
Abstract
Haploinsufficiency of FOXF1 causes alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV), a lethal neonatal lung developmental disorder. We describe two similar heterozygous CNV deletions involving the FOXF1 enhancer and re-analyze FOXF1 missense mutation, all associated with an unexpectedly mitigated disease phenotype. In one case, the deletion of the maternal allele of the FOXF1 enhancer caused pulmonary hypertension and histopathologically diagnosed MPV without the typical ACD features. In the second case, the deletion of the paternal enhancer resulted in ACDMPV rather than the expected neonatal lethality. In both cases, FOXF1 expression in lung tissue was higher than usually seen or expected in patients with similar deletions, suggesting an increased activity of the remaining allele of the enhancer. Sequencing of these alleles revealed two rare SNVs, rs150502618-A and rs79301423-T, mapping to the partially overlapping binding sites for TFAP2s and CTCF in the core region of the enhancer. Moreover, in a family with three histopathologically-diagnosed ACDMPV siblings whose missense FOXF1 mutation was inherited from the healthy non-mosaic carrier mother, we have identified a rare SNV rs28571077-A within 2-kb of the above-mentioned non-coding SNVs in the FOXF1 enhancer in the mother, that was absent in the affected newborns and 13 unrelated ACDMPV patients with CNV deletions of this genomic region. Based on the low population frequencies of these three variants, their absence in ACDMPV patients, the results of reporter assay, RNAi and EMSA experiments, and in silico predictions, we propose that the described SNVs might have acted on FOXF1 enhancer as hypermorphs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alveolar capillary dysplasia with misalignment of pulmonary veins (ACDMPV, MIM#265380) is a rare (~ 1/100,000 births) developmental lung disorder (Bishop et al. 2011; Galambos et al. 2015; Janney et al. 1981; Langston 1991; Sen et al. 2004; Slot et al. 2018). Affected newborns manifest progressive hypoxemic respiratory failure and severe pulmonary arterial hypertension (PAH) often associated with additional malformations of the cardiovascular, gastrointestinal, or genitourinary systems. Most ACDMPV infants are born at term with normal weight, develop symptoms within the first 24–48 h of life, and almost uniformly die within the first month of life. Very rarely, ACDMPV patients survive beyond the neonatal period (Abdallah et al. 1993; Ahmed et al. 2008; Edwards et al. 2019; Ito et al. 2015; Kodama et al. 2012; Michalsky et al. 2005; Shankar et al. 2006; Szafranski et al. 2014; Towe et al. 2018). Histopathologically, ACDMPV is characterized by thickening of intra-alveolar septa, a reduced number of pulmonary capillaries, the majority of which do not make contact with the alveolar epithelium, medial hypertrophy of small peripheral pulmonary arteries and arterioles, and intrapulmonary vascular anastomoses. In the vast majority of cases, ACDMPV has been causatively linked to heterozygous loss-of-function single nucleotide variants (SNVs) in FOXF1 or heterozygous copy-number variant (CNV) deletions involving FOXF1 or its lung-specific enhancer (Sen et al. 2013a, b; Stankiewicz et al. 2009; Szafranski et al. 2013, 2016a).
FOXF1 (MIM#601089) encodes a fork-head family transcription factor (TF) (Murphy et al. 1994; Pierrou et al. 1994). During lung development, FOXF1 is primarily expressed in mesoderm-derived tissues (Mahlapuu et al. 1998) where it mediates paracrine SHH signaling from epithelial cells of developing airways (Astorga and Carlsson 2007; Fernandes-Silva et al. 2017; Ho and Wainwright 2017; Kalinichenko et al. 2001; Lim et al. 2002; Mahlapuu et al. 2001; Sen et al. 2014). The FOXF1 distant lung-specific enhancer maps ~ 270 kb upstream to the gene (chr16:86,212,040–86,271,919; hg19) (Dello Russo et al. 2015; Seo et al. 2016; Szafranski et al. 2013, 2016a, b). Recently, we have narrowed this enhancer to an approximately 15 kb interval essential for normal lung development (Szafranski et al. 2016b). In cultured human fetal lung fibroblasts, IMR-90, the enhancer features histone modifications, H3K4me1 and H3K27ac (http://genome.ucsc.edu/ENCODE), which are typically found in nucleosomes around active transcriptional enhancers. As determined by ChIP-seq analysis, it binds numerous TFs and chromatin structure regulators, including, among others, GLI2, CEBP/p300, CTCF, RAD21, and YY1 (http://genome.ucsc.edu/ENCODE). In addition, this region encodes fetal lung-expressed long non-coding RNAs: LINC01081, LINC01082, and RP11-805I24.3 (http://genome.ucsc.edu/ENCODE). We showed that expression of LINC01081 and the binding of GLI2 within this enhancer positively regulated the expression of FOXF1 in cultured IMR-90 cells (Szafranski et al. 2013, 2014). However, many of the elements required for the functioning of the enhancer remain unknown.
