
Abstract
Clinically significant copy-number variants (CNVs) known to cause human diseases are routinely detected by chromosomal microarray analysis (CMA). Recently, genome sequencing (GS) has been introduced for CNV analysis; however, sequencing depth (determined by sequencing read-length and read-amount) is a variable parameter across different laboratories. Variating sequencing depths affect the CNV detection resolution and also make it difficult for cross-laboratory referencing or comparison. In this study, by using data from 50 samples with high read-depth GS (30×) and the reported clinically significant CNVs, we first demonstrated the optimal read-amount and the most cost-effective read-length for CNV analysis to be 15 million reads and single-end 50 bp (equivalent to a read-depth of 0.25-fold), respectively. In addition, we showed that CNVs at mosaic levels as low as 30% are readily detected, furthermore, CNVs larger than 2.5 Mb are also detectable at mosaic levels as low as 20%. Herein, by conducting a retrospective back-to-back comparison study of low-pass GS versus routine CMA for 532 prenatal, miscarriage, and postnatal cases, the overall diagnostic yield was 22.4% (119/532) for CMA and 23.1% (123/532) for low-pass GS. Thus, the overall relative improvement of the diagnostic yield by low-pass GS versus CMA was ~ 3.4% (4/119). Identification of cryptic and clinically significant CNVs among prenatal, miscarriage, and postnatal cases demonstrated that CNV detection at higher resolutions is warranted for clinical diagnosis regardless of referral indications. Overall, our study supports low-pass GS as the first-tier genetic test for molecular cytogenetic testing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Copy-number variants (CNVs) are defined as gains or losses of genomic segments, the majority of which contribute to human genomic diversity (1000 Genomes Project Consortium 2015; Iafrate et al. 2004). However, some are known to cause human diseases (Trost et al. 2018) such as DiGeorge syndrome (also known as 22q11.2 recurrent deletion syndrome; OMIM# 188400) and Charcot-Marie-Tooth disease (17p12 duplication; OMIM# 118220) due to the involvement of dosage sensitive genes (e.g., haploinsufficiency for TBX1 and triplosensitivity for PMP22). Chromosomal microarray analysis (CMA) has been recommended by various professional societies as the first-tier genetic test for detecting clinically significant CNVs in prenatal (Hay et al. 2018), miscarriage (Foyouzi et al. 2012), and postnatal (Miller et al. 2010) applications. However, CMA platforms have different detection resolutions attributed by different probe densities and detection methods (Lin et al. 2015; Wang et al. 2019). In addition, in some applications, the experimental repeat rate is as high as 4.5% due to inherent factors of particular sample types such as amniotic fluid samples (Wang et al. 2020a).
The advantages of next-generation sequencing-based CNV analysis include lower costs and higher throughputs, thus leading to the development of different clinical applications (such as non-invasive prenatal screening and invasive genetic diagnostic testing). As such, low-coverage and high-throughput (low-pass) genome sequencing (GS) has been introduced for CNV analysis in various applications (Chen et al. 2017; Dong et al. 2016, 2019a; Gross et al. 2019; Redin et al. 2017; Wang et al. 2018, 2020b). However, there are various sequencing platforms utilizing different sequencing parameters including sequencing read-amount (ranging from 2 million reads to > 15) and read-length (from 26 to > 150 bp). In addition, detection resolutions for CNVs across different platforms and laboratories are also variable, resulting in difficulties for clinical implementation, genetic counseling and cross-laboratory referencing. Up to date, the majority of published studies focus on the diagnostic yield of NGS-based CNV analysis alone (Chen et al. 2017; Wang et al. 2018, 2020b). In a few studies with limited sample sizes, verification of CNV detection accuracy of low-pass GS-based CNV analysis was compared with CMA in retrospective analyses (Dong et al. 2016; Gross et al. 2019; Li et al. 2014; Liang et al. 2014). Furthermore, no study has evaluated the accuracy of detecting mosaic CNVs. This warrants a study to benchmark and standardize sequencing parameters to ensure accurate and precise detection of CNVs across different laboratories.
In our previous study, simulation of “various CNVs” from YH cell line (Dong et al. 2016) concluded that the minimal read-amount for detecting CNVs at a resolution of 50 kb was 15 million reads, which was equivalent to 0.25-folds (50 bp sequencing read-length) (Dong et al. 2016). Although previously, the accuracy of our detection method was validated on Illumina (Dong et al. 2016) and BGISEQ-500 sequencing platforms (Wang et al. 2020a), in this study, we aimed to determine the optimal read-amount, sequencing read-length and the minimal mosaic threshold for CNV detection. In addition, our previous prospective back-to-back study demonstrated low-pass GS provided a 1.7% additional diagnostic yield of clinically significant CNVs (smallest 19 kb) in 1023 pregnancies referred for invasive prenatal diagnostic testing. Herein, we evaluate the performance of low-pass GS in prenatal, products of conception and postnatal cohorts in a back-to-back comparison with different designs of CMA assays.
Materials and methods
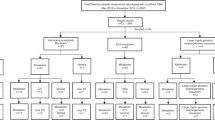
Samples analyzed
The study was approved by the institutional review boards of each collaborative site. Written consent for sample storage and genetic analyses of peripheral blood and invasive diagnostic samples was obtained from each participant. DNA was previously extracted from prenatal [chorionic villus sampling (CVS), amniotic fluid (AF), cord blood (CB)], products of conceptions (POC) and postnatal samples [peripheral blood (PB)] for CMA testing. The extracted DNA samples were retrieved retrospectively for low-pass GS. Routine quantitative fluorescence polymerase chain reaction (QF-PCR) with short tandem repeat (STR) markers was performed prior to the CMA experiment for exclusion of maternal cell admixture and polyploidies as described (Cheng et al. 2013).
