
Abstract
Mutations in the OTOF gene are a common cause of hereditary hearing loss and the main cause of auditory neuropathy spectrum disorder (ANSD). Although it is reported that most of the patients with OTOF mutations have stable, congenital or prelingual onset severe-to-profound hearing loss, some patients show atypical clinical phenotypes, and the genotype–phenotype correlation in patients with OTOF mutations is not yet fully understood. In this study, we aimed to reveal detailed clinical characteristics of OTOF-related hearing loss patients and the genotype–phenotype correlation. Detailed clinical information was available for 64 patients in our database who were diagnosed with OTOF-related hearing loss. As reported previously, most of the patients (90.6%) showed a “typical” phenotype; prelingual and severe-to-profound hearing loss. Forty-seven patients (73.4%) underwent cochlear implantation surgery and showed successful outcomes; approximately 85–90% of the patients showed a hearing level of 20–39 dB with cochlear implant and a Categories of Auditory Performance (CAP) scale level 6 or better. Although truncating mutations and p.Arg1939Gln were clearly related to severe phenotype, almost half of the patients with one or more non-truncating mutations showed mild-to-moderate hearing loss. Notably, patients with p.His513Arg, p.Ile1573Thr and p.Glu1910Lys showed “true” auditory neuropathy-like clinical characteristics. In this study, we have clarified genotype–phenotype correlation and efficacy of cochlear implantation for OTOF-related hearing loss patients in the biggest cohort studied to date. We believe that the clinical characteristics and genotype–phenotype correlation found in this study will support preoperative counseling and appropriate intervention for OTOF-related hearing loss patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hearing loss is one of the most common sensory disorders, with around 466 million people suffering from hearing loss (World Health Organization 2021). In developed countries, it is reported that 1 out of 500 newborns has bilateral hearing loss and approximately 80% of cases are due to genetic etiologies (Shearer et al. 2017). OTOF is reported to be the causative gene of DFNB9 and one of the common causes of non-syndromic recessive sensorineural hearing loss. The prevalence of OTOF mutations has been reported to be 1.4–3.2% of non-syndromic hearing loss cases (Rodríguez-Ballesteros et al. 2008; Choi et al. 2009; Wang et al. 2010a; Mahdieh et al. 2012) and 1.72% in Japan (Iwasa et al. 2019). To date, more than 220 mutations in OTOF have been reported (Vona et al. 2020). Although it is reported that most of the patients with OTOF mutations have stable, congenital or prelingual onset severe-to-profound hearing loss, some patients show atypical clinical phenotypes, such as mild-to-moderate progressive cases (Chiu et al. 2010). The genotype–phenotype correlation in the patients with OTOF mutations is, subsequently, not yet fully understood.
OTOF is also reported to be a main cause of auditory neuropathy spectrum disorder (ANSD), which is a specific form of hearing loss with an abnormal auditory brainstem response (ABR) and the presence of otoacoustic emissions (OAEs) (Starr et al. 1996). Both genetic and environmental factors cause ANSD, such as hyperbilirubinemia, thiamine deficiency, hypoxia, and noise-induced and age-related hearing loss (Shearer 2019). OTOF-related ANSD is the most prevalent form of ANSD, and it is reported that 23–90.9% of pediatric cases of ANSD are caused by OTOF mutations (Rodríguez-Ballesteros et al. 2008; Matsunaga et al. 2012; Zhang et al. 2016; Kim et al. 2018). OTOF is mainly expressed in the inner hair cells (IHCs), and mutations in the OTOF gene cause dysfunction of synaptic exocytosis at the ribbon synapse (Roux et al. 2006). OTOF-related ANSD is also known as auditory synaptopathy, presenting with impaired synaptic function between inner hair cells and spiral ganglion neurons, to differentiate it from auditory neuropathy (i.e., “true” auditory neuropathy) (Moser and Starr 2016). Due to the presence of an OAE response, there are some clinical problems with ANSD patients; they inappropriately pass the newborn hearing screening (NBHS) in countries where OAE is used as a screening tool, resulting in the late diagnosis of hearing loss. As early intervention by hearing aid (HA), cochlear implant (CI) and language rehabilitation are necessary for patients with hearing impairment to develop their speech skills, late diagnosis is a disadvantage for such hearing loss patients. Moreover, the presence of an OAE response makes it difficult for clinicians to decide the appropriate timing of cochlear implantation, as OAE positivity means the outer hair cells (OHCs) are functional and the efficacy of CI could be limited in cases in which the patient has “true” auditory neuropathy. Therefore, the genetic diagnosis of OTOF-related hearing loss in ANSD patients can encourage clinicians to recommend cochlear implantation for the patients, as the performance of CI for OTOF-related ANSD is reported to be excellent (Zheng and Liu 2020). However, each report was based on only a limited number of patients, and different evaluation tools were used for assessing the performance of CIs in each report.
