
Abstract
Cerebral cavernous malformations (CCM) are vascular malformations consisting of collections of enlarged capillaries occurring in the brain or spinal cord. These vascular malformations can occur sporadically or susceptibility to develop these can be inherited as an autosomal dominant trait due to mutation in one of three genes. Over a decade ago, we described a 77.6 Kb germline deletion spanning exons 2–10 in the CCM2 gene found in multiple affected individuals from seemingly unrelated families. Segregation analysis using linked, microsatellite markers indicated that this deletion may have arisen at least twice independently. In the ensuing decades, many more CCM patients have been identified with this deletion. In this present study we examined 27 reportedly unrelated affected individuals with this deletion. To investigate the origin of the deletion at base pair level resolution, we sequenced approximately 10 Kb upstream and downstream from the recombination junction on the deleted allele. All patients showed the identical SNP haplotype across this combined 20 Kb interval. In parallel, genealogical records have traced 11 of these individuals to five separate pedigrees dating as far back as the 1600-1700s. These haplotype and genealogical data suggest that these families and the remaining “unrelated” samples converge on a common ancestor due to a founder mutation occurring centuries ago on the North American continent. We also note that another gene, NACAD, is included in this deletion. Although patient self-reporting does not indicate an apparent phenotypic consequence for heterozygous deletion of NACAD, further investigation is warranted for these patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cerebral Cavernous Malformations (CCM) are a common vascular anomaly of the central nervous system consisting of clusters of dilated, fragile venous vessels. Although sometimes benign, hemorrhage from these lesions can cause headaches, neurologic deficits, seizures and hemorrhagic stroke (Zabramski et al. 1994; Horne et al. 2016; Al-Shahi Salman et al. 2012). Typically, CCMs occur either sporadically, presenting as a solitary lesion, or as an inherited, autosomal dominant condition characterized by multiple lesions. Heterozygous germline mutations in KRIT1 (Sahoo et al. 1999; Laberge-le Couteulx et al. 1999), CCM2 (Liquori et al. 2003; Denier et al. 2004), and PDCD10, (Bergametti et al. 2005) are the cause of familial CCM.
Since the discovery of the three CCM genes, diagnostic DNA testing of patients has revealed a large number of potential mutations in each of the three genes (https://www.ncbi.nlm.nih.gov/clinvar), all consistent with loss-of-function. As with other diseases caused by loss-of-function mutations, most mutations in the CCM genes are specific to a single family. There are cases of mutations occurring in multiple, apparently unrelated families, but these have been shown instead to be families that are distantly related to an ancestral mutation founder. Examples include the so-called “Common Hispanic Mutation” in KRIT1 in families of Mexican–American heritage (Sahoo et al. 1999), the Sardinian KRIT1 mutation in CCM families from that geographical location (Cau et al. 2009) and the Ashkenazi Jewish mutation in CCM2 that is only found in Jewish CCM families (Gallione et al. 2011).
A large deletion in CCM2, first described in 8 CCM families, was also posited to possibly be a founder mutation (Liquori et al. 2007). This 77.6 Kb deletion has excised exons 2–10 of CCM2 and appears to have been seeded by the illegitimate recombination of two distant Alu repeats. However, the limited family histories available at that time did not reveal any common ancestors among the nuclear families. Although all 8 families were self-reported of European descent, there did not appear to be a common ethnic origin that might further support a founder mutation. Segregation analysis of linked, microsatellite markers in these families revealed a single deletion haplotype proximal to the deletion, and two different haplotypes distal to the deletion. The authors concluded that this mutation may have arisen at least twice in the 8 CCM families but were uncertain on the question of whether this was indeed an authentic founder mutation.
Since the initial identification of this deletion, numerous other CCM patients and families have been found to carry the apparently identical deletion. In this study, we examined high resolution SNP haplotypes flanking the deletion in the original 8 (Liquori et al. 2007) and 19 newly identified families. In parallel, we investigated deeper family histories through examination of genealogical records. Using this combination of “genetic genealogy”, we were able to solve the question as to whether this deletion mutation is yet another example of a founder CCM gene mutation.