Chromosomal microarray analysis
Two CMA platforms were used in this study. The 8X60K Fetal DNA Chip v2.0 (Agilent Technologies) including array comparative genomic hybridization (aCGH) and single-nucleotide polymorphism (SNP) probes was used for prenatal diagnosis and POC testing (Chau et al. 2019; Huang et al. 2014). The CMA used for postnatal genetic analysis was the 4X180K postnatal array with higher probe densities (Dharmadhikari et al. 2014; Gu et al. 2016). Both were performed according to the manufacturers’ protocols and the CNVs were analyzed via the CytoGenomics 5.0 software (Chau et al. 2019; Huang et al. 2014). A minimum of three consecutive probes were required for a positive call on the Fetal DNA Chip, while at least five consecutive probes were required for the postnatal array (Brown et al. 2012). Based on the guidelines of manufacturers, the derivative log ratio spread (DLRS) value of larger than 0.2 was considered a technical failure that required repeat testing (Wang et al. 2020a). Cases with absence of heterozygosity (AOH) detected by CMA platforms were excluded from this study as AOH detection is out of the scope of the current protocol of low-pass GS (0.25-fold), which is unable to obtain genotype information.
Low-pass genome sequencing
Library construction for low-pass GS was performed with a modified protocol as described in our previous study (Wang et al. 2020a). In short, 50 ng of genomic DNA was fragmented to 200–300 bp by fragmentation end-repair restriction enzyme (MGI Tech Co., Ltd., Shenzhen, China), followed by adaptor ligation and PCR amplification. The concentration of each library was quantified using the Qubit dsDNA HS Assay Kit (Invitrogen, Carlsbad, California, USA) and pooled with equal molality (20–24 samples per lane) and sequenced with a minimum of 15 million reads per sample (single-end 50 bp) on the BGISEQ-500 platform (MGI Tech Co., Ltd., Shenzhen, China) (Wang et al. 2020a). The read depth is estimated to be 0.25-fold, which is determined by multiplying the reads (15 million) and the read length (50 bp), divided by the size of human reference genome (3 Gb).
Data simulation
In this study, high read-depth GS data (30-fold, paired-end 100 bp) from 50 fetuses with increased nuchal translucency published in our previous study (Choy et al. 2019) were downloaded and used for the evaluation of low-pass GS sequencing parameters. Among them, nine pathogenic or likely pathogenic (P/LP) CNVs and nine variants of uncertain significance (VUS) were reported previously. The detection of these CNVs under different simulated sequencing parameters were used to benchmark the optimal read-length and read-amount for low-pass GS. Read 1 (or fastq file 1) from each sample was used as single-end sequencing data (single-end 100 bp, SE100). For shorter read-lengths such as 50 bp and 35 bp, each read was trimmed to the target size. Evaluation of CNV detection using different read amounts was performed by in silico filtering through a process of random sampling to generate datasets with the target read-amounts (60, 45, 30, 15 and 10 millions). In addition, for evaluating the mosaic resolution of CNV detection, the nine P/LP CNVs (from eight cases) were analyzed under different simulated mosaic levels. Reads from each sample were pooled with a sex-matched normal control totaling to 15 million reads with single-end 50 bp according to a series of percentages (10%, 20%, 30%, 40% and 50%) to simulate different mosaic levels for subsequent CNV analyses.
Data quality control and CNV analysis
The data QC was performed as previously described (Wang et al. 2020a). After QC filtering, sequencing reads were aligned to the human reference genome (GRCh37/hg19) using the Burrows–Wheeler Aligner (BWA) (Li and Durbin 2009) with “Aln” and “Samse” alignment modules. Uniquely aligned reads were selected and deposited into adjustable sliding windows (50 kb in length with 5 kb increments) and adjustable non-overlapping windows (5 kb), respectively. The coverage of each window was calculated by the sum of read-amounts undergoing a two-step bias correction (GC correction and population-scale normalization). Genome-wide standard deviation (SD) of the copy ratios, calculated from the mean of SD of all windows with the exception of those located on chromosomes with numerical abnormalities, was used as a quality control statistic. In this study, QC cut-off value (genome-wide SD) was also set as 0.1 based on the validation result in our previous study (Wang et al. 2020a). Detection of constitutional and mosaic aneuploidies, homozygous/hemizygous/heterozygous deletions and duplications/triplications was performed with parameters consistent as our reported study (Wang et al. 2020a). The resolution for CNV detection for homozygous/hemizygous deletions was set as 10 kb; and for the other CNVs, it was set at 50 kb. Detection of homozygous/hemizygous deletions was performed by identifying nonoverlapping windows lacking aligned reads. A homozygous/hemizygous deletion would be reported if there are two or more consecutive nonoverlapping windows without aligned reads (copy ratio < 0.1). Therefore, the resolution of homozygous/hemizygous deletion detection is equal to the size of two windows (approximately 10 kb).
Detection of mosaic aneuploidies and copy number variants
Mosaic levels of aneuploidies and CNVs were estimated by the differences of copy-ratio compared with a normal copy ratio (expected as 1). In brief, the mosaic level (percentage) of a duplication with a copy-ratio N was calculated by (N−1)/M, where M is the copy-ratio of one copy in autosomal or sex chromosomes (M equals to 0.5 when the CNV located in autosomal chromosomes or X chromosome in a female, while M equals to 1 when the CNV is located on the X or Y chromosomes in a male). For a mosaic deletion, the mosaic level (percentage) with a copy-ratio N was calculated by (1−N)/M. For mosaic CNV detection, the minimal mosaic level would be set as 20% when the genome-wide SD of windows’ copy-ratios was 0.1 (Wang et al. 2020a).
Variant interpretation
Variant interpretation was performed in accordance to the recently published guideline of the American College of Medical Genetics and Genomics (ACMG) (Riggs et al. 2019) using the criteria, methods, and references to online and in-house datasets such as Database of Genomic Variants (DGV). CNVs were classified into five categories including pathogenic, likely pathogenic, benign, likely benign and variants of uncertain significance (VUS) based on the scoring system recommended by the joint consensus recommendation of the ACMG and the Clinical Genome Resource (ClinGen) (Riggs et al. 2019). The additional findings (P/LP/VUS) from low-pass GS were selected for further verification.