In Japan, social health insurance-based genetic testing for the patients with hereditary hearing loss was approved in 2012 and, from 2015, massively parallel DNA sequencing technology was combined with genetic testing, so that excellent mutation data from Japanese hearing loss patients has been accumulated over the years in the whole-Japanese database. In this study, we investigated the clinical data of the patients diagnosed with OTOF-related hearing loss registered in our database, and aimed to reveal detailed clinical characteristics of OTOF-related hearing loss patients and the genotype–phenotype correlation in the patients with OTOF mutations.
Subjects and methods
Subjects
A total of 12,137 patients were registered in our database between February 2012 and December 2020 from 96 otolaryngology departments from across Japan. Seventy-four patients had two or more mutations in the OTOF gene. Detailed clinical information was available for 66 of 74 patients, and their clinical characteristics were analyzed retrospectively through the review of their medical records, and 2 of them were excluded because the clinical phenotype was completely incompatible with OTOF-related hearing loss: One of them had two mutations of uncertain significance and this case had late-onset ski-slope hearing loss, with the other having three mutations of uncertain significance and acute-onset unilateral mild hearing loss. Hearing level was evaluated using pure-tone audiometry (PTA) classified by a pure-tone average over 500, 1000, 2000 and 4000 Hz in the better hearing ears. For children who could not undergo PTA, we used an average over 500, 1000, 2000 and 4000 Hz in conditioned oriented reflex audiometry (COR). Severity of hearing loss was classified as follows; normal hearing, < 25 dB; mild hearing loss, 25–39 dB; moderate hearing loss, 40–69 dB; severe hearing loss, 70–89 dB; and profound hearing loss, greater than 90 dB. Written informed consent was obtained from all subjects (or from their next of kin, caretaker, or guardian on the behalf of minors/children) prior to enrollment in the project. This study was approved by the ethical committees of Shinshu University and each of the other participating institutions.
MPS sequencing
Amplicon resequencing with MPS
Amplicon libraries were prepared using an Ion AmpliSeq Custom Panel (Applied Biosystems, Life Technologies), in accordance with the manufacturer’s instructions, for 68 genes reported to cause non-syndromic hereditary HL. The detailed sample preparation protocol has been described elsewhere (Iwasa et al. 2019). Sequencing was performed in accordance with the manufacturer’s instructions. Massively Parallel Sequencing (MPS) was performed with an Ion Torrent Personal Genome Machine (PGM) system, Ion Proton System or Ion S5 system using an Ion PGM 200 Sequencing Kit with an Ion 318 Chip (Life Technologies) or Ion HiQ Chef Kit with an Ion P1 chip or Ion 540 Chip kit-Chef. The sequence data were mapped against the human genome sequence (build GRCh37/hg19) with a Torrent Mapping Alignment Program. After sequence mapping, the DNA variants were detected with Torrent Variant Caller plug-in software. After variant detection, their effects were analyzed using ANNOVAR software (Wang et al. 2010b). The missense, nonsense, insertion/deletion, and splicing variants were selected from among the identified variants. Variants were further selected as less than 1% of (1) the 1000 genome database, (2) the 6500 exome variants, (3) the Human Genetic Variation Database (dataset for 1208 Japanese exome variants), and (4) the 333 in-house Japanese normal hearing controls by using our database software. All the mutations found in this study were confirmed by Sanger sequencing using exon-specific custom primers. The pathogenicity of the candidate variants was interpreted based on the standards and guidelines of the American College of Medical Genetics (ACMG) and ClinGen HL-CDWG expert specification (Oza et al. 2018; Brandt et al. 2019).