Methods
Study participants
All persons participating in this study were enrolled after informed consent and under the approval of the institutional review boards. The cohort of 27 people was all non-Hispanic individuals of European descent and all of the participants had been diagnosed with CCM through either a positive MRI and/or they had received a positive clinical test for the CCM2 exons 2–10 deletion. Eight of these individuals were from the original cohort (Liquori et al. 2007). The remaining 19 were recruited by Angioma Alliance through two channels: 1) an Angioma Alliance Facebook group specifically dedicated to families with CCM2 exon 2–10 deletion, and 2) the Angioma Alliance patient contact registry. By interviewing patients about previous research participation, researchers were assured there was no duplication between these 19 Angioma Alliance recruits and the original cohort. None of these families were known to each other before their diagnosis.
DNA was extracted from either blood (Gentra PureGene Blood Kit, Qiagen) or saliva (Oragene, DNA Genotek Inc) samples according to manufacturers’ instructions.
Deletion confirmation
The participants were screened for the CCM2 deletion using the primers and conditions described previously (Liquori et al. 2007).
Sequencing of affected alleles
LongAmp Taq (NEB) was used for the long-range amplification on the affected alleles according to the manufacturer’s directions. PCR reactions were electrophoresed on 1% SYBR Green agarose gels and products of the expected sizes were excised from the gels and prepared for either Sanger sequencing or PacBio long range sequencing using GeneClean Turbo (MPBio). Sanger sequencing was performed using the BigDye Terminator v1.1 cycle sequencing kit (Applied Biosystems) and run on an ABI Prism 3130. Sequences were analyzed using Sequencher v.5.4.6 (GeneCodes). PacBio SMRT sequencing for 13 samples covering approximately 5 Kb in both directions from the recombination junction was performed according to the manufacturer’s instructions and was carried out at the Duke Sequencing and Technologies Core Facility.
Genealogy
In 2012, members of four families with the CCM2 exons 2–10 deletion created a private Facebook group to share information with other families with this deletion. As more patients associated with Angioma Alliance were identified with this mutation, they joined the group. Several families came to the group with extensive family genealogy, some of which had been professionally prepared. In all cases, sources and conclusions were independently confirmed or corrected by the researchers. Per Board for Certification of Genealogists’ standards (Genealogy Standards 2019), research was conducted using vital records (birth, death, marriage), wills, court records, military records, and land transactions where available. These were obtained in online databases (Ancestry.com, FamilySearch.com) and at the Family History Library in Salt Lake City, UT. As research progressed, it became evident that families shared a common ancestral geography, primarily in the Southeastern states, but it was not until 2019 when a newly identified family joined the Facebook group, that the crucial, connecting genealogical link was discovered. Initially, this family and two others already in the group were connected through a common ancestor, a couple born in the 1760s in Anson County, North Carolina.
After this initial connection, a more concentrated effort was made to determine if and how other CCM2 deletion families were related. Because of the confidential nature of the information being collected, a PC-based password- and VPN-protected genealogy database was created using RootsMagic 7.0. Living family member names were not included and only one researcher had access to the entire data set. Families submitted their personal family trees and access was granted to their online genealogy on the Ancestry.com website. Diagnosed individuals were also encouraged to submit saliva samples to AncestryDNA for analysis, which would uncover shared historic family names.
Haplotype frequency
The haplotype frequency was calculated using the LDHap program (ldlink.nci.nih.gov) with the CEU (Utah residents (CEPH) from North and West Europe) and GBR (British in England and Scotland) filters as these most closely represented the reported backgrounds of the families. All of the examined SNPs were tested on the program in numerous combinations to determine the range and to look for SNPs that were in linkage disequilibrium. From these results, we chose 11 tagging SNPs to determine the final haplotype frequency of the region approximately 10 Kb upstream and downstream of the recombination junction.