Verification of CNVs and parental confirmation
Quantitative PCR (qPCR) was performed with primer-pairs specific to the candidate regions designed with Primer 3 Web, Primer-Blast (NCBI) and In-Silico PCR (UCSC) based on the GRCh37/hg19 human reference genome (Suppl. Table 1). Melt curve analysis was carried out for each primer-pair to ensure specificity of the PCR amplification and the standard curve method was used to determine PCR efficiency (ranging from 95 to 105%). Each reaction was performed in triplicate in 10-μl reaction mixtures in cases and control (in-house normal male and female controls) on a 7900HT Real-Time PCR System (Applied Biosystems, Foster City, CA, United States) with SYBR Select Master Mix (Applied Biosystems) with the default thermal cycling conditions. The copy number was determined by using the ΔΔCt method, which compares the difference in Ct (cycle threshold) of the candidate region with a reference primer-pair targeting the universally conserved element in the cases and the difference in the controls. Two independent primer pairs (Suppl. Table 1) were used in triplicate for verification of each candidate CNV. Confirmation of parental inheritance was performed whenever parental DNA samples were available.
Results
Sequencing parameters evaluation
To evaluate the optimal read-amount required for the CNV analysis at a resolution of 50 kb (10 kb for homozygous/hemizygous deletion), we generated different datasets with different read-amounts (Fig. 1A) from 50 GS data with nine P/LP and nine VUS ranging from 240 kb to 47 Mb reported by CMA and GS from our previous study (Choy et al. 2019). As SD of the copy-ratios among all windows from each sample is one of the major factors affecting the CNV detection accuracy as demonstrated before (Dong et al. 2016), we calculated the SDs from each read-amount among these 50 cases (mean values and SDs shown in Fig. 1A). Our previous study demonstrated that the cut off of SD values would be 0.1 based on the back-to-back validation data with CMA (Wang et al. 2020a). Thus, the results showed the minimal read-amount for our CNV analysis to be 15 million reads, consistent with the results based on the “simulated CNVs” from YH cell line data (Dong et al. 2016). Although all reported P/LP and VUS were detected by the dataset at 10 million reads, a number of false positive CNVs were called. For example, a homozygous deletion (resolution set as 10 kb) was falsely called due to the limited read-amount aligned to each window (Fig. 1B), demonstrating the minimal read-amount for CNV analysis to be 15 million.

Evaluation of sequencing parameters for CNV Detection. A Distribution of SDs among 50 samples with different read-amounts. Each error bar represents the standard deviation among these 50 samples when filtered to the indicated read-amounts. The X axis represents different read-amounts, while the Y axis indicates the genome-wide SD values. The red line indicates the cutoff SD threshold validated by our previous prospective study (Wang et al. 2020a), while the dotted green frame shows the optimal read-amount (15 millions) concluded. B Distribution of copy-ratios on chromosome 11q14.3q22.3 among different read-amounts simulated in the same sample. The X axis shows the genomic location, while the Y axis shows the copy-number. In each figure, read amount is shown at the top of the figure, while each black dot indicates an adjustable sliding window (50 kb in size with 5 kb increment). Blue arrows indicate duplication or copy number gains detected, while red arrow indicates a homozygous deletion identified. C Comparison of the percentages of total aligned and uniquely aligned reads over total reads (15 million) among the datasets with 100 bp, 50 bp and 35 bp read-lengths, respectively. The average percentages of uniquely aligned reads for different read-lengths are shown in each bar. Significant difference of the probabilities by Mann–Whitney tests between datasets with 100 bp and 50 bp, and between 50 bp and 35 bp are shown in the top panel (**** indicates P < 0.0001). D Distributions of SDs among 50 samples with 15 million reads, sequencing length of 50 bp (SE50) and 35 bp (SE35), respectively. Each error bar represents the SDs among these 50 samples at the indicated read-amounts. The probability of Mann–Whitney test between datasets with 50 bp and 35 bp is shown in the top panel (** indicates P < 0.01)
The sequencing read-lengths utilized by other published studies varied from 26 to 150 bp. We simulated different read-lengths (100 bp, 50 bp and 35 bp) with the same read-amount (15 million) for the CNV analysis. Although the dataset with shortest read-length provided the highest alignment rate, the read-utility (considered as the percentage of uniquely aligned reads over the raw reads) in the dataset with 35 bp was the lowest (Mann–Whitney test, P < 0.0001), at approximately 10% less than the dataset with 100 bp (Fig. 1C). However, in theory, longer sequencing read-lengths require longer turn-around-times and higher costs. Compared with dataset utilizing 35 bp reads, the dataset utilizing 50 bp reads would be able to provide a significantly lower SD (Mann Whitney test, P = 0.0042, Fig. 1D) due to the significantly higher read-utility obtained (Mann Whitney test, P < 0.0001, Fig. 1C). Although all P/LP CNVs and VUS were detected by each dataset, meaning different read-lengths did not affect the sensitivity of P/LP CNV detection, false positives shown in data with 10 million reads as described above (Fig. 1B) demonstrated the false positive detection would be contributed by higher SDs. Thus, it indicated that dataset generated using 50 bp reads would be the most cost-effective read-length for CNV analysis when comparing the performance among datasets with various read-lengths (100 bp, 50 bp and 35 bp).