Results
Mutation analysis
Detailed mutational information from 64 patients whose clinical information was available is shown in Table 1. Twenty-seven (42.2%) of the 64 patients with biallelic OTOF mutations had homozygous OTOF: NM_001287489:c.5816G > A: p.Arg1939Gln mutations. Twenty-nine (45.3%) were compound heterozygote with p.Arg1939Gln and another mutation. Eight patients (12.5%) had other non- p.Arg1939Gln mutations. Two patients excluded from the study were found to possess compound heterozygotes with p.[Pro309Leu];[Arg813Trp] and p.[Asp217Gly];[Ala1802Val];c.[3127-5G > A], respectively. The pathogenicity of the mutations found in the study were categorized in accordance with the ACMG criteria (Suppl. Table 1). Six mutations (c.2437C > T (p.Arg813Trp), c.3126 + 5G > A, c.3570 + 5G > A, c.5838G > A (p.Trp1946Ter), c.5728G > A (p.Glu1910Lys) and c.5500delG (p.Asp1834fs)) were novel.
Clinical characteristics
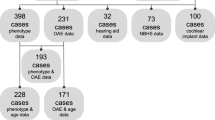
A summary of clinical information is shown in Fig. 1. Although most of the patients with OTOF-related hearing loss show congenital severe-to-profound hearing loss (90.6%) (Fig. 1a), NBHS only detects 45.3% of these cases of hearing loss (Fig. 1b). The rest of them were found to have hearing loss due to a lack of response to sound (26.6%), delayed language development (9.4%) or other reasons. Forty-seven patients (73.4%) underwent CI surgery; 9 patients (14.1%) were implanted simultaneously in both ears, 24 of them (37.5%) were implanted sequentially and 14 of them (21.9%) were implanted unilaterally (Fig. 1c). Figure 2 shows the detailed information for NBHS; 29 patients (45.3%) failed NBHS, 14 (21.5%) passed and 12 (18.5%) did not undergo NBHS (Fig. 2a). Among the 44 patients for whom information regarding NBHS was available, the detection rate of OTOF-related hearing loss was 65.9% (29 out of 44 patients). AABR and OAE was performed in 32 and 12 patients, respectively. All of the 12 patients examined by OAE improperly passed NBHS, while 3 out of 32 patients (9.4%) passed AABR screening (Fig. 2b, c). All 3 cases who passed AABR screening had homozygous p.Arg1939Gln mutations.

Clinical characteristics of the OTOF-related hearing loss patients in this study. a Hearing level in PTA or COR in very young children. b Detection of hearing loss. c Intervention for hearing loss. NA not applicable, n number of patients

Detailed information of newborn hearing screening (NBHS). a Results of NBHS for all patients in this study. b, c Result of NBHS by each screening method (AABR and OAE)
Performance of cochlear implantation
In this study, the performance of cochlear implantation was evaluated using PTA and the Categories of Auditory Performance (CAP) scale for 36 patients who had used CI for over 2 years after the initial implantation. Cochlear implantations were performed unilaterally or bilaterally depending on the decisions made at each institution (Table 1). Hearing threshold data with CI in the better hearing ear were available for all of 36 patients and are shown in Fig. 3a: 20–29 dB in 17 patients (47.2%), 30–39 dB in 15 patients (41.6%) and 40–49 dB in 4 patients (11.1%). The CAP scale data were available for 33 patients and are shown in Fig. 3b: CAP scale level 7 in 2 patients (6.1%), level 6 in 26 patients (87.8%), level 5 in 3 patients (9.1%), level 4 in 1 patient (3%) and level 3 in 1 patient (3%).

