range interactions among regulatory DNA elements. Our study combined traditional reporter assay (Fig. 2E) with CRISPR-mediated G to A switch in human HepG2 cells (Fig. 3) to demonstrate the causality of rs9653945 in controlling hLMR1 expression. We noticed that the basal expression level of hLMR1 significantly increased when HepG2 cells were transfected with CRISPR base editors. We speculate that the high basal expression of hLMR1 in base editing condition explains the less fold induction in Fig. 3C comparing with Fig. 1A. Furthermore, although we achieved about 50% editing efficiency in HepG2 cells, screening for HepG2 subclones that are homozygous of rs9653945-A after editing will be helpful to further characterize rs9653945 in regulating hLMR1. Due to the reprogramed gene expression/metabolism in HepG2 cells, it is difficult for us to observe the downstream effects of the rs9653945-hLMR1 axis in regulating the expression of genes in the cholesterol synthesis pathway and circulation lipids. Future work performing high-efficiency CRISPR base editing using viral vectors in highly repopulated humanized liver mice will be able to map the full causality chain from the genetic variant to target genes to phenotype/diseases.
hLMR1 as a therapeutic target for cardiometabolic diseases
GWAS SNPs have been traditionally assigned to the closest protein-coding genes in linear distance. Even though lncRNAs are important regulators of many biological processes and frequently harbor GWAS SNPs, their roles in mediating the effects of GWAS SNPs are understudied. In our study, we provide evidence that rs9653945 is causal and acts through regulating the expression of hLMR1, but not PAQR9, the closest protein-coding gene that is also highly expressed in the liver (Fig. 3D). rs9653945 may still affect other genes in long distance, RNA-seq analysis to systemically define differentially expressed genes in the setting of rs9653945 G-to-A editing and analyses, including trans-eQTL calculation (Vosa et al. 2021), and SNP-gene 3D interaction (Orozco et al. 2022) can be performed to prioritize these differentially expressed genes for further functional studies. Nevertheless, as hLMR1 is highly and specifically expressed in human livers, with experimental data supporting its role in promoting cholesterol synthesis in humanized liver mice (Ruan et al. 2021), and the new data we provided in this study establishing the causality of insulin/glucose-rs9653945-hLMR1 cascade in hyperlipidemia and NAFLD, targeting hLMR1 using pharmacologically approved strategies like anti-sense DNA oligos could be promising for treating/managing cardiometabolic diseases in patients homozygous for rs9653945-G.
Methods
Cell culture, treatment, and transfection
HepG2 cells (ATCC Cat# HB-0865) and 293A cells (ThermoFisher Cat# R70507) were cultured in DMEM (ThermoFisher Cat# 11,965,118) supplemented with 10% FBS (ThermoFisher Cat# 26,140,079) at 37 °C in a 5% CO2 atmosphere. For the insulin/glucose treatment, HepG2 cells were cultured on a 24-well plate overnight until reaching a confluence of ~ 70%. After incubation, cells were washed two times with PBS pH 7.4 and incubated overnight in DMEM without glucose (ThermoFisher Cat# 11,966,025) supplemented with 2.5 mM of glucose (Sigma Cat# G8270-100G) at 37 °C in a 5% CO2 atmosphere. The next day, cells were washed with PBS pH 7.4 and incubated in DMEM without glucose supplemented with 2.5 mM of glucose or 25 mM of glucose, these treatments were performed with or without 100 nM insulin (Millipore Cat# I0516-5 mL), as stated for each experiment. After the incubation, cells were washed with ice-cold PBS, and RNA was extracted and used for gene expression analysis. HepG2 cells were transfected with 20 nM of siRNA-B (Control; SCBT Cat# sc-44230) or siRNA-SREBP1 (Dharmacon Cat# L-006891–00) using Lipofectamine RNAiMax (ThermoFisher Cat# 13,778–150), following the manufacturer’s instructions.