Although in our previous study, mosaic CNVs were detected and reported with mosaic level ranging from 20 to 40% (Wang et al. 2020a), objective evaluation of the minimal cutoff for mosaic CNV detection is still required. We simulated mosaic CNVs at different levels by pooling each of the eight cases with nine P/LP CNVs ranging from 751 kb to 47 Mb in a series of different artificially reconstituted mosaic levels (from 10 to 50% at 10% intervals) with a sex-matched control sample and performed CNV detection. The results showed that none of the CNVs were detected in datasets at a 10% mosaic levels (Fig. 2A), while only four CNVs out of nine (larger than 2.5 Mb) were detected in datasets at 20% mosaic levels (Fig. 2A). All nine simulated mosaic CNVs were detected when the mosaic levels were at 30% or higher (Fig. 2A). We further replicated the data simulation twice for the datasets with a mosaic level of 30% and all “mosaic CNVs” were detected. The mosaic levels estimated by our methods for all nine P/LP CNVs among the three datasets (one from original and two from replications) were on average 29.7% with a SD of 2.3% (Fig. 2Ab). In addition, the deviation of estimated mosaic levels for each sample across three datasets had a median value of 0.9% (Fig. 2Ac). Furthermore, of P/LP CNVs selected for data simulation, the smallest was 751 kb. By setting the resolution as 500 kb [based on the minimum size (751 kb) of the P/LP CNVs used as reference] for mosaic CNVs analysis, the specificity of mosaic CNVs detection was 100%.

Evaluation of mosaic CNVs detection. Aa Detection rate for nine “mosaic” pathogenic/likely pathogenic CNVs with different expected “mosaic” levels. Ab Distribution of the estimated mosaic levels in each dataset when the mosaic level is simulated as 30%. The dataset includes the original simulation and two replicates. Ac SD of the mosaic levels detected among three simulated datasets from each case when the mosaic level was set as 30%. Detection of a 5.5 Mb deletion (B) and a 47 Mb duplication (C) in different mosaic levels with exact genomic coordinates shown in the top panel. B and C show the distributions of copy-ratios with each black dot indicating the copy number of an adjustable sliding window (50 kb in size with 5 kb increment). The X axis shows the genomic location, while the Y axis shows the copy number. In each figure, expected mosaic level is shown at the top of each figure, while estimated mosaic level from the dataset is indicated at the bottom. Blue lines indicate mosaic duplication or copy number gains detected, while red lines indicate mosaic deletion or copy number loss identified
Prenatal samples
Among the 208 prenatal cases, low-pass GS not only identified all 13 P/LP CNVs reported by CMA (Suppl. Tables 2 and 3), but also provided two additional P/LP CNV findings. First, low-pass GS identified a homozygous deletion of HBA1 and HBA2 in a fetus (15BA0039) who presented with multiple ultrasound anomalies, consistent with the Southeast Asian (SEA) type ɑ-thalassemia deletion, which was missed by CMA due to insufficient probe coverage (Fig. 3A). In addition, low-pass GS revealed a likely pathogenic CNV in another fetus (15C0667) with left-hand pre-axial polydactyly. A de novo 122.0 kb intragenic duplication seq[GRCh37] dup(18)(q21.2)dn chr18:g.52903795_53025806dup (Fig. 3B) involving exons 2–10 of gene TCF4 was detected. Loss of function variants in TCF4 are known to cause Pitt-Hopkins syndrome in an autosomal dominant manner (OMIM# 610954) (Pontual et al. 2009). In addition, 53% of the reported patients with Pitt-Hopkins syndrome had finger and toe anomalies including over-riding toes, syndactyly, and polydactyly (Goodspeed et al. 2018), consistent with the malformation in this case. This duplication was classified as a likely pathogenic variant. Among reported postnatal cases, patients often suffer from intellectual disability and epilepsy (Goodspeed et al. 2018) warranting a follow-up study. After investigation, even though there were five probes located in the duplicated region reported by low-pass GS, targeted reanalysis with the CMA software still failed to report this duplication. Overall, low-pass GS provided a 1.0% additional finding of P/LP CNVs (Tables 1 and 2) and one more VUS (Suppl. Table 4) compared with CMA in the prenatal group.

Additional findings reported by low-pass GS. Low-pass GS identified A a homozygous deletion seq[GRCh37] del(16)(p13.3) chr16:g.216050_229978del involving HBA1 and HBA2, known as the Southeast Asian (SEA) type ɑ-thalassemia deletion (15BA0039), B a de novo intragenic duplication seq[GRCh37] dup(18)(q21.2)dn chr18:g.52903795_53025806dup affecting gene TCF4 in a fetus (case: 15C0667), C a heterozygous deletion seq[GRCh37] del(16)(q24.3) chr16:g.89264235_89335546del involving partial of ANKRD11 in a POC 18BA0296, and D a de novo heterozygous deletion seq[GRCh37] del(2)(q23.1)dn chr2:g.148755869_148864775del in a postnatal case (18C0925). Aa, Ba, Ca and Da Distributions of copy-ratios with each black dot indicating the copy number of an adjustable sliding window (50 kb in size with 5 kb increment). The X axis shows the genomic location, while the Y axis shows the copy-number. In the figure, blue arrows indicate duplication or copy number gain, while red arrows indicate heterozygous deletion or one copy number loss. In Aa the distribution of copy-ratio with adjustable non-overlapping windows (5 kb) is also shown with the homozygous deletion region highlighted by yellow box. Ab The gap PCR results indicating the SEA deletion. Bb, Cb and Db Bar charts show the results of qPCR validation. Two independent pairs of primers residing in the candidate region were used for CNV verification and all showed consistent findings (duplication or deletion) with low-pass GS compared with a normal control sample. Ac, Bc, Cc and Dc Probe distribution on the CMA platform with the candidate region reported by low-pass GS highlighted in yellow
Products of conception
Among 221 POCs, CMA identified aneuploidies and P/LP CNVs in 98 cases and VUS in five cases. In contrast, low-pass GS consistently reported all abnormalities identified by CMA and also detected three additional findings including one LP CNV (Table 2) and two cases with VUS (Suppl. Table 4). In an early miscarriage case (18BA0296) with normal CMA results, low-pass GS reported a 71.3 kb heterozygous deletion (Fig. 3C) seq[GRCh37] del(16)(q24.3) chr16:g.89264235_89335546del involving the gene ANKRD11. Disruptions in ANKRD11 is known to cause KBG syndrome in an autosomal dominant manner (OMIM # 148050). Studies have demonstrated heterozygous premature terminations would result in reduction of RNA expression. This CNV was classified as likely pathogenic based on the current joint ACMG and ClinGen guideline (Riggs et al. 2019). However, the parental samples were not available for further confirmation of inheritance. In another fetus with fetal death at 19 weeks of gestation, low-pass GS detected a heterozygous deletion seq[GRCh37] del(8)(q21.13) chr8:g.82352080_82389112del involving the entire gene PMP2 (Suppl. Table 4), whereas the CMA was normal. PMP2 mutations reported in the literature cause Charcot-Marie-Tooth disease, demyelinating, type 1G (OMIM # 618279) through a toxic gain of function mechanism (Motley et al. 2016). This heterozygous deletion was classified as a VUS as there was no such deletion reported in the DGV database and our in-house datasets and no parental samples were available for confirmation of inheritance. It is uncertain whether whole gene deletion of PMP2 would result in any phenotypic consequences. All of the three additional findings were due to insufficient probe densities at the targeted regions, hence were not reported by CMA (Suppl. Figure 1 and Suppl. Table 4).