Results
Deletion testing
We collected DNA from 27 apparently unrelated individuals who by diagnostic testing were reported to exhibit the previously published CCM2 exons 2–10 deletion (Liquori et al. 2007). To ensure that these individuals did indeed harbor this deletion, we re-tested each sample using PCR amplification primers designed to only amplify a product if the deletion is present (Liquori et al. 2007) (Fig. 1). All members of this cohort exhibited the expected deletion-spanning PCR product (Online Resource 1).

Schematic diagram of the CCM2 exons 2–10 deletion. The 77.6 Kb deletion begins in an AluSx site in intron 1 of CCM2 (orange bar) and extends to an AluSg site in the intergenic region between NACAD and TBRG4 (yellow bar). The red arrows represent the primers used to test for the deletion in the samples (Liquori et al. 2007). The green and purple arrows represent amplification primers for sequencing. Due to the size of the deletion, only the affected allele will amplify in these reactions
Deletion-associated haplotype analysis
We then determined if these individuals shared the same deletion SNP haplotype by sequencing approximately 10 Kb proximally and distally across the recombination site of the deletion. The SNP haplotype across the 20 Kb deletion interval is identical in all 27 patient samples (Fig. 2). Note that each 10 Kb half of this haplotype is normally separated by 77.6 Kb so the extent of this haplotype identity is even more striking. Most of the 62 SNPs present exhibit the major (most frequent) allele, with only one SNP, rs7792895, exhibiting the minor allele (Online Resource 2). There were no private polymorphisms identified in any members of the cohort.

SNPs evaluated approximately 10 Kb upstream and downstream of the AluSx/AluSg recombination junction. The disease haplotype is noted above the SNP names. The base pair location on Chromosome 7 is indicated under the line (Hg38) and the small marks indicate 1 Kb increments
We used the LDHap program (ldlink.nci.nih.gov) to determine the frequency of this SNP haplotype. Using the SNP allele frequencies for the CEU and GBR populations based on the self-identified ethnic backgrounds of our cohort, we chose a tagging SNP set to determine the haplotype frequency (Online Resource 3). These SNPs showed the deletion-associated SNP haplotype frequency to be 12.9%. Choosing a different set of tagging SNPs had no appreciable difference in the haplotype frequency. Thus, this deletion is found on a modestly-common SNP haplotype.
Alu/Alu-mediated recombination
This deletion occurs due to a recombination event between an AluSx sequence and an AluSg sequence which while highly homologous (83.1%, LALIGN36), are not identical. We used the interval level prediction algorithm for Alu/Alu-mediated rearrangements (AAMR) (Song et al. 2018); (http://alualucnvpredictor.research.bcm.edu:3838/) to determine the likelihood for these two specific Alu sites to recombine. These two sites were not predicted to pair together during recombination. This analysis strengthens the argument that the specific recombination event leading to this CCM2 exon 2–10 deletion would be infrequent.
Additional gene loss discovered
Comparing the updated version of the human genome reference sequence (Dec. 2013, GRCH38/hg38) to the version from when this CCM2 deletion was first described (Mar. 2006, NCBI136/hg18), we noticed that an entire gene, NACAD, located distal to the last exon of the CCM2 gene, is included in the deletion. NACAD is Nascent-Polypeptide-Associated Complex, alpha domain, named for its putative role in trafficking of non-secretory proteins as they exit the ribosome and for its sequence homology to the NACA protein. There are no published reports on NACAD’s function and no reported loss-of-function mutations in this gene in the biomedical literature or in ClinVar. A recent, single report has suggested a long-Covid related phenotypic consequence of an in-frame deletion polymorphism in NACAD present at 4% in the population (Reddy et al. 2021). Since the CCM patients harboring the CCM2 2–10 deletion are haploid for NACAD, we were curious whether the heterozygous loss of this gene had any phenotypic consequences. NACAD is expressed in the brain and neuronal tissues (http://biogps.org) and GWAS studies have identified SNPs near NACAD for various phenotypes including eosinophil counts (Vuckovic et al. 2020; Chen et al. 2020; Kichaev et al. 2019) and colorectal cancer (Sakaue et al. 2021; Huyghe et al. 2019). However, a preliminary analysis of self-reported clinical information comparing patients with point mutations in CCM2 to these CCM2 deletion patients did not reveal any notable differences between these groups (Online Resource Table 1). Patients did not report colon cancer or any immunological phenotypes.