Performance of cochlear implantation for OTOF-related hearing loss. a Hearing threshold with cochlear implants was available for 36 patients who had used CIs for more than 2 years. b The CAP scale was available for 33 patients
Disappearance of OAE
Figure 4A shows the timing of the disappearance of OAE response in the ear in which the OAE response remained longer; 20 patients (30.8%) lost OAE response at CI implantation, 22 patients (33.8%) were tested only once and showed OAE, 6 patients (9.2%) showed no response at the first visit and 2 patients (3.1%) still had a positive OAE at the time of this survey. Accurate timing of OAE disappearance was available only in one patient (between two and half years to three years old). Figure 4b shows the OAE results for the ears in which OAE response remained longer at the last OAE testing. No patients showed OAE passed 5 years of age.

Detailed information on the disappearance of OAE response. a Timing of the disappearance of OAE response in the ear in which OAE response remained longer or the current status of OAE response. b Results at the last OAE testing
Discussion
In this study, we investigated the clinical characteristics of 64 patients who were diagnosed with OTOF-related hearing loss. One of the main aims was to identify genotype–phenotype correlations. As reported previously, most of the patients (90.6%) participating in this study showed a “typical” phenotype; congenital or prelingual onset and severe-to-profound hearing loss (Fig. 1). All of the patients who revealed a homozygous c.5816G > A (p.Arg1939Gln) and compound heterozygote with p.Arg1939Gln and a truncating mutation, and 1 patient with two truncating mutations (c.1422 T > A (p.Tyr474Ter) and c3126 + 5G > A) showed profound hearing loss. Therefore, it is plausible that p.Arg1939Gln and a truncating mutation are related to severe-to-profound hearing loss based on these results. On the other hand, the phenotype of patients with non-truncating mutations is more complicated. Figure 5a shows a summary of genotype–phenotype correlations in this and previous studies, in which the patients’ hearing level and mutation information were available (Tekin et al. 2005; Varga et al. 2006; Rodríguez-Ballesteros et al. 2008; Santarelli et al. 2009; Chiu et al. 2010; Zadro et al. 2010; Mahdieh et al. 2012; Matsunaga et al. 2012; Yildirim-Baylan et al. 2014; Zhang et al. 2016; Kim et al. 2018; Wang et al. 2018). There is a clear relationship between either a homozygous or compound heterozygous state with p.Arg1939Gln and a severe phenotype. Truncating mutations including c.2485C > T (p.Gln829Ter) are also clearly related to a severe phenotype. Among patients having one or more non-truncating mutation, almost half showed mild-to-moderate hearing loss (Fig. 5c, d), even when possessing p.Arg1939Gln or a truncating mutation on the second allele. In cases with non-truncating mutations, the severity of clinical phenotype appears to vary depending on the functional alterations caused by each mutation. For instance, p.Arg1939Gln is clearly related to a severe phenotype according to clinical evidence to date, suggesting that the impact caused by the mutation strongly affects the function of otoferlin. However, no functional analysis has been reported, as with the most of other mutations. Genotype–phenotype correlation in non-truncating mutations remains partially unclear: the same mutation can cause different phenotypes, suggesting that other factors may affect the phenotype, such as mutations in other genes or epigenetic changes. Further research is needed on the issue.