Postnatal samples
Among the group of 103 postnatal cases, low-pass GS identified all eight P/LP CNVs reported by CMA, and one additional pathogenic CNV missed by CMA. A 2.5-year-old girl with developmental delay, failure to thrive and speech delay had a normal CMA report. Low-pass GS identified a de novo 103 kb heterozygous deletion (Fig. 3D) seq[GRCh37] del(2)(q23.1)dn chr2:g.148755869_148864775del involving gene MBD5 (Table 2). Disruptions of MBD5 causes mental retardation, autosomal dominant 1 (OMIM # 156200) in an autosomal dominant manner. The reported patients commonly suffer from delay of motor development, severe psychomotor retardation and language impairment, which were consistent with the presentation of our patient. In addition, seizures were reported in some of the patients. Although our patient did not suffer from epilepsy, further follow-up and precautionary management for seizures would be beneficial for the patient care. The reason behind the missed detection by CMA was again, insufficient probe density at the targeted region (Fig. 3Dc).
Together, among all 532 samples, low-pass GS detected all P/LP CNVs reported by previous CMA testing and provided an overall 0.8% increased diagnostic yield of P/LP CNVs compared with routine CMA.
Discussion
In the present study, we not only provided the critical sequencing parameters for constitutional and mosaic CNV detection, but also demonstrated that low-pass GS is able to provide an overall ~ 3.4% relative improvement of additional diagnostic yield regardless of referral indications compared with routine CMA.
Our evaluation first demonstrated the optimal read-amount for CNV analysis to be 15 million reads at a resolution of 50 kb (10 kb for homozygous/hemizygous deletion). We also show decrease in read-amounts led to an increase in the rate of false-positive calling (Fig. 1A, B). In addition, although read-length would not affect the sensitivity of P/LP CNV detection in this study, longer read-length would provide significantly higher read utility (Fig. 1C) and significantly lower genome-wide SD (Fig. 1B, D), which would minimize the probability of false calling, particularly for the small CNVs. However, due to longer sequencing turn-around-times and higher costs for longer read-lengths, 50 bp would be the most cost-effective read-length for CNV analysis when comparing with read-lengths of 100 bp and 35 bp. Furthermore, this study also demonstrated that mosaic CNVs can be readily detected using our validated sequencing parameters (15 million reads and single-end 50 bp) for mosaic CNVs at levels as low as 30%, while there were four out of nine CNVs (larger than 2.5 Mb) detected in datasets expected to have 20% mosaic level (Fig. 2). In our previous study, there were two prenatal cases with normal CMA results but mosaic CNVs cryptic to CMA, which indicated that low-pass GS had a higher sensitivity in detecting clinically significant mosaic variants compared to the CMA platforms used.
We further conducted a retrospective back-to-back comparison detecting clinically significant CNVs by low-pass GS (15 million reads, single-end sequencing 50 bp, 0.25-fold) among 532 samples including 208 fetuses, 221 POCs and 103 postnatal cases, compared with CMA. For prenatal and POC applications, the routine CMA platform used was demonstrated to be able to detect > 98% of the known P/LP CNVs reported in the literature for prenatal diagnosis (Chau et al. 2019), while for postnatal analysis, the routine CMA platform employed had higher probe density (Dharmadhikari et al. 2014; Gu et al. 2016). Nonetheless, low-pass GS yielded an overall diagnostic rate of 23.1% and was able to provide additional findings among different referral cohorts. Since the baseline diagnostic yield by CMA was 22.4%, this resulted in an overall relative improvement of diagnostic yield by low-pass GS to be ~ 3.4% (4/119). However, our previous study reported a 1.7% increase in diagnostic yield in a prenatal cohort of 1,023 cases, which is different from that of our current study (0.75%) among 523 cases. This may be explained by the various referral cohorts (prenatal, postnatal and products of conception) and the sample size of prenatal cohort is limited (n = 208). Interestingly, among the four additional P/LP CNVs identified by low-pass GS, insufficient probe coverage on the CMA was the reason for missed detection in three of them. An intragenic duplication with 122.0 kb in size was missed by CMA even though there were five probes located in the targeted region reported by low-pass GS. After targeted reanalysis of the raw data, the CMA software still reported normal results, suggesting potential flaws exist in the current CMA platforms or software. Furthermore, variant interpretation might be reclassified or changed based on the continuously published updates and guidelines (Riggs et al. 2019). As the variant should receive the same variant classification (P, LP, VUS, etc.), regardless of the clinical phenotype or presentation in the patient, the 71.3 kb heterozygous deletion detected in a POC case, involving gene ANKRD11, was classified as a LP CNV. As such, since small clinically significant CNV exists in POC, our study demonstrates POC analysis may require investigation at higher resolution (Chen et al. 2017; Wang et al. 2020b).
