
Abstract
Mutations of coding regions and splice sites of SLC26A4 cause Pendred syndrome and nonsyndromic recessive hearing loss DFNB4. SLC26A4 encodes pendrin, a transmembrane exchanger of anions and bases. The mutant SLC26A4 phenotype is characterized by inner ear malformations, including an enlarged vestibular aqueduct (EVA), incomplete cochlear partition type II and modiolar hypoplasia, progressive and fluctuating hearing loss, and vestibular dysfunction. A thyroid iodine organification defect can lead to multinodular goiter and distinguishes Pendred syndrome from DFNB4. Pendred syndrome and DFNB4 are each inherited as an autosomal recessive trait caused by biallelic mutations of SLC26A4 (M2). However, there are some EVA patients with only one detectable mutant allele (M1) of SLC26A4. In most European-Caucasian M1 patients, there is a haplotype that consists of 12 variants upstream of SLC26A4, called CEVA (Caucasian EVA), which acts as a pathogenic recessive allele in trans to mutations affecting the coding regions or splice sites of SLC26A4. This combination of an M1 genotype with the CEVA haplotype is associated with a less severe phenotype than the M2 genotype. The phenotype in EVA patients with no mutant alleles of SLC26A4 (M0) has a very low recurrence probability and is likely to be caused by other factors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hereditary hearing loss can be either syndromic, which includes abnormalities affecting other organs and tissues, or nonsyndromic, which is not associated with other signs and symptoms. Nonsyndromic hearing loss phenotypes and loci can be categorized as autosomal dominant (termed DFNA), autosomal recessive (DFNB), X-linked (DFNX), or mitochondrial. DFNA, DFNB, and DFNX phenotypes and loci are numbered in the order the genetic loci were first reported. The causative genes have been identified for more than 70% of the reported loci (Van Camp and Smith 2021).
The same gene can underlie both syndromic and nonsyndromic hearing loss. SLC26A4 (solute carrier family 26, member 4), formally known as PDS, is the causative gene for Pendred syndrome and DFNB4 (Everett et al. 1997; Li et al. 1998). Pendred syndrome was originally described by Vaughan Pendred in 1896 as an autosomal recessive disorder comprised of goiter (thyroid gland enlargement) and severe congenital deafness (Pendred 1896). DFNB4 was first reported in 1995 as nonsyndromic congenital recessive hearing loss mapping to chromosome 7q31 (Baldwin et al. 1995). Both Pendred syndrome and DFNB4 are associated with enlargement of the vestibular aqueduct (EVA), which is also referred to as dilated or large vestibular aqueduct (DVA or LVA, respectively). DFNB4 is thus a form of nonsyndromic EVA (NSEVA). The term “large vestibular aqueduct syndrome (LVAS)”, introduced by Valvassori and Clemis in 1978, refers to the auditory phenotype associated with Pendred syndrome, DFNB4/NSEVA, and other forms of nonsyndromic hearing loss of unknown etiology associated with EVA. Large vestibular aqueduct syndrome is not actually a syndrome, as defined above, although it can be associated with a hearing loss syndrome, such as Pendred syndrome. In this review, we refer to Pendred syndrome and DFNB4 as SLC26A4-related hearing loss (Fig. 1).

Classification of diseases with hearing loss and an enlarged vestibular aqueduct
Mutations of SLC26A4 are one of the most common causes of hereditary hearing loss worldwide (Park et al. 2003). SLC26A4 encodes pendrin, which is expressed in a restricted tissue distribution that includes the inner ear, thyroid, and kidney (Everett et al. 1997, 2001; Royaux et al. 2003). Pendrin functions as a nonspecific exchanger of anions (e.g., Cl− and I−) and bases (e.g., HCO3− and OH−) across apical plasma membranes of epithelial cells.
Clinical phenotype
SLC26A4-related hearing loss is associated with inner ear malformations, hearing loss, vestibular dysfunction, and thyroid abnormalities.
Inner ear malformations
EVA is a completely penetrant feature of SLC26A4-related hearing loss. The vestibular aqueduct, a bony canal surrounding the endolymphatic duct and a portion of the endolymphatic sac, is abnormally dilated in EVA ears. There are two commonly used radiologic definitions of EVA. The original (Valvassori) criterion of a midpoint VA diameter > 1.5 mm (Valvassori and Clemis 1978) has largely been replaced by the “Cincinnati criteria” of a midpoint diameter ≥ 1.0 mm or an operculum diameter ≥ 2.0 mm (Fig. 2A) (Boston et al. 2007; Vijayasekaran et al. 2007). Enlargement of the endolymphatic duct and sac are also observed by magnetic resonance imaging (MRI) of ears with SLC26A4-related hearing loss (Fig. 2B). SLC26A4-related hearing loss and EVA can be bilateral or unilateral. In a North American EVA cohort, 25% of subjects with unilateral EVA had one or two mutant alleles of SLC26A4 (Chattaraj et al. 2013).

Reproduced from https://www.nidcd.nih.gov/health/enlargedvestibular-aqueducts-and-childhood-hearing-loss
Radiologic imaging of an enlarged vestibular aqueduct. Axial computed tomography (CT) scan (top) and magnetic resonance image (MRI) (bottom) of the same right ear with an enlarged vestibular aqueduct. Arrows in the top and bottom panels indicate the same location corresponding to the enlarged endolymphatic sac and duct represented by isodensity on CT and high signal intensity on MRI.
Another characteristic inner ear deformity in SLC26A4-related hearing loss is incomplete partition type II (IP-II). IP-II is a deformity in which the middle and apical turns of the cochlear duct coalesce to form a cystic apex, resulting in 1.5 turns instead of the normal 2.5 turns (Sennaroglu and Saatci 2002). A combination of IP-II and EVA (Mondini dysplasia) was present in the temporal bones of the 8-year-old deaf boy reported by Carlo Mondini in 1791 (Mondini 1997). The incidence of IP-II in EVA ears ranges from 21.7 to 73.9% (Forli et al. 2020; King et al. 2010; Mey et al. 2019b). The more common cochlear anomaly in SLC26A4-related hearing loss is hypoplasia or deficiency of the cochlear modiolus, which can be detected by MRI or computed tomography (CT) (Goldfeld et al. 2005; Lemmerling et al. 1997).