Genealogical analysis
In addition to the genetic work supporting a founder mutation, we used genealogical tools to try to connect the families with the CCM2 2–10 deletion. Beginning in 2012, a group of four separate families started a private Facebook group for patients with the CCM2 2–10 deletion. In 2016, the CCM patient support group Angioma Alliance began offering free diagnostic testing for individuals exhibiting multiple CCMs on MRI, a strong indicator of a germline mutation (Morrison and Akers 1993). In the first year that such testing was offered, 5/18 families tested exhibited the CCM2 deletion, a surprising outcome for a single mutation. These families were unknown to each other and did not know any of the previously identified CCM2 deletion families. After 2016, Angioma Alliance began making special efforts to find more CCM2 deletion families. This resulted in an increase to 29 families joining the Facebook group. In addition, other families harboring the CCM2 deletion are known to Angioma Alliance that have not joined the group.
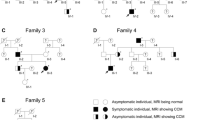
In 2019, shared information on the Facebook group led to members of three of the families being able to connect their ancestors to a couple born in Anson County, North Carolina in the 1760s. With this discovery, efforts to trace the other CCM2 deletion families increased. Using genealogical research methods described above, additional connections between families were discovered. While no other families in the 2019 Facebook group could be traced to the 1760s North Carolina couple, researchers identified four common ancestral couples connecting eight additional families (2 present-day families per 1 ancestral couple) including a connection in 1800s Missouri, two unique connections in 1700s South Carolina, and a connection in 1600s Varina, Henrico, Virginia. The pedigrees of the five largest families tracing back at least 200 years each as well as the pedigrees of the smaller families are shown in Fig. 3. Note that two individuals from the original study (Liquori et al. 2007) have been connected in one of the large families (Family R/H, Fig. 3). The pedigrees include only family members who were included in haplotype analysis. Since 2019, four new families have entered the Facebook group who can be traced to the original 1760s North Carolina couple but were not included in this study cohort.

Family pedigrees. The trimmed pedigrees of the families represented in the sequencing cohort. The five larger families are noted with the founders’ birth centuries and locations. The individuals included in this study are indicated by numbers below the dark symbols. The dark blue represents a clinical diagnosis of CCM, the light blue represents inferred CCM, and the dot represents a positive deletion test. Individuals from the original cohort (Liquori et al. 2007) are marked with an asterisk*. Note that two of these are now connected through genealogical records in Family R/H. The smaller families are trimmed to show first degree relatives with a positive MRI for CCM. The additional 11 patients sequenced were individuals not known to be connected to any of these families nor to each other
Interestingly and possibly coincidentally, the common ancestor in Varina, Henrico, Virginia shares a surname with one of the South Carolina ancestors from the 1700s. We have traced three families in this study to the same period of 1600s Varina, Henrico, Virginia. Based on wills and land transactions, these ancestral families were acquainted, but we have not yet found the specific family connection between them.
Strikingly, none of the families in our cohort are from the Northeast US although there are currently family members in many Southern, Midwestern and Western states. Figure 4 follows the spread of family members from the five large pedigrees across the US based on their location and half-decade of birth. While this mutation has spread across the US there are no reported cases in the UK, the EU or Brazil (Jonathan Berg, University of Dundee, Scotland; Elisabeth Tournier-Lasserve, INSERM, Paris, France; Jorge Marcondes de Souza, UFRJ, Rio de Janeiro, Brazil) consistent with this CCM2 deletion mutation having a US founder.