Here, we propose that rare non-coding SNVs, mapping within a 2 kb segment of the enhancer core, might have delayed the onset of ACDMPV or prevented development of lethal ACD features caused by FOXF1 deficiency.
Materials and methods
DNA and RNA isolation
Peripheral blood, saliva, and frozen or formalin-fixed paraffin-embedded (FFPE) lung biopsy or autopsy samples were received after obtaining written informed consent from the patients’ parents. DNA was extracted from blood and saliva using Gentra Purgene Blood Kit (Qiagen, Germantown, MD, USA), and from frozen lung tissue using DNaesy Blood and Tissue Kit (Qiagen). RNA from frozen lung samples, and cultured IMR-90 fibroblasts (ATCC, Manassas, VA, USA) was extracted using miRNeasy Mini Kit (Qiagen). RNA from FFPE lung tissues obtained by biopsy or acquired at autopsy was extracted using Quick-RNA FFPE Kit (Zymo Research, Irvine, CA, USA).
Array comparative genomic hybridization
For pt 165.3, clinical array comparative genomic hybridization (array CGH) was done with a NimbleGen CGX-12 microchip containing 135 K oligo probes (Roche NimbleGen, Madison, WI, USA). Array CGH for pt 179.3 was performed using a custom designed chromosome 16-specific microarray (Agilent, Santa Clara, CA, USA). For the family 180, Affymetrix CytoScan HD platform (Affymetrix, Santa Clara, CA, USA) was used. DNA labelling and hybridizations were completed according to manufacturers’ protocols.
Sequencing of deletion breakpoints
Deletion junctions were amplified by long-range PCR using LA Taq polymerase (Takara Bio., Madison, WI, USA). The PCR-amplified products were treated with ExoSAP-IT (USB, Cleveland, OH, USA) and directly sequenced by the Sanger method. Sequences were assembled using Sequencher v4.8 (Gene Codes, Ann Arbor, MI, USA).
Parental origin of deletion- or FOXF1 missense mutation-bearing chromosome 16
The parental origin of the pathogenic CNV deletions on chromosome 16 was determined using informative SNVs or microsatellite polymorphisms. In cases with FOXF1 pathogenic single nucleotide variants, a PCR-amplified DNA fragment, containing both a mutation and an informative non-pathogenic SNV, was cloned in pGEM-T Easy vector (Promega, Madison, WI, USA). At least three transformed DH5α clones were used for plasmid isolation with the PureLink Quick Plasmid Miniprep Kit (Invitrogen, Carlsbad, CA, USA) and Sanger sequencing of the cloned insert.
Whole-genome sequencing
Whole-genome sequencing (WGS) was done for pts 165.3 and 179.3. The libraries were prepared with a TruSeq Nano DNA HT Library Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol, followed by sequencing on the HiSeqX platform (Illumina) at CloudHealth Genomics (Shanghai, China). The sequencing depth was 30x. The raw sequencing data were processed according to the specification of the bcl2fastq package from Illumina. Short reads obtained during sequencing were processed using Trimmomatic (Bolger et al. 2014) to remove adapter sequences. Data were aligned and mapped to the human genome reference sequence using the BWA 0.7.12 tool (Li and Durbin 2009). Variants were called using the GATK 3.7 software (McKenna et al. 2010) and the Atlas2 suit (Challis et al. 2012).
Transcript quantification by real time PCR
RNA samples were reverse-transcribed using SuperScript III First-Strand Synthesis System (Invitrogen). TaqMan primers and probes were obtained from Applied Biosystems (Foster City, CA, USA). Transcript levels were normalized to GAPDH and ACTB. qPCR was done using TaqMan Universal PCR Master Mix (Applied Biosystems). qPCR conditions included 40 cycles of heating the reaction mixtures at 95 °C for 15 s and 60 °C for 1 min. For relative quantification of the transcripts, the comparative CT method was used. RNA from healthy lungs of four infants at the age of 0–6 days or from IMR-90 cells transfected with control DNA constructs were used as calibrators.