Audiological phenotype
The stereotypical presentation of SLC26A4-related hearing loss is a fluctuating or progressive sensorineural hearing loss (SNHL) with a pre-, peri- or even postlingual onset (Jackler and De La Cruz 1989; Levenson et al. 1989). The severity, laterality, and age of onset of SNHL are highly variable. Audiometry of SLC26A4-related hearing loss often shows air-bone gaps (indicative of conductive hearing loss) at low frequencies which results in mixed hearing loss (both sensorineural and conductive) in the presence of a normal middle ear. This is attributed to a “third-window” effect of EVA, in which the power of sound transmission within the labyrinth is shunted away from the cochlea (Merchant et al. 2007). The fluctuation or progression of the SNHL can occur in a stepwise incremental fashion and can be precipitated by minor head trauma or barotrauma. A study of 109 patients in Denmark with SLC26A4-related hearing loss demonstrated that the average level of hearing loss progressed and reached 80 dBHL by 6 years of age, whereas almost one half of the patients passed neonatal hearing screening (Mey et al. 2019a). This observation highlights the postnatal onset of SNHL in many ears with EVA.
Vestibular phenotype
Vestibular dysfunction also occurs in SLC26A4-related hearing loss. The reported prevalence of vestibular symptoms is 4–47% among patients with EVA (Antonelli et al. 1998; Berrettini et al. 2005; Grimmer and Hedlund 2007; Jackler and De La Cruz 1989; Jung et al. 2016; Valvassori and Clemis 1978; Yang et al. 2016; Zalewski et al. 2015). The symptoms can include episodic rotatory vertigo, clumsiness, head-tilting and vomiting, and a delayed onset (> 18 months of age) of independent ambulation. Abnormal results of vestibular function tests, including the caloric test, rotational chair test, video head impulse test (vHIT), and the cervical vestibular evoked myogenic potential (cVEMP) test, have been reported for some patients with EVA (Jung et al. 2016, 2017; Sheykholeslami et al. 2004; Yang et al. 2016; Zalewski et al. 2015; Zhou and Gopen 2011; Zhou et al. 2017). In a North American cohort of patients with EVA, 21 (32.8%) of 64 patients had caloric abnormalities, six (25.0%) of 24 patients had abnormal results of rotational chair testing, and two (22.2%) of 9 patients had abnormal cVEMP results (Zalewski et al. 2015). There was no correlation between the severity of hearing loss and vestibular signs and symptoms or abnormal vestibular test results. In a cohort of 31 Korean patients with biallelic mutations in SLC26A4, 16 patients (45.2%) had unilateral caloric weakness and two patients (6.4%) had bilateral caloric weakness (Jung et al. 2016). Among 10 patients undergoing both the caloric test and vHIT in the Korean cohort, four patients (40%) had unilateral caloric weakness and one patient (10%) had abnormal vHIT results (Jung et al. 2017). These results suggest that the pathophysiologic mechanism of SLC26A4 mutations in vestibular systems may differ from that in the auditory system.
Thyroid phenotype
Pendred syndrome is characterized by the presence of thyroid abnormalities. Goiter is incompletely penetrant and does not typically present until adolescence. Thus, Pendred syndrome often presents prior to the onset of goiter as nonsyndromic hearing loss (Madeo et al. 2009). Ultrasonography can detect thyroid structural abnormalities with a higher sensitivity and specificity than manual palpation. The underlying, more penetrant thyroid phenotype is a deficiency of iodine organification in the biosynthesis of thyroid hormone. This is thought to reflect deficient transport of iodide anions by pendrin in the luminal plasma membrane of thyroid follicular cells. The perchlorate discharge test is the most sensitive clinical diagnostic method to detect the iodide organification defect (Madeo et al. 2009). Fraser clinically defined Pendred syndrome as congenital deafness, goiter, and a positive perchlorate discharge test (Fraser 1965). The perchlorate discharge test was previously used to distinguish between Pendred syndrome and DFNB4/NSEVA before the availability of reliable SLC26A4 mutation testing. Subclinical or frank hypothyroidism in Pendred syndrome ranges from 0 to 79% (Ladsous et al. 2014; Madeo et al. 2009; Reardon et al. 1999; Soh et al. 2015). Thyroid serologic testing is not useful as an initial diagnostic screen for Pendred syndrome but is helpful for the management of thyroid-related symptoms.
Genetics
Pendred syndrome and DFNB4 are each inherited as an autosomal recessive trait. Thus, a conclusive genetic test for those phenotypes is expected to detect two mutant alleles (M2), either as homozygous or compound heterozygous mutations, of SLC26A4. However, there are some EVA patients with only one detectable mutant allele (M1), or with no mutant alleles (M0), identified through sequence analysis of the coding regions and adjacent splice sites of SLC26A4. The relative proportion of M2, M1, and M0 genotypes is variable in different populations. In North American Caucasian EVA patients, approximately 25% of patients are M2, another 25% are M1, and the other 50% are M0 (Choi et al. 2009a, b; Pryor et al. 2005). In East Asia, including China, Korea, or Japan, 67–90% of EVA patients have M2 genotypes and 8–21% have M1 genotypes (Choi et al. 2009c; Miyagawa et al. 2014; Reyes et al. 2009; Wang et al. 2007; Zhao et al. 2013). This observation suggested the existence of one or more etiologic factors for EVA that are more common in European-Caucasian populations than in east Asian populations.
The prevalence of EVA among siblings of M1 probands is approximately 0.25, which is expected for an autosomal recessive trait (Choi et al. 2009a). This observation, as well as co-segregation of EVA with SLC26A4-linked markers on both chromosomes 7, suggested the existence of a second undetected pathologic variant in noncoding regions of SLC26A4 in M1 patients (Choi et al. 2009a). In contrast, the probability of EVA in a non-twin sibling of an M0 EVA patient is nearly zero, indicating that EVA in M0 patients is not inherited as a Mendelian trait (Choi et al. 2009a).
Recently, a shared haplotype comprised of 12 variants located in introns or intergenic regions upstream of SLC26A4 was identified as a recessive mutant allele when present in trans to a pathogenic variant of SLC26A4 in Caucasian M1 families (Fig. 3) (Chattaraj et al. 2017). The SLC26A4-linked haplotype, termed CEVA (Caucasian EVA), was present on 7 of 10 mutation-negative chromosomes in a North American Caucasian M1 EVA cohort and 6 of 6 mutation-negative chromosomes in a Danish M1 EVA cohort. In contrast, the observed prevalence of the CEVA haplotype among Caucasian control chromosomes is 28 of 1006, which is significantly lower than among M1 patients (p < 0.0001). Thus, the association of CEVA with EVA is statistically significant and CEVA is likely to act as a pathogenic recessive allele in M1 patients. The CEVA haplotype was also found on 11 of 126 chromosomes in a North American Caucasian M0 EVA cohort, which is significantly higher (p = 0.0042) than the prevalence among Caucasian controls. The CEVA haplotype does not co-segregate with EVA in M0 patients, indicating that CEVA is not the major etiologic factor of EVA in M0 patients. Although several M0 patients are heterozygous for variants of unknown significance (p.Met775Thr and p.Glu29Gln) and homozygous for the CEVA haplotype, the pathogenic role of CEVA in these patients remains unclear.