RNA extraction, cDNA synthesis, and quantitative PCR
The RNA was purified using the Qiagen RNeasy Mini Kit (Qiagen Cat# 74,106) following the manufacturer’s instructions and performing the on-column DNA digestion. To obtain the cDNA, 500 ng of RNA was used for the reverse transcription using the SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen Cat# 11,752,050). The cDNA was diluted tenfold before qPCR, and then, the Power SYBR Green PCR Master Mix (ThermoFisher Cat# 4,368,702) was used for qPCR analysis. The qPCR was performed in a ThermoFisher Quantstudio 7 Flex using a 386-well plate (ThermoFisher Cat# 4,309,849) using ΔΔCT for gene quantitation in a Standard run. The PCR program was 2 min 30 s at 95 °C for enzyme activation, 40 cycles of 15 s at 95 °C, and 1 min at 60 °C. Melting curve analysis was performed to confirm the real-time PCR products. qPCR primers for hLMR1 and all other genes are included in Supplementary Table 2, and human RPL13A gene is used for normalization.
RNA decay assay in HepG2 cells
The pcDNA3.1 plasmid with the hLMR1 sequence carrying rs9653945-A (hLMR1-A) was obtained from (Ruan et al. 2021). hLMR1-G was prepared using directed point mutagenesis by PCR with the oligonucleotides as listed in Supplementary Table 2.
HepG2 cells were transfected with 500 ng of pcDNA3.1 hLMR1-A or hLMR1-G using GenJet In Vitro DNA Transfection Reagent for HepG2 Cells. 24 h After transfection, the cells were washed with PBS pH 7.4 and incubated in DMEM supplemented with 10% FBS and 0.5 µg/mL of Actinomycin D (Millipore Cat# A9415-10MG) as indicated, and RNA was extracted and used for gene expression analysis.
Luciferase reporter assay
A 120-base pair DNA sequence flanking rs9653945-A or -G was synthesized from IDT (Supplementary Table 2) and cloned into a destination vector pGL4.23-GW using Gateway cloning. HepG2 cells were cultured on a 24-well plate and transfected with 300 ng of the rs9653945-A or -G reporter, 100 ng of the vector control or an active form of SREBP1c, in combination with 25 ng pRL-TK renilla luciferase vectors. The luciferase reporter activity was measured using the Dual-Luciferase Reporter Assay System (Promega Cat# E1960) following the manufacturer’s instructions.
Genotyping of rs9653945 in HepG2 cells
HepG2 cells genotype was determined using a TaqMan probe assay (ThermoFisher Cat# 4,371,353), following the manufacturer’s protocol. Briefly, the DNA from the cells was purified using the Allprep DNA/RNA/Protein Mini Kit (QIAGEN Cat# 80,004). Then, 10 ng of DNA from the cells was used for the genotyping reaction using rs9653945 custom probes (ThermoFisher Cat# 4,331,349). The G-allele probe was tagged with the Vic dye label and the A-allele probe with the Fam dye label. As a control for the reaction, we used 1 ng of pcDNA3.1 plasmid containing the sequence of hLMR1-A or hLMR1-G (Supplementary Table 3).
CRISPR-Cas9 base editing of rs9653945 in HepG2 cells
The base editing of the target SNP rs9653945 in HepG2 cells was performed following the protocol in (Huang et al. 2021). Briefly, the sgRNA was cloned into the plasmid pFYF1320 (Addgene Cat# 47,511) using the USER cloning method (NEB Cat# M5505). The insertion of the sgRNA into the pFYF1320 plasmid was confirmed using SANGER sequencing. HepG2 cells were plated in a 24 well-plate and left to grow overnight until reaching a confluence of ~ 70%. Then, the transfection was performed using the GenJet In Vitro DNA Transfection Reagent for HepG2 Cells (SignaGen Cat# SL100488-HEPG2), following the manufacturer’s transfection protocol. 500 ng of target pFYF1320 plasmid and 1ug of the base editor plasmid pBT376 (Addgene Cat# 125,617) were used for the cell’s transfection using 4.5 µL of GenJet reagent. After performing the CRISPR-Cas9 Base Editing of rs9653945 in HepG2 cells, the cells were washed twice with PBS pH 7.4 and incubated overnight in DMEM without glucose supplemented with 2.5 mM of glucose. The next morning, cells were washed with PBS pH 7.4 and incubated in DMEM without glucose supplemented with 2.5 mM of glucose or 25 mM of glucose and 100 nM insulin. After 24 h of incubation, cells were washed with ice-cold PBS, and RNA was extracted and used for gene expression analysis and DNA was extracted for PCR amplification-Sanger Sequencing for validating designed base editing.