Luciferase reporter assay
The 2.8 kb FOXF1 promoter region (chr16: 86,541,532–86,544,295; hg19), including the ChIP-seq-determined binding site for RNA PolII, was amplified from normal human DNA using primers 5′-atagctagcGCATGAAGTGTGCACTTAAACCAAAGT-3′ and 5′-atactcgagGTTGGTCTTCTTGGCCTTGGAC-3′ that contained the restriction enzyme sites for NheI and XhoI, respectively. PCR was done using LA Taq DNA Pol (Takara Bio.) in the presence of 16% betaine (Sigma-Aldrich, St. Louis, MO, USA), applying 30 cycles of incubation at 94 °C for 30 s, 58 °C for 30 s and 72 °C for 3 min. The amplified FOXF1 promoter was cut with NheI and XhoI and cloned into NheI–XhoI site of the multiple cloning site of a promoter-less vector pGL4.10 (Promega). ChIP-seq-determined 1.3 kb-large TF binding region of the FOXF1 enhancer (chr16:86,257, 438–86,258,766, hg19), bearing SNP rs150502618, was amplified from normal human DNA and from pt 165.3 DNA using primers 5′-aatggtaccTACAATTTCTGGGAGCTTGGGATCA-3′ and 5′-atagctagcAAAGGGAGTGACCCTTGGTCAGA-3′ that contained sites for KpnI and NheI, respectively. PCR was done using Phusion DNA Pol (New England Biolabs, Ipswich, MA, USA), applying 30 cycles of incubation at 94 °C for 30 s, 58 °C for 30 s and 72 °C for 1 min. The amplified fragments were digested with KpnI and NheI and cloned in KpnI-NheI site of FOXF1 promoter vector upstream of the promoter in the orientation as they appear on chromosome 16.
Fibroblasts IMR-90, derived from normally developing lungs of a 16-week-old fetus were grown in EMEM medium with l-glutamine supplemented with sodium pyruvate and 10% FBS. Transfections were done using sub-confluent cells in Opti-MEM medium (Invitrogen) on 12-well plates. The cells were co-transfected with 1 µg per well of Photinus luciferase (luc2) reporter plasmid pGL4.10 (Promega) or its recombinant derivative and 0.1 µg of pGL4.75 (Promega), constitutively expressing Renilla luciferase (Rluc), using Lipofectamine 3000 (4 µl/well) with P3000 reagent. They were lysed in Qiazol (Qiagen) 48 h after transfection. Luciferase gene expression was determined by measuring its transcript levels applying qPCR. Rluc transcript levels were used to normalize the transfection efficiency of each sample. Transfections were conducted in triplicates.
RNAi-based knockdown
The knockdown experiments were done applying siRNA methodology in IMR-90 cells. Silencer Select siRNAs, targeting transcripts of TFAP2A (exons 4 and 6), TFAP2C (exon 3), and two randomized Silencer Select siRNA negative controls were obtained from Ambion (Foster City, CA, USA) (24 pmols of siRNA duplexes per well). Forward transfections were done in 12-well plates using Lipofectamine RNAiMAX (Invitrogen) (3 µl per well). Cells for RNA preparation were lysed in Qiazol (Qiagen) 48 h after the transfection.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was performed with recombinant full length CTCF and two different SNV-bearing DNA probes using SYBR Green-based EMSA Kit (Invitrogen). As DNA probes, we used either synthetic 50 bp duplex oligonucleotide (chr16: 86, 257, 720–86,257, 770, hg19) (IDT, San Diego, CA, USA) or 230 bp PCR-amplified fragment (chr16: 86, 257, 700–86, 257, 929, hg19), including larger part of ChIP-seq-determined TFAP2/CTCF binding site. In a 10 µl reaction mixture, 0.25 pmol of 50 bp probe or 1 pmol of 230 bp probe was combined respectively with 0.5 or 1 pmol of CTCF (Novus Biologicals, Centennial, CO, USA) in a buffer containing 10 mM Tris (pH 7.5), 150 mM KCl, 1 mM ZnSO4, 0.5 mM DTT, and 0.1 mM EDTA. Reaction mixtures were incubated for 30 min at 25 °C and electrophoresed in 6% (acrylamide:bis-acrylamide 20:1) non-denaturing polyacrylamide gel at 10 V/cm in 0.5 × TBE. The gel was stained with SYBR Green. DNA bands were visualized under UV and pictured using a CCD camera. DNA band intensities were subsequently measured at non-saturated pixel densities using ImageJ (http://imagej.nih.gov/ij). To compare intensities of DNA-CTCF bands, these were expressed as a fraction of total DNA (CTCF-bound and unbound DNA). The experiments were performed in triplicates.