Genetic map of the SLC26A4-linked CEVA (Caucasian EVA) haplotype. Twelve uncommon variants in intergenic regions or introns of genes including PIK3CG, PRKAR2B, HBP1, COG5, DUS4L, and BCAP29, are located within a linkage disequilibrium block spanning a 613-kb region upstream of SLC26A4. Two variants (rs199667576 and rs199915614) are single-nucleotide deletions and ten are single-nucleotide substitutions
Digenic inheritance of a heterozygous mutation in SLC26A4 in combination with a heterozygous mutation in either the FOXI1 or KCNJ10 gene was proposed for M1 nonsyndromic EVA patients (Yang et al. 2007, 2009), but other studies have not been able to replicate or reproduce these findings (Chen et al. 2012; Jonard et al. 2010; Landa et al. 2013; Pique et al. 2014). A more recent study proposed digenic inheritance of point mutations in EPHA2 and SLC26A4 in two Japanese patients in Pendred syndrome (Li et al. 2020).
Genotype–phenotype correlation
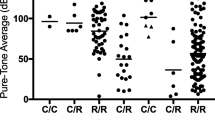
The number of mutant alleles of SLC26A4 is correlated with inner ear morphology, auditory, and thyroid phenotypes in Caucasian population (Table 1).
The M2 genotype is tightly correlated with bilateral EVA (Azaiez et al. 2007; Choi et al. 2009b; Pryor et al. 2005). King et al. could detect no significant relationship between EVA size and the number of mutant alleles of SLC26A4. In contrast, a 2019 study suggested that the M2 genotype is associated with enlarged size and presence of “high-protein” contents in the endolymphatic sac seen on MRI (Mey et al. 2019b). Others have reported a significant association of the number of mutant alleles of SLC26A4 with the presence of IP-II (Forli et al. 2020; King et al. 2010; Mey et al. 2019b).
The severity of hearing loss in EVA ears is greater in M2 patients than in M0 or M1 patients (Albert et al. 2006; King et al. 2010; Mey et al. 2019a, b; Rose et al. 2017) The prevalence of cochlear implantation is lower in M0 or M1 ears in comparison to M2 ears (Rose et al. 2017). Hearing loss reaches severe to profound levels (> 80 dB HL) earlier in M0 patients (3.2 years of age) than in all patients with SLC26A4-related hearing loss (6.0 years of age) (Mey et al. 2019a). However, the prevalence of hearing fluctuation does not seem to be associated with the number of mutant alleles of SLC26A4 (Rose et al. 2017).
The iodine organification defect, as detected by the perchlorate discharge test, is significantly correlated with the M2 genotype (Madeo et al. 2009; Pryor et al. 2005). Thyroid function measured by serologic testing is not associated with the number of mutant alleles (Madeo et al. 2009).
M1 patients carrying the CEVA haplotype have a normal thyroid phenotype and less severe hearing loss in comparison to M2 patients (Chao et al. 2019). In M0 patients, hearing loss was more severe in patients who carry CEVA in comparison to non-carriers (Chao et al. 2019). This result suggested that CEVA might act as a genetic modifier of the EVA phenotype, although it is not the cause of EVA, in M0 patients.
The genotype–phenotype correlation is different in East Asian populations. Auditory phenotypes are more strongly associated with the type, rather than the number of mutant alleles, of SLC26A4 in a Korean cohort (Rah et al. 2015). Other studies in Japanese and Chinese populations have not detected an association of the type or number of mutant alleles with the auditory phenotype (Miyagawa et al. 2014; Reyes et al. 2009; Wu et al. 2010). Intrafamilial variability in individuals with the same biallelic SLC26A4 mutations have been reported (Miyagawa et al. 2014; Song et al. 2014).
Pathophysiological mechanism
SLC26A4 is comprised of 21 exons that encode a 780-amino acid (86-kDa) protein called pendrin (Everett et al. 1997). Pendrin is a nonspecific exchanger of anions and bases that is expressed in the plasma membrane of epithelial cells (Choi et al. 2009b; Wasano et al. 2020). The coding regions of human SLC26A4 and the mouse ortholog Slc26a4 share 85% nucleotide identity and the predicted amino acid sequences are 87% identical (Everett et al. 1999). In the mouse inner ear, pendrin is expressed in the apical membrane of outer sulcus and spindle cells located in the lateral wall of the cochlea, transitional cells of the vestibular organs, and mitochondria-rich cells (MRCs) of the endolymphatic sac (Dou et al. 2004; Royaux et al. 2003; Wangemann et al. 2004). During development, pendrin expression is first initiated in the endolymphatic sac at embryonic day (E) 11.5 (Kim and Wangemann 2011). The onset of expression in the cochlea and the vestibular organs occurs at E13.5–E16.5 (Kim and Wangemann 2011).
A mouse model lacking pendrin (Slc26a4Δ/Δ) has profound hearing loss, vestibular dysfunction, and massively enlarged endolymphatic spaces from the cochlear duct to the endolymphatic sac (Everett et al. 2001). The endolymphatic spaces begin to enlarge at E14.5 and reach an approximately tenfold increase in size of the cochlear lumen at E18.5 in comparison to Slc26a4Δ/+ littermate controls (Kim and Wangemann 2010). Acidification of the endolymph occurs in the cochlea at E15.5 and in the endolymphatic sac at E17.5 due to the failure of HCO3− secretion by pendrin (Kim and Wangemann 2011). The two primary pathological alterations, luminal enlargement and acidification, may be followed by secondary consequences including increased K+ secretion by marginal cells, oxidative stress in the stria vascularis, loss of Kcnj10 in intermediate cells, a loss of endocochlear potential, increase of Ca2+ concentration in the endolymph, formation of giant otoconia, and a degeneration of sensory cells and the stria vascularis (Jabba et al. 2006; Li et al. 2013b; Nakaya et al. 2007; Singh and Wangemann 2008; Wangemann et al. 2004, 2007).