Chromatin immunoprecipitation (ChIP)
ChIP assays in HepG2 cells were performed using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology) according to the manufacturer’s protocol. Briefly, HepG2 cells were cultured in 24 well plates and transfected with CRISPR base editors as indicated following 24 h treatment of 25 mM glucose plus 100 nM insulin. Cells were crosslinked using formaldehyde at a final concentration of 1% for 15 min. After crosslinking, cells from each group were pooled and processed following the the manufacturer’s protocol.
Each IP was performed using 5 µg chromatin with 5 µg antibody as indicated. The antibodies used are Rabbit IgG from the kit (Cell Signaling Cat #2729) and SREBP1 antibody (Cell Signaling Technology, Cat #95879S). The immunoprecipitation (IP) was performed in a total volume of 500 µL overnight at 4 °C with rotation, and 30 µL of Protein G magnetics beads #9006 were added with the same IP conditions. After the incubation, the beads were washed 3 times with low salt buffer and 1 time with high salt buffer, and they were incubated at 65 °C for 30 min for elution. The elution was treated with Proteinase K for 2 h, and the DNA was purified using the QIAquick PCR purification kit (Qiagen Cat# 28,105) eluting the DNA into 30 µL of Nuclease-free water (Cat #AM9932). The DNA in each ChIP were determined by qPCR analysis using primers as indicated in Supplementary Table 2. Please note, as the region amplified by hLMR1 qPCR primers covers rs9653945 DNA region, the sample primer set was also used for determining SREBP1 enrichment at rs9653945 locus. The relative enrichment was calculated by normalizing the amount of ChIP DNA to input DNA and comparing with the IgG control as fold enrichment.
RNA-seq analysis
The RNA-seq dataset (BioProject No. PRJNA523510) (Suppli et al. 2019) was downloaded from NCBI. This dataset contains 57 samples from healthy normal-weight (n = 14) and obese (n = 12) individuals, NAFL (n = 15), and NASH (n = 16) patients. The Fastq files of the dataset were downloaded and prepared using the SRA Toolkit (version 2.11.1) [https://www.ncbi.nlm.nih.gov/books/NBK569238/]. Briefly, the prefetch command was used to download the SRA files of the dataset and convert them into FASTQ format using the fasterq-dump command. The quality of the Fastq files was evaluated using FastQC (version 0.11.8) [http://www.bioinformatics.babraham.ac.uk/projects/fastqc/], and adapters and low-quality reads were trimmed or removed using Trimmomatic (version 0.39) (Bolger et al. 2014). The filtered reads were then mapped to the GRCh38 Ensembl human genome using STAR (version 2.7.8a) (Dobin et al. 2013). Genotypes near rs9653945 were determined using the mpileup and call commands from BCFtools (version 1.9) (Li 2011). Gene-level counts were generated using featureCounts from Subread (version 2.0.0) (Liao et al. 2013) by using uniquely mapped reads in the union exon region of the human genome (Ensembl human annotation, GRCh38 release 106). The raw count data was normalized using DESeq2 (version 1.36.0) (Love et al. 2014), and normalized counts of genes were extracted using the plotCounts function from DESeq2 for drawing the normalized count–genotype plots.
For plotting hLMR1 expression after low-carb diet intervention in obese patients, NCBI dataset GSE107650 (Mardinoglu et al. 2018) was reanalyzed by modifying the GEO2R script to retrieve normalized counts for hLMR1 (ENSG00000243818).
Oligonucleotides and plasmids
All oligonucleotides and gene sequences used for cloning were ordered from IDT and are listed in Supplementary Table 2. All the plasmids used for this work and their source are listed in Supplementary Table 3.
Statistical analysis
One-way ANOVA statistical analysis and Tukey HSD test was performed for Fig. 1A, B, Fig. 2E, 3C and D, 4A and Supplementary Fig. 1B. Fisher's exact test was performed for the counts of genotype x phenotype contingency table in Fig. 4B. The Paired Samples t Test was performed for Supplementary Fig. 1A.