Summary of genotype–phenotype correlations in this and previous studies. a Mutation type and hearing severity in this and previous studies. b Hearing severity of patients with two truncating mutations. c Hearing severity of patients with truncating and non-truncating mutations (excluding p.Arg1939Gln). d Hearing severity of patients with two non-truncating mutations (excluding p.Arg1939Gln)
Among the moderate cases in our cohort, there were three representative cases who showed “true” auditory neuropathy-like clinical characteristics (AK6640, AL8610 and AH0083 were compound heterozygote with c.[5816G > A];[1538A > G] (p.[Arg1939Gln];[His513Arg], c.[5815C > T];[5728G > A](p.[Arg1939Trp];[Glu1910Lys]) and c.[4718 T > C];[4129_4138del](p.[Ile1573Thr];[Ala1377Argfs*142], respectively)); the PTA of all patients showed moderate hearing loss while their ABR showed no response. p.His513Arg is located near the p.Ile515Thr and p.Gly541Ser mutations in the C2C domain, which are reported as mutations related to temperature-sensitive auditory neuropathy (TS-AN) and milder hearing loss in a non-febrile state (Varga et al. 2006; Matsunaga et al. 2012). OtofIle515Thr/Ile515Thr mice exhibited moderate hearing impairment with lower otoferlin levels and enlarged synaptic vesicles in the IHCs (Strenzke et al. 2016), suggesting that mutations around the area in the C2C domain may cause a milder phenotype. p.Ile1573Thr was reported as the cause of ANSD with mild-to-moderate hearing loss in a previous report (Yildirim-Baylan et al. 2014), which is compatible with the present case. We consider that p.His513Arg and p.Ile1573Thr are related to a milder phenotype. p.Arg1939Trp was reported previously, and the patient with homozygous p.Arg1939Trp showed severe-to-profound hearing loss (Choi et al. 2009). p.Glu1910Lys is novel mutation. As p.Arg1939Trp causes the same amino acid change as p.Arg1939Gln, p.Glu1910Lys might cause moderate hearing loss. Other than these mutations, it has been reported that p.Glu1700Gln was related to a progressive and milder phenotype (Chiu et al. 2010), and p.Arg1607Trp and p.Gly1804del were related to TS-AN (Marlin et al. 2010; Wang et al. 2010a). Interestingly, all of the patients with the aforementioned mutations related to a milder and non-typical phenotype showed no ABR response, just like “true” auditory neuropathy. Electrophysiological testing, such as ABR, is important, especially when estimating the hearing threshold of very young children. However, when determining the hearing level for OTOF-related hearing loss, the results of audiological testing should be carefully interpreted, considering that there is a discrepancy between PTA threshold and ABR response, especially when the PTA thresholds are mild-to-moderate.
Different mutation spectra and recurrent mutations have been reported in each population. p.Gln829Ter is quite frequently found in the Spanish population, being present in about 3% of all cases of recessive prelingual deafness (Migliosi et al. 2002). c.2905_2923delinsCTCCGAGCGGCA in Argentinean (Rodríguez-Ballesteros et al. 2008), p.Val1778Phe in Ashkenazi Jewish (Fedick et al. 2016), p.Glu57Ter and p.Arg1792His in Saudi Arabian (Almontashiri et al. 2018) and p. Glu1700Gln in Taiwanese populations (Chiu et al. 2010) are also common mutations in the respective populations as well. In this study, 59 of 65 patients (90.8%) had at least one p.Arg1939Gln mutation, which was reported to be a founder mutation (Matsunaga et al. 2012), suggesting that it is indeed a recurrent mutation in Japanese.
This study revealed that the detection rate of OTOF-related hearing loss in NBHS was 65.9% among the 44 patients for whom NBHS information was available in this study, which means one-third of OTOF-related hearing loss patients improperly pass screening in Japan. The presence of OAE is the main reason for missing OTOF-related ANSD at NBHS. Similar to our result, it was reported that 75% of OTOF-related patients improperly passed NBHS in a previous study (Wu et al. 2019). The risk of using OAE for NBHS has been discussed from the point of view of its inability to detect ANSD. Nevertheless, OAE is still used for NBHS in most countries and there are only a few countries where both OAE and AABR are used for NBHS (Wroblewska-Seniuk et al. 2017). In Japan, as is in some other countries, the screening method differs depending on the clinic and prefecture. All the patients in this study screened by OAE passed NBHS improperly, and their hearing loss was detected by their unawareness to sound or delayed language development. For better language development, early detection of HL is important. We strongly believe that NBHS should be performed by AABR or combination of OAE and AABR for reliable detection of ANSD.