The molecular mechanism of endolymphatic enlargement is likely to be impaired absorption of luminal Na+, Cl− and water by the endolymphatic sac during development (Choi et al. 2011; Honda et al. 2017; Li et al. 2013a). MRCs comprise about 30% of endolymphatic sac epithelial cells (Dahlmann and von Düring 1995; Honda et al. 2017). MRCs have plentiful mitochondria and numerous apical microvilli, and express ion transport genes including Slc26a4, Atp6v0a4, Atp6v1b1, Cftr and the forkhead transcription factor Foxi1 that regulates expression of those genes (Honda et al. 2017; Hulander et al. 2003; Raft et al. 2014; Vidarsson et al. 2009). This structure and gene expression profile are features shared with other ionocytes such as intercalated cells of the kidney, narrow and clear cells in the epididymis, and recently identified pulmonary ionocytes (Blomqvist et al. 2004, 2006; Montoro et al. 2018; Plasschaert et al. 2018; Scudieri et al. 2020). Human orthologs ATP6V0A4 and ATP6V1B1 encoding V-ATPase subunits are causal genes for distal renal tubal acidosis with deafness and EVA (Vidarsson et al. 2009). Atp6v0a4Δ/Δ mice, Atp6v1b1 mutant mice, and Foxi1Δ/Δ mice all have an inner ear phenotype similar to that of Slc26a4Δ/Δ mice (Hulander et al. 2003; Lorente-Canovas et al. 2013; Tian et al. 2017). MRCs, acting as “the inner ear ionocyte”, appear to be engaged in absorption of Na+ Cl−, and thus water, by the endolymphatic sac and regulate the balance between secretion and absorption of the endolymph during inner ear development. Disruption of this pathway may be the underlying mechanism for enlargement of the endolymphatic sac, cochlea, and vestibular aqueduct (Honda et al. 2017).
Slc26a4 knockout and knockin mouse models result in auditory and inner ear phenotypes that are more severe than those observed in patients with SLC26A4-related hearing loss (Dror et al. 2010; Hu et al. 2021; Lu et al. 2011, 2014; Wen et al. 2019). A doxycycline-induced Slc26a4-insufficient mouse model has fluctuation of hearing associated with fluctuation of the endocochlear potential and more closely approximates the phenotype observed in human patients. In this model, the loss of hearing is closely correlated with loss of the endocochlear potential and structural abnormalities and degeneration of the stria vascularis (Ito et al. 2014; Jabba et al. 2006). Expression of Slc26a4 in the endolymphatic sac, but not the cochlea, is required for the acquisition of normal ABR thresholds in mice, suggesting that the cochlear abnormalities occur secondarily to the endolymphatic sac dysfunction (Li et al. 2013a). The pathogenic link between endolymphatic sac and cochlea dysfunction is unknown but may include alterations of pH, ionic composition, size, or osmotic pressure of endolymph. Elucidation of this pathophysiologic mechanism should inform our understanding and management of SLC26A4-related hearing loss.
Conclusions
Hearing loss associated with enlargement of the vestibular aqueduct is a penetrant feature of SLC26A4-related mutant phenotypes. The phenotypes range from nonsyndromic recessive sensorineural hearing loss DFNB4 to sensorineural hearing loss as part of Pendred syndrome. Some EVA patients, especially those of European or Caucasian ancestry, will only have one SLC26A4 allele with a detectable mutation affecting the coding regions or splice sites. The phenotype associated with one mutant allele (M1) is generally less severe than that associated with two mutant alleles (M2). Recent studies have revealed the existence of an uncommon haplotype, called CEVA, located upstream of SLC26A4 that acts as a pathogenic recessive allele in trans-configuration with the mutated SLC26A4 gene in a majority of European-Caucasian M1 EVA patients. CEVA is associated with a milder phenotype than mutations affecting the coding regions or splice sites of SLC26A4. The pathogenesis of EVA in patients with no detectable mutations of SLC26A4 is likely caused by other mechanisms. Studies of Slc26a4-insufficient or -null mice indicate that disruption of sodium chloride and fluid absorption by the developing endolymphatic sac is the precipitating developmental event in the pathogenesis of SLC26A4-related hearing loss.
Declarations:
Availability of data and materials
Not applicable.
Code availability
Not applicable.
References
Albert S, Blons H, Jonard L, Feldmann D, Chauvin P, Loundon N, Sergent-Allaoui A, Houang M, Joannard A, Schmerber S, Delobel B, Leman J, Journel H, Catros H, Dollfus H, Eliot MM, David A, Calais C, Drouin-Garraud V, Obstoy MF, Tran Ba Huy P, Lacombe D, Duriez F, Francannet C, Bitoun P, Petit C, Garabedian EN, Couderc R, Marlin S, Denoyelle F (2006) SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet 14:773–779. https://doi.org/10.1038/sj.ejhg.5201611
Antonelli PJ, Nall AV, Lemmerling MM, Mancuso AA, Kubilis PS (1998) Hearing loss with cochlear modiolar defects and large vestibular aqueducts. Am J Otol 19:306–312
Azaiez H, Yang T, Prasad S, Sorensen JL, Nishimura CJ, Kimberling WJ, Smith RJ (2007) Genotype-phenotype correlations for SLC26A4-related deafness. Hum Genet 122:451–457. https://doi.org/10.1007/s00439-007-0415-2
Baldwin CT, Weiss S, Farrer LA, De Stefano AL, Adair R, Franklyn B, Kidd KK, Korostishevsky M, Bonné-Tamir B (1995) Linkage of congenital, recessive deafness (DFNB4) to chromosome 7q31 and evidence for genetic heterogeneity in the Middle Eastern Druze population. Hum Mol Genet 4:1637–1642. https://doi.org/10.1093/hmg/4.9.1637