Results
Case presentations
Patient 165.3
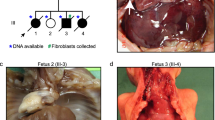
Patient 165.3 is an 8-year-old girl who had a clinical diagnosis of moderate to severe PAH at the age of 14 months. A lung biopsy was performed at 2 years and 2 months of age (Fig. 1a). It revealed a pattern of alveolar development that appeared appropriate for her age. Lobular septa were present with dilated lymphatics, but lacked veins. The walls of the pulmonary arterioles were markedly thick, and the smooth muscle extended peripherally. Lymphatics were also dilated around the bronchovascular bundles. There were misaligned veins accompanying the arteries, which were confirmed by immunostaining for lymphatics and vascular endothelium. The number of alveolar capillaries and their position with respect to the alveolar surface appeared normal. The patient’s condition has remained stable over time with the administration of pulmonary vasodilators and oxygen during sleep and exercise.

Lung histopathology of formalin-fixed, paraffin-embedded biopsy and autopsy tissue stained with hematoxylin (H&E), CD 34 or CD31 endothelial markers. a Patient 165.3, with lung biopsy at 2 years of age shows the misalignment of the vein (v) adjacent to the normally positioned artery (a), lymphatics (l) and bronchiole (b) (100 ×). There is peripheral extension of the pulmonary arteriolar smooth muscle (arrow) and normal alveolar septa (H and E, 400 ×). The CD34 endothelial marker highlights the normal appearing alveolar capillaries (CD34, 400 ×). b Patient 179.3 with classic ACDMPV, deceased at 10 days. The arteries (a), veins (v) and lymphatics (l) are adjacent to the bronchi (b). CD31 stains the endothelium of the artery, vein and lymphatics. The alveoli are small and simplified (astreik). The alveolar septa are markedly thickened with few larger centrally placed capillaries (arrows) (H and E and CD31, 400 ×)
Patient 179.3
Patient 179.3 was born at 39 weeks gestation to a 37-year-old G5P3 mother with a past medical history of ankylosing spondylitis and Hashimoto’s disease. The infant was initially admitted to the newborn nursery for routine care until a cyanotic event needing resuscitation at 12 h of age. The blood gas on admission to the neonatal intensive care unit was significant for a mixed metabolic and respiratory acidosis. The infant had to be emergently cannulated to veno-arterial extracorporeal membrane oxygenation (ECMO). Approximately 72 h after her ECMO de-cannulation, she developed progressive hypoxemia that was not amenable to maximal medical therapy, and died at 10 days of age. The gross examination on autopsy (Fig. 1b) was significant for abnormal lobation including a mono-lobate left lung and a right lung with posterior fusion of the upper and lower lobes. Histology was significant for diffusely underdeveloped alveoli with a reduced number of centrally placed capillaries. Lobular septa were thickened and pulmonary veins were identified within the bronchovascular bundle. The final clinical diagnosis following autopsy was ACDMPV.
Patient 180.3
In this case, the pregnancy was terminated because of the presence of a hypoplastic left heart with a double outlet right ventricle and a ventricular septal defect in the fetus. Moreover, the aorta was small, and nuchal translucency thickness was 4 mm. No autopsy was performed.
Family 72.1-8
In 2013, we reported a family with two aborted fetuses and four children, three of whom were histopathologically diagnosed with ACDMPV (Supplemental Fig. S1) (Sen et al. 2013a) caused by a heterozygous FOXF1 missense variant c.416G>T (p.Arg139Leu). This variant was inherited from their apparently healthy non-mosaic carrier mother, whose only past medical history was trachea narrowing.
Sequence characteristics of the 16q23.3q24.1 pathogenic variants
Sanger sequencing of FOXF1 exons and the flanking ~ 100 bp intronic regions in patients 165.3, 179.3, and 180.3 did not reveal any pathogenic SNVs or indel variants. However, array CGH analysis revealed in each case a heterozygous CNV deletion at 16q23.3q24.1 that removed one allele of the FOXF1 enhancer, leaving the FOXF1 gene intact (Fig. 2a, Supplemental Table S1, Supplemental Fig. S2). In pts 165.3 and 179.3, the 2.6 Mb deletions were almost identical in size and location to each other as well as to already reported ACDMPV-causing deletion in pts 60.4 (Szafranski et al. 2016a). These were mediated by highly homologous and directly oriented LINE1 (L1) elements, L1HS (chr16:83,670,858–83,676,901, hg19) and L1PA3 (chr16:86,266,902–86,272,916, hg19), or L1PA2 (chr16:86,295,780–86,301,803, hg19), respectively. The two L1PA elements are integral part of the recently described genomic instability hotspot at 16q24.1 (Szafranski et al. 2018). The L1HS element, harboring the proximal deletion breakpoint at 16q23.3, defines another putative genomic instability hotspot since four ACDMPV-causative deletions (pts 60.4, 165.3, 179.3, and the one reported by Dello Russo et al. (2015), DR2015) were found to have their proximal breakpoints located in this L1 element (Fig. 2a). In support of this prediction, query of the Database of Genomic Variants (DGV) of polymorphic CNVs (http://dgv.tcag.ca/dgv/app/home) revealed 10 deletions, and five duplications (none of which included FOXF1 or its enhancer), all having one of their breakpoints apparently mapping (they were not sequenced) within the L1HS instability locus.