The efficacy of cochlear implantation for ANSD has been controversial due to the heterogenicity of ANSD in terms of its etiology: ANSD is a disorder which includes auditory synaptopathy and neuropathy. Generally, prior to cochlear implantation, genetic testing should be considered to clarify which part of the auditory pathway is impaired and in order to be able to predict the outcomes of CI (Miyagawa et al. 2016). Once a patient is diagnosed with OTOF-related ANSD, a good CI outcome is expected because the auditory nerve remains intact. Indeed, past reports suggested that patients with OTOF-related ANSD are good candidates for CI (Zheng and Liu 2020). In this study, we investigated the efficacy of CI for OTOF-related hearing loss in the biggest cohort studied to date. Most of the patients who underwent cochlear implantation showed successful outcomes: approximately 85–90% of patients showed a hearing level of 20–39 dB with CI and a CAP scale level 6 or better, which means the OTOF-related hearing loss patients who underwent CI can understand conversation without lip reading. These data will support preoperative counseling for OTOF-related ANSD patients who are considered to be CI candidates.
In this study, we also investigated the timing of OAE disappearance. Although the exact timing of OAE disappearance was not available for most of the patients, some patients showed a positive OAE even at 3–4 years of age (Fig. 3b). Given no patients showed a positive OAE passed 5 years of age, we presume that the OAE response in OTOF-related ANSD disappears by 4–5 years of age at most, a little longer than previously thought. Related to the disappearance of OAE, some reports suggest that the disappearance of OAE could occur due to OHC damage originating from hearing aid use (Rouillon et al. 2006; Vona et al. 2020). On the other hand, it is also suggested that the use of hearing aids is not a definitive factor for deterioration of OAE (Kitao et al. 2019). In our study, some patients showed similar OAE responses even after wearing HAs until CI surgery, and some lost OAE response regardless of HA use; twenty patients lost OAE response due to CI, not due to wearing HAs (Fig. 4a), and three patients (AH9534, AG6481 and AK6678) lost OAE response under one year of age with or without a very short period wearing HAs (Fig. 4b). Therefore, it is unclear whether the disappearance of OAE is related to wearing HAs or is part of the natural course of OTOF-related hearing loss. As early intervention of HA for deaf patients is important for language development, there is no evidence for not recommending HAs for patients with OTOF-related ANSD with the aim of maintaining OAE response.
Although CI provide excellent hearing performance in cases of OTOF-related ANSD, it does not reach normal hearing level. Therefore, there has been a strong desire for a curative therapy, such as gene therapy. Recently, two reports have shown that cochlear gene therapy mediated by adeno-associated virus (AAV) successfully improved the prognosis of hearing impairment in Otof−/− mice (Al‐Moyed et al. 2018; Akil et al. 2019). A new era of next-generation treatments for hereditary hearing loss, such as gene therapy, is certainly approaching; however, we should not forget that accurate treatment is based on accurate diagnosis and understanding of the clinical course. With the improvements in molecular biological diagnostic method, the etiologies of hereditary hearing loss have gradually become better understood. Although genotype–phenotype correlations have been obscure, the accumulation of patient data is gradually revealing the clinical characteristics of OTOF-related hearing loss. We believe that the clinical characteristics and genotype–phenotype correlation found in this study will support appropriate intervention and future treatment for OTOF-related hearing loss patients.