Currently, the resolution for CNV detection suitable for paediatric application is still under debate in previously reported studies with higher read-depth methods (Gross et al. 2019; Trost et al. 2018; Zhang et al. 2019). In this study, although the overall resolution of CNV detection was set at 50 kb as in the application in POC and prenatal samples, low-pass GS was still able to provide 1.0% increased diagnostic yield by comparing with high probe density (4X180K) CMA in the application of postnatal cases. As studies with genotyping SNP arrays show a higher sensitivity in detecting mosaicisms (< ~ 10%) (Markello et al. 2012), it is higher than low-pass GS. However, as the CMA platforms used in this study were not SNP-based arrays, no such comparison could be evaluated. Nonetheless, such study is warranted in future for further evaluation of the performance of low-pass GS.
Finally, by comparing with various genome-wide CNV analysis methods including CMA (aCGH and SNP-array) (Choy et al. 2019)] and GS with different genome read-depths and library construction methods [small-insert (Trost et al. 2018) or mate-pair/large-insert (Dong et al. 2019a, b)], low-pass GS (with single-end 50 bp and small-insert library) described in this study would be the most cost-effective approach (Table 3) as the first-tier genetic testing for CNV analysis from different referral indications. This table does not include data review and variant interpretation time and efforts. In fact, the average number of raw CNVs detected by low-pass GS are more than that of CMA (48 versus 16 in our cohort). However, after filtering the variants against our in-house database, the number of remaining variants that require clinical interpretation are not significantly higher (on average 4.3). Furthermore, there were only three additional VUS identified by low-pass GS in this study, providing the additional yield of VUS as 0.6%. As shown in our previous study, we highlight the importance of referencing public databases such as DGV and constructing a local reference dataset to filter out variants that are not of clinical significance but otherwise due to polymorphisms.
Conclusion
In summary, our study demonstrated the optimal read-amount and the most cost-effective read-length for CNV analysis to be 15 million reads and 50 bp, respectively. In addition, we also demonstrated CNVs with a mosaic level of 30% or higher can be readily detected, while CNVs larger than 2.5 Mb were also detectable at mosaic levels as low as 20%. Our study demonstrated that low-pass GS provided an overall 0.8% of additional diagnostic yield (3.4% relative improvement) of clinically significant CNVs in different applications including prenatal, miscarriage and postnatal cases compared with routine CMA. With its robust performance, our study supports that low-pass GS is suitable and should be recommended as the first-tier genetic test for CNV detection in the various applications.
Availability of data and material
The genome sequencing data in this study have been made available on the CNGB Nucleotide Sequence Archive (CNSA: https://db.cngb.org/cnsa) under the accession number CNP0000901. Code availability: All relative programs are available at https://sourceforge.net/projects/increment-ratio-of-coverage-v2/files/Increment_Ratio_of_Coverage_V2.0.tar.gz/download.
References
1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature 526:68–74. https://doi.org/10.1038/nature15393
Brown KH, Dobrinski KP, Lee AS, Gokcumen O, Mills RE, Shi X, Chong WW, Chen JY, Yoo P, David S, Peterson SM, Raj T, Choy KW, Stranger BE, Williamson RE, Zon LI, Freeman JL, Lee C (2012) Extensive genetic diversity and substructuring among zebrafish strains revealed through copy number variant analysis. Proc Natl Acad Sci USA 109:529–534. https://doi.org/10.1073/pnas.1112163109
Chau MHK, Cao Y, Kwok YKY, Chan S, Chan YM, Wang H, Yang Z, Wong HK, Leung TY, Choy KW (2019) Characteristics and mode of inheritance of pathogenic copy number variants in prenatal diagnosis. Am J Obstet Gynecol 221(493):e1–493. https://doi.org/10.1016/j.ajog.2019.06.007
Chen Y, Bartanus J, Liang D, Zhu H, Breman AM, Smith JL, Wang H, Ren Z, Patel A, Stankiewicz P, Cram DS, Cheung SW, Wu L, Yu F (2017) Characterization of chromosomal abnormalities in pregnancy losses reveals critical genes and loci for human early development. Hum Mutat 38:669–677. https://doi.org/10.1002/humu.23207
Cheng YK, Wong C, Wong HK, Leung KO, Kwok YK, Suen A, Wang CC, Leung TY, Choy KW (2013) The detection of mosaicism by prenatal BoBs. Prenat Diagn 33:42–49. https://doi.org/10.1002/pd.4006
Choy KW, Wang H, Shi M, Chen J, Yang Z, Zhang R, Yan H, Wang Y, Chen S, Chau MHK, Cao Y, Chan OYM, Kwok YK, Zhu Y, Chen M, Leung TY, Dong Z (2019) Prenatal diagnosis of fetuses with increased nuchal translucency by genome sequencing analysis. Front Genet 10:761. https://doi.org/10.3389/fgene.2019.00761