Berrettini S, Forli F, Bogazzi F, Neri E, Salvatori L, Casani AP, Franceschini SS (2005) Large vestibular aqueduct syndrome: audiological, radiological, clinical, and genetic features. Am J Otolaryngol 26:363–371. https://doi.org/10.1016/j.amjoto.2005.02.013
Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, Brown R, Persson AE, Bergstrom GG, Enerback S (2004) Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J Clin Invest 113:1560–1570. https://doi.org/10.1172/jci20665
Blomqvist SR, Vidarsson H, Söder O, Enerbäck S (2006) Epididymal expression of the forkhead transcription factor Foxi1 is required for male fertility. EMBO J 62:4131–4141. https://doi.org/10.1038/sj.emboj.7601272
Boston M, Halsted M, Meinzen-Derr J, Bean J, Vijayasekaran S, Arjmand E, Choo D, Benton C, Greinwald J (2007) The large vestibular aqueduct: a new definition based on audiologic and computed tomography correlation. Otolaryngol Head Neck Surg 136:972–977. https://doi.org/10.1016/j.otohns.2006.12.011
Chao JR, Chattaraj P, Munjal T, Honda K, King KA, Zalewski CK, Chien WW, Brewer CC, Griffith AJ (2019) SLC26A4-linked CEVA haplotype correlates with phenotype in patients with enlargement of the vestibular aqueduct. BMC Med Genet 20:118. https://doi.org/10.1186/s12881-019-0853-4
Chattaraj P, Reimold FR, Muskett JA, Shmukler BE, Chien WW, Madeo AC, Pryor SP, Zalewski CK, Butman JA, Brewer CC, Kenna MA, Alper SL, Griffith AJ (2013) Use of SLC26A4 mutation testing for unilateral enlargement of the vestibular aqueduct. JAMA Otolaryngol Head Neck Surg 139:907–913. https://doi.org/10.1001/jamaoto.2013.4185
Chattaraj P, Munjal T, Honda K, Rendtorff ND, Ratay JS, Muskett JA, Risso DS, Roux I, Gertz EM, Schaffer AA, Friedman TB, Morell RJ, Tranebjaerg L, Griffith AJ (2017) A common SLC26A4-linked haplotype underlying non-syndromic hearing loss with enlargement of the vestibular aqueduct. J Med Genet 54:665–673. https://doi.org/10.1136/jmedgenet-2017-104721
Chen K, Wang X, Sun L, Jiang H (2012) Screening of SLC26A4, FOXI1, KCNJ10, and GJB2 in bilateral deafness patients with inner ear malformation. Otolaryngol Head Neck Surg 146:972–978. https://doi.org/10.1177/0194599812439670
Choi BY, Madeo AC, King KA, Zalewski CK, Pryor SP, Muskett JA, Nance WE, Butman JA, Brewer CC, Griffith AJ (2009a) Segregation of enlarged vestibular aqueducts in families with non-diagnostic SLC26A4 genotypes. J Med Genet 46:856–861. https://doi.org/10.1136/jmg.2009.067892
Choi BY, Stewart AK, Madeo AC, Pryor SP, Lenhard S, Kittles R, Eisenman D, Kim HJ, Niparko J, Thomsen J, Arnos KS, Nance WE, King KA, Zalewski CK, Brewer CC, Shawker T, Reynolds JC, Butman JA, Karniski LP, Alper SL, Griffith AJ (2009b) Hypo-functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: genotype-phenotype correlation or coincidental polymorphisms? Hum Mutat 30:599–608. https://doi.org/10.1002/humu.20884
Choi BY, Stewart AK, Nishimura KK, Cha WJ, Seong MW, Park SS, Kim SW, Chun YS, Chung JW, Park SN, Chang SO, Kim CS, Alper SL, Griffith AJ, Oh SH (2009c) Efficient molecular genetic diagnosis of enlarged vestibular aqueducts in East Asians. Genet Test Mol Biomark 13:679–687. https://doi.org/10.1089/gtmb.2009.0054
Choi BY, Kim HM, Ito T, Lee KY, Li X, Monahan K, Wen Y, Wilson E, Kurima K, Saunders TL, Petralia RS, Wangemann P, Friedman TB, Griffith AJ (2011) Mouse model of enlarged vestibular aqueducts defines temporal requirement of Slc26a4 expression for hearing acquisition. J Clin Invest 121:4516–4525. https://doi.org/10.1172/jci59353
Dahlmann A, von Düring M (1995) The endolymphatic duct and sac of the rat: a histological, ultrastructural, and immunocytochemical investigation. Cell Tissue Res 282:277–289. https://doi.org/10.1007/BF00319118
Dou H, Xu J, Wang Z, Smith AN, Soleimani M, Karet FE, Greinwald JH Jr, Choo D (2004) Co-expression of pendrin, vacuolar H+-ATPase alpha4-subunit and carbonic anhydrase II in epithelial cells of the murine endolymphatic sac. J Histochem Cytochem 52:1377–1384. https://doi.org/10.1369/jhc.3A6228.2004
Dror AA, Politi Y, Shahin H, Lenz DR, Dossena S, Nofziger C, Fuchs H, Hrabe de Angelis M, Paulmichl M, Weiner S, Avraham KB (2010) Calcium oxalate stone formation in the inner ear as a result of an Slc26a4 mutation. J Biol Chem 285:21724–21735. https://doi.org/10.1074/jbc.M110.120188
Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED (1997) Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 17:411–422. https://doi.org/10.1038/ng1297-411
Everett LA, Morsli H, Wu DK, Green ED (1999) Expression pattern of the mouse ortholog of the Pendred’s syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci U S A 96:9727–9732. https://doi.org/10.1073/pnas.96.17.9727
Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED (2001) Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet 10:153–161. https://doi.org/10.1093/hmg/10.2.153
Forli F, Lazzerini F, Auletta G, Bruschini L, Berrettini S (2020) Enlarged vestibular aqueduct and Mondini Malformation: audiological, clinical, radiologic and genetic features. Eur Arch Otorhinolaryngol. https://doi.org/10.1007/s00405-020-06333-9
Fraser GR (1965) Assoiation of congenital deafness with goitre (Pendred’s syndrome): a study of 207 families. Ann Hum Genet 28:201–249. https://doi.org/10.1111/j.1469-1809.1964.tb00479.x
Goldfeld M, Glaser B, Nassir E, Gomori JM, Hazani E, Bishara N (2005) CT of the ear in Pendred syndrome. Radiology 235:537–540. https://doi.org/10.1148/radiol.2352031583
Grimmer JF, Hedlund G (2007) Vestibular symptoms in children with enlarged vestibular aqueduct anomaly. Int J Pediatr Otorhinolaryngol 71:275–282. https://doi.org/10.1016/j.ijporl.2006.10.010