Migration map of the five large CCM2 deletion families. The birth locations of members of the five large families beginning with the proposed founders and ending with the samples included in the sequencing cohort. The colors represent the half-centuries of the birth years of the family members shown in the trimmed pedigrees in Fig. 3
Discussion
There are numerous cases of the identical “founder” mutation occurring more than once in the same or different populations. For example, a three-bp deletion in DFNA5 causing familial deafness is a founder mutation in the East Asian population yet is also found in a family of European ancestry with no East Asian background (Booth et al. 2020). In Lynch Syndrome, a missense mutation in MLH1, c.2059C > T, p.Arg687Trp is a founder mutation in the Swedish population but the same mutation is seen in families from other countries on different haplotypes indicating that the mutation occurred independently in those cases (Salome et al. 2017). Another mutation in MLH1, c.589-2A > G, was determined to be a founder mutation in the US and in Italy, however, the mutation arose on different haplotypes in those countries (Tomsic et al. 2012). In each of these cases the haplotype on which the mutation was found distinguished between the mutation being a founder or a recurrent event.
We examined the sequence approximately 10 Kb both proximally and distally from the recombination junction on the affected allele in our cohort of 27 CCM2 exons 2–10 deletion patients. All of the people sequenced shared the identical SNP haplotype across this entire region, including the presence of the minor allele for one SNP, rs7792895. As it is unlikely that the deletion would have occurred 27 times on the identical haplotype, these data are consistent with the hypothesis that the deletion had a single founder. Adding to this argument is the specific location of the recombination between an AluSx site on the proximal side and an AluSg site on the distal side. Alu/Alu rearrangements are well documented and are the cause of ~ 0.3% of human diseases (Batzer and Deininger 2002). However, a tool to calculate potential Alu/Alu recombination (Song et al. 2018) does not predict these two sites pairing together, suggesting that this would be an unusual event.
Also striking in the group of CCM2 deletion families is the geographic distribution within the US. The majority of the families live in Southern and mid-Western states; none are from the mid-Atlantic or New England. While this appears to be a somewhat limited range, it spans the entire continent. In contrast to other known founder mutations in CCM genes that are restricted to specific ethnic or geographic backgrounds such as the common Hispanic mutation in KRIT1, the CCM2 mutation in Ashkenazi Jews, and the Sardinian mutation in KRIT1 (Sahoo et al. 1999; Gallione et al. 2011; Cau et al. 2009), this CCM2 deletion has spread into the population of the United States over an expansive geographic range. To date, this deletion has only been reported in the US and not in other countries, implying that this is a US founder mutation.
Intriguingly, most of the families also report long family histories of living in the continental US, some dating back to colonial times in the 1600s. Extensive genealogical research, sparked by CCM2 deletion patients discovering shared ancestral family names via social media, uncovered five large families with founders in the 1600s–1800s in southern US states. At least two members from distant arms of each of these families were included in the sequencing cohort. The most closely related were 3rd cousins (Family R/H) and the most distantly related were 8th cousins once removed (Family F/W) (Fig. 3). At these generational distances between 0.0008 and 0.78% of the genome, or 2.5–53 Mb in total, would be predicted to be shared (Browning and Browning 2012) (isogg.org/wiki). It is perhaps not surprising then that within a family two distant cousins would share a 97 Kb SNP haplotype (10 Kb on either side of a 77 Kb deletion). What is most notable is that this identical SNP haplotype is shared between all sequenced members of this CCM2 patient cohort. Through genealogical research, many of these individuals have now been connected to one of these five multi-generational families but this same haplotype is also shared with individuals from smaller, unconnected families. Thus, the genetic and genealogical data combined support the hypothesis that this mutation arose once on this haplotype and that all of these people are related to a single, common founder. We may never be able to connect all of these individuals and families using genealogical tools due to the paucity and fragmentary nature of historical records in the United States. We are, however, able to connect these individuals genetically, based on their shared haplotype across this mutation. This combination of genetics and genealogy allows us to observe this founder mutation as it spread into the population across the country over at least the last four centuries.