de Pontual L, Mathieu Y, Golzio C, Rio M, Malan V, Boddaert N, Soufflet C, Picard C, Durandy A, Dobbie A, Heron D, Isidor B, Motte J, Newburry-Ecob R, Pasquier L, Tardieu M, Viot G, Jaubert F, Munnich A, Colleaux L, Vekemans M, Etchevers H, Lyonnet S, Amiel J (2009) Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat 30:669–676. https://doi.org/10.1002/humu.20935
Dharmadhikari AV, Gambin T, Szafranski P, Cao W, Probst FJ, Jin W, Fang P, Gogolewski K, Gambin A, George-Abraham JK, Golla S, Boidein F, Duban-Bedu B, Delobel B, Andrieux J, Becker K, Holinski-Feder E, Cheung SW, Stankiewicz P (2014) Molecular and clinical analyses of 16q24.1 duplications involving FOXF1 identify an evolutionarily unstable large minisatellite. BMC Med Genet 15:128. https://doi.org/10.1186/s12881-014-0128-z
Dong Z, Zhang J, Hu P, Chen H, Xu J, Tian Q, Meng L, Ye Y, Wang J, Zhang M, Li Y, Wang H, Yu S, Chen F, Xie J, Jiang H, Wang W, Choy KW, Xu Z (2016) Low-pass whole-genome sequencing in clinical cytogenetics: a validated approach. Genet Med 18:940–948. https://doi.org/10.1038/gim.2015.199
Dong Z, Yan J, Xu F, Yuan J, Jiang H, Wang H, Chen H, Zhang L, Ye L, Xu J, Shi Y, Yang Z, Cao Y, Chen L, Li Q, Zhao X, Li J, Chen A, Zhang W, Wong HG, Qin Y, Zhao H, Chen Y, Li P, Ma T, Wang WJ, Kwok YK, Jiang Y, Pursley AN, Chung JPW, Hong Y, Kristiansen K, Yang H, Pina-Aguilar RE, Leung TY, Cheung SW, Morton CC, Choy KW, Chen ZJ (2019a) Genome sequencing explores complexity of chromosomal abnormalities in recurrent miscarriage. Am J Hum Genet 105:1102–1111. https://doi.org/10.1016/j.ajhg.2019.10.003
Dong Z, Zhao X, Li Q, Yang Z, Xi Y, Alexeev A, Shen H, Wang O, Ruan J, Ren H, Wei H, Qi X, Li J, Zhu X, Zhang Y, Dai P, Kong X, Kirkconnell K, Alferov O, Giles S, Yamtich J, Kermani BG, Dong C, Liu P, Mi Z, Zhang W, Xu X, Drmanac R, Choy KW, Jiang Y (2019b) Development of coupling controlled polymerizations by adapter-ligation in mate-pair sequencing for detection of various genomic variants in one single assay. DNA Res. https://doi.org/10.1093/dnares/dsz011
Foyouzi N, Cedars MI, Huddleston HG (2012) Cost-effectiveness of cytogenetic evaluation of products of conception in the patient with a second pregnancy loss. Fertil Steril 98:151–155. https://doi.org/10.1016/j.fertnstert.2012.04.007
Goodspeed K, Newsom C, Morris MA, Powell C, Evans P, Golla S (2018) Pitt-Hopkins syndrome: a review of current literature, clinical approach, and 23-patient case series. J Child Neurol 33:233–244. https://doi.org/10.1177/0883073817750490
Gross AM, Ajay SS, Rajan V, Brown C, Bluske K, Burns NJ, Chawla A, Coffey AJ, Malhotra A, Scocchia A, Thorpe E, Dzidic N, Hovanes K, Sahoo T, Dolzhenko E, Lajoie B, Khouzam A, Chowdhury S, Belmont J, Roller E, Ivakhno S, Tanner S, McEachern J, Hambuch T, Eberle M, Hagelstrom RT, Bentley DR, Perry DL, Taft RJ (2019) Copy-number variants in clinical genome sequencing: deployment and interpretation for rare and undiagnosed disease. Genet Med 21:1121–1130. https://doi.org/10.1038/s41436-018-0295-y
Gu S, Szafranski P, Akdemir ZC, Yuan B, Cooper ML, Magrina MA, Bacino CA, Lalani SR, Breman AM, Smith JL, Patel A, Song RH, Bi W, Cheung SW, Carvalho CM, Stankiewicz P, Lupski JR (2016) Mechanisms for complex chromosomal insertions. PLoS Genet 12:e1006446. https://doi.org/10.1371/journal.pgen.1006446
Hay SB, Sahoo T, Travis MK, Hovanes K, Dzidic N, Doherty C, Strecker MN (2018) ACOG and SMFM guidelines for prenatal diagnosis: is karyotyping really sufficient? Prenat Diagn 38:184–189. https://doi.org/10.1002/pd.5212
Huang J, Poon LC, Akolekar R, Choy KW, Leung TY, Nicolaides KH (2014) Is high fetal nuchal translucency associated with submicroscopic chromosomal abnormalities on array CGH? Ultrasound Obstet Gynecol 43:620–624. https://doi.org/10.1002/uog.13384
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C (2004) Detection of large-scale variation in the human genome. Nat Genet 36:949–951. https://doi.org/10.1038/ng1416
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li X, Chen S, Xie W, Vogel I, Choy KW, Chen F, Christensen R, Zhang C, Ge H, Jiang H, Yu C, Huang F, Wang W, Jiang H, Zhang X (2014) PSCC: sensitive and reliable population-scale copy number variation detection method based on low coverage sequencing. PLoS ONE 9:e85096. https://doi.org/10.1371/journal.pone.0085096
Liang D, Peng Y, Lv W, Deng L, Zhang Y, Li H, Yang P, Zhang J, Song Z, Xu G, Cram DS, Wu L (2014) Copy number variation sequencing for comprehensive diagnosis of chromosome disease syndromes. J Mol Diagn 16:519–526. https://doi.org/10.1016/j.jmoldx.2014.05.002
Lin SB, Xie YJ, Chen Z, Zhou Y, Wu JZ, Zhang ZQ, Shi SS, Chen BJ, Fang Q (2015) Improved assay performance of single nucleotide polymorphism array over conventional karyotyping in analyzing products of conception. J Chin Med Assoc 78:408–413. https://doi.org/10.1016/j.jcma.2015.03.010
Markello TC, Carlson-Donohoe H, Sincan M, Adams D, Bodine DM, Farrar JE, Vlachos A, Lipton JM, Auerbach AD, Ostrander EA, Chandrasekharappa SC, Boerkoel CF, Gahl WA (2012) Sensitive quantification of mosaicism using high density SNP arrays and the cumulative distribution function. Mol Genet Metab 105:665–671. https://doi.org/10.1016/j.ymgme.2011.12.015