Honda K, Kim SH, Kelly MC, Burns JC, Constance L, Li X, Zhou F, Hoa M, Kelley MW, Wangemann P, Morell RJ, Griffith AJ (2017) Molecular architecture underlying fluid absorption by the developing inner ear. Elife 6:e26851. https://doi.org/10.7554/eLife.26851
Hu CJ, Lu YC, Yang TH, Chan YH, Tsai CY, Yu IS, Lin SW, Liu TC, Cheng YF, Wu CC, Hsu CJ (2021) Toward the pathogenicity of the SLC26A4 p.C565Y variant using a genetically driven mouse model. Int J Mol Sci. https://doi.org/10.3390/ijms22062789
Hulander M, Kiernan AE, Blomqvist SR, Carlsson P, Samuelsson EJ, Johansson BR, Steel KP, Enerback S (2003) Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development 130:2013–2025. https://doi.org/10.1242/dev.00376
Ito T, Li X, Kurima K, Choi BY, Wangemann P, Griffith AJ (2014) Slc26a4-insufficiency causes fluctuating hearing loss and stria vascularis dysfunction. Neurobiol Dis 66:53–65. https://doi.org/10.1016/j.nbd.2014.02.002
Jabba SV, Oelke A, Singh R, Maganti RJ, Fleming S, Wall SM, Everett LA, Green ED, Wangemann P (2006) Macrophage invasion contributes to degeneration of stria vascularis in Pendred syndrome mouse model. BMC Med 4:37. https://doi.org/10.1186/1741-7015-4-37
Jackler RK, De La Cruz A (1989) The large vestibular aqueduct syndrome. Laryngoscope 99:1238–1242. https://doi.org/10.1288/00005537-198912000-00006
Jonard L, Niasme-Grare M, Bonnet C, Feldmann D, Rouillon I, Loundon N, Calais C, Catros H, David A, Dollfus H, Drouin-Garraud V, Duriez F, Eliot MM, Fellmann F, Francannet C, Gilbert-Dussardier B, Gohler C, Goizet C, Journel H, Mom T, Thuillier-Obstoy MF, Couderc R, Garabedian EN, Denoyelle F, Marlin S (2010) Screening of SLC26A4, FOXI1 and KCNJ10 genes in unilateral hearing impairment with ipsilateral enlarged vestibular aqueduct. Int J Pediatr Otorhinolaryngol 74:1049–1053. https://doi.org/10.1016/j.ijporl.2010.06.002
Jung J, Seo YW, Choi JY, Kim SH (2016) Vestibular function is associated with residual low-frequency hearing loss in patients with bi-allelic mutations in the SLC26A4 gene. Hear Res 335:33–39. https://doi.org/10.1016/j.heares.2016.02.009
Jung J, Suh MJ, Kim SH (2017) Discrepancies between video head impulse and caloric tests in patients with enlarged vestibular aqueduct. Laryngoscope 127:921–926. https://doi.org/10.1002/lary.26122
Kim HM, Wangemann P (2010) Failure of fluid absorption in the endolymphatic sac initiates cochlear enlargement that leads to deafness in mice lacking pendrin expression. PLoS ONE 5:e14041. https://doi.org/10.1371/journal.pone.0014041
Kim HM, Wangemann P (2011) Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin. PLoS ONE 6:e17949. https://doi.org/10.1371/journal.pone.0017949
King KA, Choi BY, Zalewski C, Madeo AC, Manichaikul A, Pryor SP, Ferruggiaro A, Eisenman D, Kim HJ, Niparko J, Thomsen J, Butman JA, Griffith AJ, Brewer CC (2010) SLC26A4 genotype, but not cochlear radiologic structure, is correlated with hearing loss in ears with an enlarged vestibular aqueduct. Laryngoscope 120:384–389. https://doi.org/10.1002/lary.20722
Ladsous M, Vlaeminck-Guillem V, Dumur V, Vincent C, Dubrulle F, Dhaenens CM, Wemeau JL (2014) Analysis of the thyroid phenotype in 42 patients with Pendred syndrome and nonsyndromic enlargement of the vestibular aqueduct. Thyroid 24:639–648. https://doi.org/10.1089/thy.2013.0164
Landa P, Differ AM, Rajput K, Jenkins L, Bitner-Glindzicz M (2013) Lack of significant association between mutations of KCNJ10 or FOXI1 and SLC26A4 mutations in Pendred syndrome/enlarged vestibular aqueducts. BMC Med Genet 14:85. https://doi.org/10.1186/1471-2350-14-85
Lemmerling MM, Mancuso AA, Antonelli PJ, Kubilis PS (1997) Normal modiolus: CT appearance in patients with a large vestibular aqueduct. Radiology 204:213–219. https://doi.org/10.1148/radiology.204.1.9205250
Levenson MJ, Parisier SC, Jacobs M, Edelstein DR (1989) The large vestibular aqueduct syndrome in children. A review of 12 cases and the description of a new clinical entity. Arch Otolaryngol Head Neck Surg 115:54–58. https://doi.org/10.1001/archotol.1989.01860250056026
Li XC, Everett LA, Lalwani AK, Desmukh D, Friedman TB, Green ED, Wilcox ER (1998) A mutation in PDS causes non-syndromic recessive deafness. Nat Genet 18:215–217. https://doi.org/10.1038/ng0398-215
Li X, Sanneman JD, Harbidge DG, Zhou F, Ito T, Nelson R, Picard N, Chambrey R, Eladari D, Miesner T, Griffith AJ, Marcus DC, Wangemann P (2013a) SLC26A4 targeted to the endolymphatic sac rescues hearing and balance in Slc26a4 mutant mice. PLoS Genet 9:e1003641. https://doi.org/10.1371/journal.pgen.1003641
Li X, Zhou F, Marcus DC, Wangemann P (2013b) Endolymphatic Na(+) and K(+) concentrations during cochlear growth and enlargement in mice lacking Slc26a4/pendrin. PLoS ONE 8:e65977. https://doi.org/10.1371/journal.pone.0065977
Li M, Nishio SY, Naruse C, Riddell M, Sapski S, Katsuno T, Hikita T, Mizapourshafiyi F, Smith FM, Cooper LT, Lee MG, Asano M, Boettger T, Krueger M, Wietelmann A, Graumann J, Day BW, Boyd AW, Offermanns S, Kitajiri SI, Usami SI, Nakayama M (2020) Digenic inheritance of mutations in EPHA2 and SLC26A4 in Pendred syndrome. Nat Commun 11:1343. https://doi.org/10.1038/s41467-020-15198-9
Lorente-Canovas B, Ingham N, Norgett EE, Golder ZJ, Karet Frankl FE, Steel KP (2013) Mice deficient in H+-ATPase a4 subunit have severe hearing impairment associated with enlarged endolymphatic compartments within the inner ear. Dis Model Mech 6:434–442. https://doi.org/10.1242/dmm.010645