We also note that an entire gene, NACAD, is included in the deletion but was not noted previously (Liquori et al. 2007). Our preliminary investigation of patients with the large NACAD-including deletion and those with point mutations only affecting CCM2 did not uncover any phenotypic differences between these groups. More work will be needed to determine which potential clinical phenotypes may be affected by NACAD deletion, especially as a contiguous gene deletion with the CCM2 gene.
Uncovering founder mutations helps to understand the natural history of the mutation in the population, its spread and impact in affected groups. When a certain disease-causing founder mutation is known to be present at an increased level within a specific group of people or in a certain location, healthcare professionals are better able to recognize the disease and provide appropriate care for these patients and their families. This care might include targeted genetic testing and pre-symptomatic diagnosis leading to early interventions with the goal of an improved quality of life for the patients.
When making recommendations for genetic testing, the frequency of a mutation in the population undergoing genetic testing should guide the determination whether any “common” mutations will initially be evaluated before sequencing a larger panel of genes. When a specific, common mutation is present at a relatively high frequency within the testing population, it is reasonable to test for that specific mutation before proceeding to other more extensive and expensive DNA sequencing gene panels. For example, Hispanic CCM patients are generally tested first for the common Hispanic nonsense mutation in the KRIT1 gene (Sahoo et al. 1999) before undergoing DNA sequencing of the three known CCM genes. For this population, the cost benefit is quite high.
In the present case, the nature of the mutation itself increases the cost benefit of a stand-alone test. This CCM2 exons 2–10 deletion is invisible to routine DNA sequence analysis. It can only be uncovered by laboratory techniques that are generally employed only after a sample is considered “mutation-negative” by DNA sequencing. Thus, for individuals of European descent that reside in the southern United States (Fig. 4), diagnostic laboratories might consider targeted genetic testing for this deletion. Importantly, the test can be performed with a simple, cost-effective, PCR-based assay (Liquori et al. 2007) (also Online Resource 1). Furthermore, presumed affected individuals may know or believe that they are related to other individuals in this ever-expanding CCM2 deletion pedigree. Here especially, it would be cost effective to assay for this deletion before any further testing is performed. Nonetheless, each diagnostic laboratory will generate CCM gene mutation frequency data for their unique testing population, and this data can be used to determine their priorities for CCM genetic testing.
References
Al-Shahi Salman R, Hall JM, Horne MA et al (2012) Untreated clinical course of cerebral cavernous malformations: a prospective, population-based cohort study. Lancet Neurol 11:217–224. https://doi.org/10.1016/S1474-4422(12)70004-2
Batzer MA, Deininger PL (2002) Alu repeats and human genomic diversity. Nat Rev Genet 3:370–379. https://doi.org/10.1038/nrg798
Bergametti F, Denier C, Labauge P et al (2005) Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet 76:42–51
Booth KT, Azaiez H, Smith RJH (2020) DFNA5 (GSDME) c.991-15_991-13delTTC: founder mutation or mutational hotspot? Int J Mol Sci. https://doi.org/10.3390/ijms21113951
Browning SR, Browning BL (2012) Identity by descent between distant relatives: detection and applications. Annu Rev Genet 46:617–633. https://doi.org/10.1146/annurev-genet-110711-155534
Cau M, Loi M, Melis M, Congiu R, Loi A, Meloni C, Serrenti M, Addis M, Melis MA (2009) C329X in KRIT1 is a founder mutation among CCM patients in Sardinia. Eur J Med Genet 52:344–348. https://doi.org/10.1016/j.ejmg.2009.05.002
Chen MH, Raffield LM, Mousas A et al (2020) Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell 182:1198-213e14. https://doi.org/10.1016/j.cell.2020.06.045