Change history
02 November 2021
A Correction to this paper has been published: https://doi.org/10.1007/s00439-021-02392-y
References
Akil O, Dyka F, Calvet C et al (2019) Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci 116:4496–4501. https://doi.org/10.1073/pnas.1817537116
Almontashiri NAM, Alswaid A, Oza A et al (2018) Recurrent variants in OTOF are significant contributors to prelingual nonsydromic hearing loss in Saudi patients. Genet Med 20:536–544. https://doi.org/10.1038/gim.2017.143
Al-Moyed H, Cepeda AP, Jung S et al (2018) A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol Med. https://doi.org/10.15252/emmm.201809396
Brandt T, Sack LM, Arjona D et al (2019) Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet Med. https://doi.org/10.1038/s41436-019-0655-2
Chiu YH, Wu CC, Lu YC et al (2010) Mutations in the OTOF gene in Taiwanese patients with auditory neuropathy. Audiol Neurotol 15:364–374. https://doi.org/10.1159/000293992
Choi BY, Ahmed ZM, Riazuddin S et al (2009) Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin Genet 75:237–243. https://doi.org/10.1111/j.1399-0004.2008.01128.x
Fedick AM, Jalas C, Swaroop A et al (2016) Identification of a novel pathogenic OTOF variant causative of nonsyndromic hearing loss with high frequency in the Ashkenazi Jewish population. Appl Clin Genet 9:141–146. https://doi.org/10.2147/TACG.S113828
Iwasa Y-i, Nishio S-y, Sugaya A et al (2019) OTOF mutation analysis with massively parallel DNA sequencing in 2,265 Japanese sensorineural hearing loss patients. PLoS ONE 14:1–10. https://doi.org/10.1371/journal.pone.0215932
Kim BJ, Jang JH, Han JH et al (2018) Mutational and phenotypic spectrum of OTOF-related auditory neuropathy in Koreans: eliciting reciprocal interaction between bench and clinics. J Transl Med 16:1–13. https://doi.org/10.1186/s12967-018-1708-z
Kitao K, Mutai H, Namba K et al (2019) Deterioration in distortion product otoacoustic emissions in auditory neuropathy patients with distinct clinical and genetic backgrounds. Ear Hear 40:184–191. https://doi.org/10.1097/AUD.0000000000000586
Mahdieh N, Shirkavand A, Rabbani B et al (2012) Screening of OTOF mutations in Iran: a novel mutation and review. Int J Pediatr Otorhinolaryngol 76:1610–1615. https://doi.org/10.1016/j.ijporl.2012.07.030
Marlin S, Feldmann D, Nguyen Y et al (2010) Temperature-sensitive auditory neuropathy associated with an otoferlin mutation: deafening fever! Biochem Biophys Res Commun 394:737–742. https://doi.org/10.1016/j.bbrc.2010.03.062
Matsunaga T, Mutai H, Kunishima S et al (2012) A prevalent founder mutation and genotype-phenotype correlations of OTOF in Japanese patients with auditory neuropathy. Clin Genet 82:425–432. https://doi.org/10.1111/j.1399-0004.2012.01897.x
Migliosi V, Villamar M, Tellería D et al (2002) Q829X, a novel mutation in the gene encoding otoferlin. J Med Genet 39:502–507
Miyagawa M, Nishio SY, Usami SI (2016) A comprehensive study on the etiology of patients receiving cochlear implantation with special emphasis on genetic epidemiology. Otol Neurotol 37:e126–e134. https://doi.org/10.1097/MAO.0000000000000936
Moser T, Starr A (2016) Auditory neuropathy-neural and synaptic mechanisms. Nat Rev Neurol 12:135–149. https://doi.org/10.1038/nrneurol.2016.10
Oza AM, DiStefano MT, Hemphill SE et al (2018) Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat 39:1593–1613. https://doi.org/10.1002/humu.23630
Rodríguez-Ballesteros M, Reynoso R, Olarte M et al (2008) A multicenter study on the prevalence and spectrum of mutations in the otoferlin gene (OTOF) in subjects with nonsyndromic hearing impairment and auditory neuropathy. Hum Mutat 29:823–831. https://doi.org/10.1002/humu.20708
Rouillon I, Marcolla A, Roux I et al (2006) Results of cochlear implantation in two children with mutations in the OTOF gene. Int J Pediatr Otorhinolaryngol 70:689–696. https://doi.org/10.1016/j.ijporl.2005.09.006
Roux I, Safieddine S, Nouvian R et al (2006) Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell 127:277–289. https://doi.org/10.1016/j.cell.2006.08.040