Lu YC, Wu CC, Shen WS, Yang TH, Yeh TH, Chen PJ, Yu IS, Lin SW, Wong JM, Chang Q, Lin X, Hsu CJ (2011) Establishment of a knock-in mouse model with the SLC26A4 c.919–2A>G mutation and characterization of its pathology. PLoS ONE 6:e22150. https://doi.org/10.1371/journal.pone.0022150
Lu YC, Wu CC, Yang TH, Lin YH, Yu IS, Lin SW, Chang Q, Lin X, Wong JM, Hsu CJ (2014) Differences in the pathogenicity of the p.H723R mutation of the common deafness-associated SLC26A4 gene in humans and mice. PLoS ONE 8:e64906. https://doi.org/10.1371/journal.pone.0064906
Madeo AC, Manichaikul A, Reynolds JC, Sarlis NJ, Pryor SP, Shawker TH, Griffith AJ (2009) Evaluation of the thyroid in patients with hearing loss and enlarged vestibular aqueducts. Arch Otolaryngol Head Neck Surg 135:670–676. https://doi.org/10.1001/archoto.2009.66
Merchant SN, Nakajima HH, Halpin C, Nadol JB Jr, Lee DJ, Innis WP, Curtin H, Rosowski JJ (2007) Clinical investigation and mechanism of air-bone gaps in large vestibular aqueduct syndrome. Ann Otol Rhinol Laryngol 116:532–541. https://doi.org/10.1177/000348940711600709
Mey K, Bille M, Rye Rasmussen SH, Tranebjaerg L, Caye-Thomasen P (2019a) The natural history of hearing loss in pendred syndrome and non-syndromic enlarged vestibular aqueduct. Otol Neurotol 40:e178–e185. https://doi.org/10.1097/MAO.0000000000002140
Mey K, Muhamad AA, Tranebjaerg L, Rendtorff ND, Rasmussen SH, Bille M, Caye-Thomasen P (2019b) Association of SLC26A4 mutations, morphology, and hearing in pendred syndrome and NSEVA. Laryngoscope 129:2574–2579. https://doi.org/10.1002/lary.27319
Miyagawa M, Nishio SY, Usami S (2014) Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: a large cohort study. J Hum Genet 59:262–268. https://doi.org/10.1038/jhg.2014.12
Mondini C (1997) Minor works of Carlo Mondini: the anatomical section of a boy born deaf. Am J Otol 18:288–293
Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, Birket SE, Yuan F, Chen S, Leung HM, Villoria J, Rogel N, Burgin G, Tsankov AM, Waghray A, Slyper M, Waldman J, Nguyen L, Dionne D, Rozenblatt-Rosen O, Tata PR, Mou H, Shivaraju M, Bihler H, Mense M, Tearney GJ, Rowe SM, Engelhardt JF, Regev A, Rajagopal J (2018) A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560:319–324. https://doi.org/10.1038/s41586-018-0393-7
Nakaya K, Harbidge DG, Wangemann P, Schultz BD, Green ED, Wall SM, Marcus DC (2007) Lack of pendrin HCO3- transport elevates vestibular endolymphatic [Ca2+] by inhibition of acid-sensitive TRPV5 and TRPV6 channels. Am J Physiol Renal Physiol 292:F1314–F1321. https://doi.org/10.1152/ajprenal.00432.2006
Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, Kim HN, Moon SK, Abe S, Tukamoto K, Riazuddin S, Kabra M, Erdenetungalag R, Radnaabazar J, Khan S, Pandya A, Usami SI, Nance WE, Wilcox ER, Riazuddin S, Griffith AJ (2003) Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet 40:242–248. https://doi.org/10.1136/jmg.40.4.242
Pendred V (1896) Deaf-mutism and goitre. The Lancet 148:532. https://doi.org/10.1016/S0140-6736(01)74403-0
Pique LM, Brennan ML, Davidson CJ, Schaefer F, Greinwald J Jr, Schrijver I (2014) Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. PeerJ 2:e384. https://doi.org/10.7717/peerj.384
Plasschaert LW, Zilionis R, Choo-Wing R, Savova V, Knehr J, Roma G, Klein AM, Jaffe AB (2018) A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560:377–381. https://doi.org/10.1038/s41586-018-0394-6
Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA, Griffith AJ (2005) SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet 42:159–165. https://doi.org/10.1136/jmg.2004.024208
Raft S, Andrade LR, Shao D, Akiyama H, Henkemeyer M, Wu DK (2014) Ephrin-B2 governs morphogenesis of endolymphatic sac and duct epithelia in the mouse inner ear. Dev Biol 390:51–67. https://doi.org/10.1016/j.ydbio.2014.02.019
Rah YC, Kim AR, Koo JW, Lee JH, Oh SH, Choi BY (2015) Audiologic presentation of enlargement of the vestibular aqueduct according to the SLC26A4 genotypes. Laryngoscope 125:E216–E222. https://doi.org/10.1002/lary.25079
Reardon W, Coffey R, Chowdhury T, Grossman A, Jan H, Britton K, Kendall-Taylor P, Trembath R (1999) Prevalence, age of onset, and natural history of thyroid disease in Pendred syndrome. J Med Genet 36:595–598. https://doi.org/10.1136/jmg.36.8.595
Reyes S, Wang G, Ouyang X, Han B, Du LL, Yuan HJ, Yan D, Dai P, Liu XZ (2009) Mutation analysis of SLC26A4 in mainland Chinese patients with enlarged vestibular aqueduct. Otolaryngol Head Neck Surg 141:502–508. https://doi.org/10.1016/j.otohns.2009.07.004
Rose J, Muskett JA, King KA, Zalewski CK, Chattaraj P, Butman JA, Kenna MA, Chien WW, Brewer CC, Griffith AJ (2017) Hearing loss associated with enlarged vestibular aqueduct and zero or one mutant allele of SLC26A4. Laryngoscope 127:E238-e243. https://doi.org/10.1002/lary.26418
Royaux IE, Belyantseva IA, Wu T, Kachar B, Everett LA, Marcus DC, Green ED (2003) Localization and functional studies of pendrin in the mouse inner ear provide insight about the etiology of deafness in pendred syndrome. J Assoc Res Otolaryngol 4:394–404 https://doi.org/10.1007/s10162-002-3052-4
Scudieri P, Musante I, Venturini A, Guidone D, Genovese M, Cresta F, Caci E, Palleschi A, Poeta M, Santamaria F, Ciciriello F, Lucidi V, Galietta LJV (2020) Ionocytes and CFTR chloride channel expression in normal and cystic fibrosis nasal and bronchial epithelial cells. Cells. https://doi.org/10.3390/cells9092090
Sennaroglu L, Saatci I (2002) A new classification for cochleovestibular malformations. Laryngoscope 112:2230–2241. https://doi.org/10.1097/00005537-200212000-00019