Data availability
All data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Ahituv N (2016) Exonic enhancers: proceed with caution in exome and genome sequencing studies. Genome Med 8:14. https://doi.org/10.1186/s13073-016-0277-0
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Consortium EP (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. https://doi.org/10.1038/nature11247
Consortium GT (2013) The Genotype-Tissue Expression (GTEx) project. Nat Genet 45:580–585. https://doi.org/10.1038/ng.2653
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Graham SE, Clarke SL, Wu KH, Kanoni S, Zajac GJM, Ramdas S, Surakka I, Ntalla I, Vedantam S, Winkler TW et al (2021) The power of genetic diversity in genome-wide association studies of lipids. Nature 600:675–679. https://doi.org/10.1038/s41586-021-04064-3
Hasegawa M, Kawai K, Mitsui T, Taniguchi K, Monnai M, Wakui M, Ito M, Suematsu M, Peltz G, Nakamura M, Suemizu H (2011) The reconstituted “humanized liver” in TK-NOG mice is mature and functional. Biochem Biophys Res Commun 405:405–410. https://doi.org/10.1016/j.bbrc.2011.01.042
Horn CL, Morales AL, Savard C, Farrell GC, Ioannou GN (2022) Role of cholesterol-associated steatohepatitis in the development of NASH. Hepatol Commun 6:12–35. https://doi.org/10.1002/hep4.1801
Huang TP, Newby GA, Liu DR (2021) Precision genome editing using cytosine and adenine base editors in mammalian cells. Nat Protoc 16:1089–1128. https://doi.org/10.1038/s41596-020-00450-9
Johnsson P, Lipovich L, Grander D, Morris KV (2014) Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta 1840:1063–1071. https://doi.org/10.1016/j.bbagen.2013.10.035
Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, Gagnon DR, DuVall SL, Li J, Peloso GM et al (2018) Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet 50:1514–1523. https://doi.org/10.1038/s41588-018-0222-9
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27:2987–2993. https://doi.org/10.1093/bioinformatics/btr509
Li P, Ruan X, Yang L, Kiesewetter K, Zhao Y, Luo H, Chen Y, Gucek M, Zhu J, Cao H (2015) A liver-enriched long non-coding RNA, lncLSTR, regulates systemic lipid metabolism in mice. Cell Metab 21:455–467. https://doi.org/10.1016/j.cmet.2015.02.004
Liao Y, Smyth GK, Shi W (2013) The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res 41:e108. https://doi.org/10.1093/nar/gkt214
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Mardinoglu A, Wu H, Bjornson E, Zhang C, Hakkarainen A, Rasanen SM, Lee S, Mancina RM, Bergentall M, Pietilainen KH et al (2018) An integrated understanding of the rapid metabolic benefits of a carbohydrate-restricted diet on hepatic steatosis in humans. Cell Metab 27(559–571):e555. https://doi.org/10.1016/j.cmet.2018.01.005
Orozco G, Schoenfelder S, Walker N, Eyre S, Fraser P (2022) 3D genome organization links non-coding disease-associated variants to genes. Front Cell Dev Biol 10:995388. https://doi.org/10.3389/fcell.2022.995388
Pasquali L, Gaulton KJ, Rodriguez-Segui SA, Mularoni L, Miguel-Escalada I, Akerman I, Tena JJ, Moran I, Gomez-Marin C, van de Bunt M et al (2014) Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet 46:136–143. https://doi.org/10.1038/ng.2870
Qi G, Chatterjee N (2018) Heritability informed power optimization (HIPO) leads to enhanced detection of genetic associations across multiple traits. PLoS Genet 14:e1007549. https://doi.org/10.1371/journal.pgen.1007549
Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M (2008) Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet 4:e1000133. https://doi.org/10.1371/journal.pgen.1000133
Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J et al (2015) Integrative analysis of 111 reference human epigenomes. Nature 518:317–330. https://doi.org/10.1038/nature14248
Ruan X, Li P, Cangelosi A, Yang L, Cao H (2016) A long non-coding RNA, lncLGR, regulates hepatic glucokinase expression and glycogen storage during fasting. Cell Rep 14:1867–1875. https://doi.org/10.1016/j.celrep.2016.01.062