Santarelli R, Del Castillo I, Rodríguez-Ballesteros M et al (2009) Abnormal cochlear potentials from deaf patients with mutations in the otoferlin gene. JARO J Assoc Res Otolaryngol 10:545–556. https://doi.org/10.1007/s10162-009-0181-z
Shearer AE (2019) Auditory synaptopathy auditory neuropathy, and cochlear implantation. Laryngoscope Investig Otolaryngol. https://doi.org/10.1002/lio2.288
Shearer AE, Hildebrand MS and Smith RJH (2017) Hereditary hearing loss and deafness overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa, Amemiya A (eds) GeneReviews®. Seattle
Starr A, Picton TW, Sininger Y, Hood LJ, Berlin CI (1996) Auditory neuropathy. Brain 119:741–753
Strenzke N, Chakrabarti R, Al-Moyed H et al (2016) Hair cell synaptic dysfunction, auditory fatigue and thermal sensitivity in otoferlin Ile515Thr mutants. EMBO J 35:2519–2535. https://doi.org/10.15252/embj.201694564
Tekin M, Akcayoz D, Incesulu A (2005) A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am J Med Genet 138A:6–10. https://doi.org/10.1002/ajmg.a.30907
Varga R, Avenarius MR, Kelley PM et al (2006) OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. J Med Genet 43:576–581. https://doi.org/10.1136/jmg.2005.038612
Vona B, Rad A, Reisinger E (2020) The many faces of dfnb9: relating OTOF variants to hearing impairment. Genes (basel) 11:1–23. https://doi.org/10.3390/genes11121411
Wang DY, Wang YC, Weil D et al (2010a) Screening mutations of OTOF gene in Chinese patients with auditory neuropathy, including a familial case of temperature-sensitive auditory neuropathy. BMC Med Genet 11:3–5. https://doi.org/10.1186/1471-2350-11-79
Wang K, Li M, Hakonarson H (2010b) ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38:1–7. https://doi.org/10.1093/nar/gkq603
Wang Y, Lu Y, Cheng J et al (2018) Novel OTOF gene mutations identified using a massively parallel DNA sequencing technique in DFNB9 deafness. Acta Otolaryngol. https://doi.org/10.1080/00016489.2018.1476777
WHO (2021) Deafness and hearing loss. https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss
Wroblewska-Seniuk KE, Dabrowski P, Szyfter W, Mazela J (2017) Universal newborn hearing screening: methods and results, obstacles, and benefits. Pediatr Res 81:415–422. https://doi.org/10.1038/pr.2016.250
Wu CC, Tsai CY, Lin YH et al (2019) Genetic epidemiology and clinical features of hereditary hearing impairment in the Taiwanese population. Genes (basel) 10:1–20. https://doi.org/10.3390/genes10100772
Yildirim-Baylan M, Bademci G, Duman D et al (2014) Evidence for genotype-phenotype correlation for OTOF mutations. Int J Pediatr Otorhinolaryngol 78:950–953. https://doi.org/10.1016/j.ijporl.2014.03.022
Zadro C, Ciorba A, Fabris A et al (2010) Five new OTOF gene mutations and auditory neuropathy. Int J Pediatr Otorhinolaryngol 74:494–498. https://doi.org/10.1016/j.ijporl.2010.02.004
Zhang QJ, Han B, Lan L et al (2016) High frequency of OTOF mutations in Chinese infants with congenital auditory neuropathy spectrum disorder. Clin Genet 90:238–246. https://doi.org/10.1111/cge.12744
Zheng D, Liu X (2020) Cochlear implantation outcomes in patients with OTOF mutations. Front Neurosci 14:1–7. https://doi.org/10.3389/fnins.2020.00447
Acknowledgements
We thank all participant in the present study and collaborators for providing samples and clinical information.
Funding
This research was funded by a Health and Labor Sciences Research Grant for Research on Rare and Intractable diseases and Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labor and Welfare of Japan (S.U. 20FC1048), a Grant-in-Aid from Japan Agency for Medical Research and Development (AMED) (S.U.: 17kk0205010h0002, 18ek0109363h0001).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to correction in Table 1.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iwasa, Yi., Nishio, Sy., Yoshimura, H. et al. Detailed clinical features and genotype–phenotype correlation in an OTOF-related hearing loss cohort in Japan. Hum Genet 141, 865–875 (2022). https://doi.org/10.1007/s00439-021-02351-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02351-7