Sheykholeslami K, Schmerber S, Habiby Kermany M, Kaga K (2004) Vestibular-evoked myogenic potentials in three patients with large vestibular aqueduct. Hear Res 190:161–168. https://doi.org/10.1016/s0378-5955(04)00018-8
Singh R, Wangemann P (2008) Free radical stress-mediated loss of Kcnj10 protein expression in stria vascularis contributes to deafness in Pendred syndrome mouse model. Am J Physiol Renal Physiol 294:F139–F148. https://doi.org/10.1152/ajprenal.00433.2007
Soh LM, Druce M, Grossman AB, Differ AM, Rajput L, Bitner-Glindzicz M, Korbonits M (2015) Evaluation of genotype-phenotype relationships in patients referred for endocrine assessment in suspected Pendred syndrome. Eur J Endocrinol 172:217–226. https://doi.org/10.1530/eje-14-0679
Song MH, Shin JW, Park HJ, Lee KA, Kim Y, Kim UK, Jeon JH, Choi JY (2014) Intrafamilial phenotypic variability in families with biallelic SLC26A4 mutations. Laryngoscope 124:E194-202. https://doi.org/10.1002/lary.24504
Tian C, Gagnon LH, Longo-Guess C, Korstanje R, Sheehan SM, Ohlemiller KK, Schrader AD, Lett JM, Johnson KR (2017) Hearing loss without overt metabolic acidosis in ATP6V1B1 deficient MRL mice, a new genetic model for non-syndromic deafness with enlarged vestibular aqueducts. Hum Mol Genet 26:3722–3735. https://doi.org/10.1093/hmg/ddx257
Valvassori GE, Clemis JD (1978) The large vestibular aqueduct syndrome. Laryngoscope 88:723–728. https://doi.org/10.1002/lary.1978.88.5.723
Van Camp G, Smith RJ (2021) Hereditary hearing loss homepage. https://hereditaryhearingloss.org. Accessed 1 April 2021
Vidarsson H, Westergren R, Heglind M, Blomqvist SR, Breton S, Enerback S (2009) The forkhead transcription factor Foxi1 is a master regulator of vacuolar H-ATPase proton pump subunits in the inner ear, kidney and epididymis. PLoS ONE 4:e4471. https://doi.org/10.1371/journal.pone.0004471
Vijayasekaran S, Halsted MJ, Boston M, Meinzen-Derr J, Bardo DM, Greinwald J, Benton C (2007) When is the vestibular aqueduct enlarged? A statistical analysis of the normative distribution of vestibular aqueduct size. AJNR Am J Neuroradiol 28:1133–1138. https://doi.org/10.3174/ajnr.A0495
Wang QJ, Zhao YL, Rao SQ, Guo YF, Yuan H, Zong L, Guan J, Xu BC, Wang DY, Han MK, Lan L, Zhai SQ, Shen Y (2007) A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin Genet 72:245–254. https://doi.org/10.1111/j.1399-0004.2007.00862.x
Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC (2004) Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med 2:30. https://doi.org/10.1186/1741-7015-2-30
Wangemann P, Nakaya K, Wu T, Maganti RJ, Itza EM, Sanneman JD, Harbidge DG, Billings S, Marcus DC (2007) Loss of cochlear HCO3- secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am J Physiol Renal Physiol 292:F1345–F1353. https://doi.org/10.1152/ajprenal.00487.2006
Wasano K, Takahashi S, Rosenberg SK, Kojima T, Mutai H, Matsunaga T, Ogawa K, Homma K (2020) Systematic quantification of the anion transport function of pendrin (SLC26A4) and its disease-associated variants. Hum Mutat 41:316–331. https://doi.org/10.1002/humu.23930
Wen Z, Zhu H, Li Z, Zhang S, Zhang A, Zhang T, Fu X, Sun D, Zhang J, Gao J (2019) A knock-in mouse model of Pendred syndrome with Slc26a4 L236P mutation. Biochem Biophys Res Commun 515:359–365. https://doi.org/10.1016/j.bbrc.2019.05.157
Wu CC, Lu YC, Chen PJ, Yeh PL, Su YN, Hwu WL, Hsu CJ (2010) Phenotypic analyses and mutation screening of the SLC26A4 and FOXI1 genes in 101 Taiwanese families with bilateral nonsyndromic enlarged vestibular aqueduct (DFNB4) or Pendred syndrome. Audiol Neurootol 15:57–66. https://doi.org/10.1159/000231567
Yang T, Vidarsson H, Blomqvist SR, Rosengren SS, Enerback S, Smith RJ (2007) Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am J Hum Genet 80:1055–1063. https://doi.org/10.1086/518314
Yang T, Gurrola JG 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ (2009) Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet 84:651–657. https://doi.org/10.1016/j.ajhg.2009.04.014
Yang CJ, Lavender V, Meinzen-Derr JK, Cohen AP, Youssif M, Castiglione M, Manickam V, Bachmann KR, Greinwald JH (2016) Vestibular pathology in children with enlarged vestibular aqueduct. Laryngoscope 126:2344–2350. https://doi.org/10.1002/lary.25890
Zalewski CK, Chien WW, King KA, Muskett JA, Baron RE, Butman JA, Griffith AJ, Brewer CC (2015) Vestibular dysfunction in patients with enlarged vestibular aqueduct. Otolaryngol Head Neck Surg 153:257–262. https://doi.org/10.1177/0194599815585098
Zhao FF, Lan L, Wang DY, Han B, Qi Y, Zhao Y, Zong L, Li Q, Wang QJ (2013) Correlation analysis of genotypes, auditory function, and vestibular size in Chinese children with enlarged vestibular aqueduct syndrome. Acta Otolaryngol 133:1242–1249. https://doi.org/10.3109/00016489.2013.822555
Zhou G, Gopen Q (2011) Characteristics of vestibular evoked myogenic potentials in children with enlarged vestibular aqueduct. Laryngoscope 121:220–225. https://doi.org/10.1002/lary.21184
Zhou YJ, Wu YZ, Cong N, Yu J, Gu J, Wang J, Chi FL (2017) Contrasting results of tests of peripheral vestibular function in patients with bilateral large vestibular aqueduct syndrome. Clin Neurophysiol 128:1513–1518. https://doi.org/10.1016/j.clinph.2017.05.016
Funding
No funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Honda, K., Griffith, A.J. Genetic architecture and phenotypic landscape of SLC26A4-related hearing loss. Hum Genet 141, 455–464 (2022). https://doi.org/10.1007/s00439-021-02311-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-021-02311-1