Ruan X, Li P, Chen Y, Shi Y, Pirooznia M, Seifuddin F, Suemizu H, Ohnishi Y, Yoneda N, Nishiwaki M et al (2020) In vivo functional analysis of non-conserved human lncRNAs associated with cardiometabolic traits. Nat Commun 11:45. https://doi.org/10.1038/s41467-019-13688-z
Ruan X, Li P, Ma Y, Jiang CF, Chen Y, Shi Y, Gupta N, Seifuddin F, Pirooznia M, Ohnishi Y et al (2021) Identification of human long noncoding RNAs associated with nonalcoholic fatty liver disease and metabolic homeostasis. J Clin Invest. https://doi.org/10.1172/JCI136336
Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, Narita A, Konuma T, Yamamoto K, Akiyama M et al (2021) A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet 53:1415–1424. https://doi.org/10.1038/s41588-021-00931-x
Sallam T, Jones M, Thomas BJ, Wu X, Gilliland T, Qian K, Eskin A, Casero D, Zhang Z, Sandhu J et al (2018) Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat Med 24:304–312. https://doi.org/10.1038/nm.4479
Sollis E, Mosaku A, Abid A, Buniello A, Cerezo M, Gil L, Groza T, Gunes O, Hall P, Hayhurst J et al (2023) The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res 51:D977–D985. https://doi.org/10.1093/nar/gkac1010
Suppli MP, Rigbolt KTG, Veidal SS, Heeboll S, Eriksen PL, Demant M, Bagger JI, Nielsen JC, Oro D, Thrane SW et al (2019) Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am J Physiol Gastrointest Liver Physiol 316:G462–G472. https://doi.org/10.1152/ajpgi.00358.2018
Thuronyi BW, Koblan LW, Levy JM, Yeh WH, Zheng C, Newby GA, Wilson C, Bhaumik M, Shubina-Oleinik O, Holt JR, Liu DR (2019) Continuous evolution of base editors with expanded target compatibility and improved activity. Nat Biotechnol 37:1070–1079. https://doi.org/10.1038/s41587-019-0193-0
Toth PP, Potter D, Ming EE (2012) Prevalence of lipid abnormalities in the United States: the National Health and Nutrition Examination Survey 2003–2006. J Clin Lipidol 6:325–330. https://doi.org/10.1016/j.jacl.2012.05.002
Vosa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Yazar S et al (2021) Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet 53:1300–1310. https://doi.org/10.1038/s41588-021-00913-z
Wang Y, Viscarra J, Kim SJ, Sul HS (2015) Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol 16:678–689. https://doi.org/10.1038/nrm4074
Yang L, Li P, Yang W, Ruan X, Kiesewetter K, Zhu J, Cao H (2016) Integrative transcriptome analyses of metabolic responses in mice define pivotal LncRNA metabolic regulators. Cell Metab 24:627–639. https://doi.org/10.1016/j.cmet.2016.08.019
Zhao XY, Li S, Wang GX, Yu Q, Lin JD (2014) A long noncoding RNA transcriptional regulatory circuit drives thermogenic adipocyte differentiation. Mol Cell 55:372–382. https://doi.org/10.1016/j.molcel.2014.06.004
Acknowledgements
We thank Dr. Timothy Osborne for providing the active SREBP1c plasmid. We are grateful to Dr. Timothy Osborne for critical reading of the manuscript and helpful comments. This study was funded by Johns Hopkins University institutional funds for Dr. Xiangbo Ruan.
Funding
This work was supported by Johns Hopkins University institutional funds for Dr. Xiangbo Ruan.
Author information
Authors and Affiliations
Contributions
M.J. and I.P. performed the experiments and analyzed the data. S.T. performed the bioinformatics analyses. M.J., S.T. and X.R. wrote the manuscript. X.R. conceived and supervised the study.
Corresponding author
Ethics declarations
Conflict of Interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jaso-Vera, M.E., Takaoka, S., Patel, I. et al. Integrative regulation of hLMR1 by dietary and genetic factors in nonalcoholic fatty liver disease and hyperlipidemia. Hum. Genet. 143, 897–906 (2024). https://doi.org/10.1007/s00439-024-02654-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-024-02654-5