Denier C, Goutagny S, Labauge P et al (2004) Mutations within the MGC4607 gene cause cerebral cavernous malformations. Am J Hum Genet 74:326–337
Gallione CJ, Solatycki A, Awad IA, Weber JL, Marchuk DA (2011) A founder mutation in the Ashkenazi Jewish population affecting messenger RNA splicing of the CCM2 gene causes cerebral cavernous malformations. Genet Med 13:662–666. https://doi.org/10.1097/GIM.0b013e318211ff8b
Genealogy Standards (2019) Turner Publishing Company, Nashville
Horne MA, Flemming KD, Su IC et al (2016) Clinical course of untreated cerebral cavernous malformations: a meta-analysis of individual patient data. Lancet Neurol 15:166–173. https://doi.org/10.1016/S1474-4422(15)00303-8
Huyghe JR, Bien SA, Harrison TA et al (2019) Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet 51:76–87. https://doi.org/10.1038/s41588-018-0286-6
Kichaev G, Bhatia G, Loh PR, Gazal S, Burch K, Freund MK, Schoech A, Pasaniuc B, Price AL (2019) leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet 104:65–75. https://doi.org/10.1016/j.ajhg.2018.11.008
Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier-Lasserve E (1999) Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet 23:189–193
Liquori CL, Berg MJ, Siegel AM et al (2003) Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet 73:1459–1464
Liquori CL, Berg MJ, Squitieri F, Leedom TP, Ptacek L, Johnson EW, Marchuk DA (2007) Deletions in CCM2 are a common cause of cerebral cavernous malformations. Am J Hum Genet 80:69–75
Morrison L, and Akers A (1993) Cerebral cavernous malformation, familial. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G , Amemiya G (eds). GeneReviews((R)), Seattle
Reddy R, Efimenko I, Chertman W, Kohn T, Diaz P, Seetharam D, Khodamoradi K, Kresch E, Ramasamy R (2021) Whole exome sequencing identifies a rare mutation in NACAD as a possible cause of COVID orchitis in brothers. Urology. https://doi.org/10.1016/j.urology.2021.09.021
Sahoo T, Johnson EW, Thomas JW et al (1999) Mutations in the gene encoding KRIT1, a Krev-1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Hum Mol Genet 8:2325–2333
Sakaue S, Kanai M, Tanigawa Y et al (2021) A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet 53:1415–1424. https://doi.org/10.1038/s41588-021-00931-x
Song X, Beck CR, Du R et al (2018) Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res 28:1228–1242. https://doi.org/10.1101/gr.229401.117
Tomsic J, Liyanarachchi S, Hampel H et al (2012) An American founder mutation in MLH1. Int J Cancer 130:2088–2095. https://doi.org/10.1002/ijc.26233
von Salome J, Boonstra PS, Karimi M, Silander G, Stenmark-Askmalm M, Gebre-Medhin S, Aravidis C, Nilbert M, Lindblom A, Lagerstedt-Robinson K (2017) Genetic anticipation in Swedish Lynch syndrome families. PLoS Genet 13:e1007012. https://doi.org/10.1371/journal.pgen.1007012
Vuckovic D, Bao EL, Akbari P et al (2020) The polygenic and monogenic basis of blood traits and diseases. Cell 182:1214-31e11. https://doi.org/10.1016/j.cell.2020.08.008
Zabramski JM, Wascher TM, Spetzler RF, Johnson B, Golfinos J, Drayer BP, Brown B, Rigamonti D, Brown G (1994) The natural history of familial cavernous malformations: results of an ongoing study. J Neurosurg 80:422–432
Acknowledgements
We thank the patients who participated in both the genealogical and genetic arms of this study, the patient support group, Angioma Alliance, the CCM2 Exon 2-10 Deletion Families Facebook Group, and our funders: NIH P01-NS092521 (DAM). We also thank Dr. Christine Beck (University of Connecticut Health Center and The Jackson Laboratory for Genomic Medicine) for helpful discussions.
Funding
This work was supported by NIH P01-NS092521 (DAM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gallione, C.J., Detter, M.R., Sheline, A. et al. Genetic genealogy uncovers a founder deletion mutation in the cerebral cavernous malformations 2 gene. Hum Genet 141, 1761–1769 (2022). https://doi.org/10.1007/s00439-022-02458-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-022-02458-5