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH (2010) Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86:749–764. https://doi.org/10.1016/j.ajhg.2010.04.006
Motley WW, Palaima P, Yum SW, Gonzalez MA, Tao F, Wanschitz JV, Strickland AV, Loscher WN, De Vriendt E, Koppi S, Medne L, Janecke AR, Jordanova A, Zuchner S, Scherer SS (2016) De novo PMP2 mutations in families with type 1 Charcot-Marie-Tooth disease. Brain 139:1649–1656. https://doi.org/10.1093/brain/aww055
Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, Hanscom C, Pillalamarri V, Seabra CM, Abbott MA, Abdul-Rahman OA, Aberg E, Adley R, Alcaraz-Estrada SL, Alkuraya FS, An Y, Anderson MA, Antolik C, Anyane-Yeboa K, Atkin JF, Bartell T, Bernstein JA, Beyer E, Blumenthal I, Bongers EM, Brilstra EH, Brown CW, Bruggenwirth HT, Callewaert B, Chiang C, Corning K, Cox H, Cuppen E, Currall BB, Cushing T, David D, Deardorff MA, Dheedene A, D'Hooghe M, de Vries BB, Earl DL, Ferguson HL, Fisher H, FitzPatrick DR, Gerrol P, Giachino D, Glessner JT, Gliem T, Grady M, Graham BH, Griffis C, Gripp KW, Gropman AL, Hanson-Kahn A, Harris DJ, Hayden MA, Hill R, Hochstenbach R, Hoffman JD, Hopkin RJ, Hubshman MW, Innes AM, Irons M, Irving M, Jacobsen JC, Janssens S, Jewett T, Johnson JP, Jongmans MC, Kahler SG, Koolen DA, Korzelius J, Kroisel PM, Lacassie Y, Lawless W, Lemyre E, Leppig K, Levin AV, Li H, Li H, Liao EC, Lim C, Lose EJ, Lucente D, Macera MJ, Manavalan P, Mandrile G, Marcelis CL, Margolin L, Mason T, Masser-Frye D, McClellan MW, Mendoza CJ, Menten B, Middelkamp S, Mikami LR, Moe E, Mohammed S, Mononen T, Mortenson ME et al (2017) The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet 49:36–45. https://doi.org/10.1038/ng.3720
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, Pineda-Alvarez D, Aradhya S, Martin CL (2019) Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. https://doi.org/10.1038/s41436-019-0686-8
Trost B, Walker S, Wang Z, Thiruvahindrapuram B, MacDonald JR, Sung WWL, Pereira SL, Whitney J, Chan AJS, Pellecchia G, Reuter MS, Lok S, Yuen RKC, Marshall CR, Merico D, Scherer SW (2018) A comprehensive workflow for read depth-based identification of copy-number variation from whole-genome sequence data. Am J Hum Genet 102:142–155. https://doi.org/10.1016/j.ajhg.2017.12.007
Wang J, Chen L, Zhou C, Wang L, Xie H, Xiao Y, Zhu H, Hu T, Zhang Z, Zhu Q, Liu Z, Liu S, Wang H, Xu M, Ren Z, Yu F, Cram DS, Liu H (2018) Prospective chromosome analysis of 3429 amniocentesis samples in China using copy number variation sequencing. Am J Obstet Gynecol 219:287e1–287e18. https://doi.org/10.1016/j.ajog.2018.05.030
Wang JC, Radcliff J, Coe SJ, Mahon LW (2019) Effects of platforms, size filter cutoffs, and targeted regions of cytogenomic microarray on detection of copy number variants and uniparental disomy in prenatal diagnosis: results from 5026 pregnancies. Prenat Diagn 39:137–156. https://doi.org/10.1002/pd.5375
Wang H, Dong Z, Zhang R, Chau MHK, Yang Z, Tsang KYC, Wong HK, Gui B, Meng Z, Xiao K, Zhu X, Wang Y, Chen S, Leung TY, Cheung SW, Kwok YK, Morton CC, Zhu Y, Choy KW (2020a) Low-pass genome sequencing versus chromosomal microarray analysis: implementation in prenatal diagnosis. Genet Med 22:500–510. https://doi.org/10.1038/s41436-019-0634-7
Wang Y, Li Y, Chen Y, Zhou R, Sang Z, Meng L, Tan J, Qiao F, Bao Q, Luo D, Peng C, Wang YS, Luo C, Hu P, Xu Z (2020b) Systematic analysis of copy-number variations associated with early pregnancy loss. Ultrasound Obstet Gynecol 55:96–104. https://doi.org/10.1002/uog.20412
Zhang L, Bai W, Yuan N, Du Z (2019) Comprehensively benchmarking applications for detecting copy number variation. PLoS Comput Biol 15:e1007069. https://doi.org/10.1371/journal.pcbi.1007069
Funding
This project is supported by the National Natural Science Foundation of China (31801042, 81741004 and 81741009), the Health and Medical Research Fund (04152666) and the Health Department of Guangxi Province (Z20190782). Zirui Dong thanks the Hong Kong Obstetrical & Gynaecological Trust Fund for the support.
Author information
Authors and Affiliations
Contributions
ZD, MHKC, HW, YL, TYL, YK, LX, YZ, WW, and KWC designed the study. HW, YL, TYL, JPWC, SCC, YZ, LX and WW collected the samples and followed up. MHKC, HW, YL, FX, YT, YW and ZC performed CMA and low-pass GS. ZD, MHKC and YK performed the analysis and data interpretation. MHKC, HW and YL conducted the validation. ZD, MHKC, HW, YL, and KWC wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest or competing interest.
Ethics approval
The study was approved by the institutional review boards of each collaborative site. Written consent for sample storage and genetic analyses of peripheral blood and invasive diagnostic samples was obtained from each participant.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chau, M.H.K., Wang, H., Lai, Y. et al. Low-pass genome sequencing: a validated method in clinical cytogenetics. Hum Genet 139, 1403–1415 (2020). https://doi.org/10.1007/s00439-020-02185-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02185-9