The 16q23.3q24.1 deletions and rare SNVs involving the FOXF1 distant enhancer. a Similar by size and location CNV deletions, resulting in ACDMPV of variable expressivity. Parental origin of the deletion-bearing chromosome 16: red, maternal; blue, paternal. Abbreviations: DR2015, deletion reported by Dello Russo et al. (2015); HLH, hypoplastic left heart; PCH, pulmonary capillary hemangiomatosis; VM, vascular malformations; VSD, ventricular septal defect. b Putatively hypermorphic SNVs within the FOXF1 enhancer that likely mitigated the lethal ACDMPV phenotype. Histone modifications identified in IMR-90 lung fibroblasts are shown (http://genome.ucsc.edu/ENCODE)
The larger 3.8 Mb deletion in the pt 180.3 included both FOXF1 and its upstream enhancer. Breakpoints of this deletion mapped to unique sequences, and the deletion junction contained an insertion of AAG, both findings suggesting NHEJ as the most likely mechanism of deletion formation.
To determine whether the deletions in patients arose de novo, we have performed PCR on parental and patient’s DNA samples with primers flanking the deletions identified in the patients (Campbell et al. 2014). We have not amplified any deletion junction in the parental DNA samples, thus excluding somatic mosaicism in the parents. Moreover, in family 180, using array CGH analysis of parental samples, we have not found any evidence of the deletion present in the fetus (180.3). Analysis of the informative SNV markers within the deletion regions showed that in pts 179.3 and 180.3, the deletion arose on the paternal chromosome 16, whereas in pt 165.3 it arose on maternal chromosome 16 (Supplemental Fig. S3).
Previously, we have interpreted the transmission of the FOXF1 pathogenic SNV c.416G>T (p.Arg139Leu) in the family 72, inherited from the healthy carrier mother, as being consistent with a model of paternal imprinting of FOXF1 (Sen et al. 2013a). We have now studied parental origin of a chromosome bearing the apparently de novo pathogenic FOXF1 variants in seven ACDMPV newborns from unrelated families using clonal co-segregation of the investigated mutations with the neighboring informative SNVs. We found that two mutations arose on the maternal allele and five others on the paternal allele (Supplemental Table S2). Thus, paternal genomic imprinting may not be responsible for the lack of penetrance of pathogenic variants in FOXF1.
FOXF1 expression in the lungs of patients with CNV deletions of the FOXF1 enhancer
Comparative RT-qPCR analysis of pt 60.4, 165.3, and 179.3 transcriptomes showed that whereas FOXF1 transcript levels in pt 179.3 and 60.4 lungs were significantly reduced in comparison with age-matched normal lung controls (one-way ANOVA P = 0.0004 and 0.00005, respectively), the FOXF1 transcript level in pt 165.3 lung biopsy performed at 2.2 years of age was in the range found in normal newborn lungs and higher than in normal lung autopsy of a 2-year-old child used as an age-matched control (t test P = 0.01) (Fig. 3a). This suggests that the absence of typical ACD features in pt 165.3 and the development of ACDMPV instead of the expected prenatal lethality in pt 179.3 might have resulted from a compensatory increase and lesser decrease of FOXF1 expression, respectively.

Analyses of FOXF1 expression. aFOXF1 transcript levels in the lungs from the patients with the heterozygous 16q23.3q24.1 CNV deletions. b Luciferase reporter assay showing the increase of FOXF1 promoter activity by SNV rs150502618-A. c, d Regulation of FOXF1 expression by TFAP2s in IMR-90 lung fibroblasts. Depletion of TFAP2C by siRNA resulted in a significant decrease of FOXF1 transcript levels. Data represent means of triplicate experiments ± SD (t test *P ≤ 0.01). Cn normal lung controls used as calibrator, C2yo normal lung of 2 year-old child for comparison with pt 165.3
Low population frequency non-pathogenic SNVs in the FOXF1 enhancer
WGS and Sanger sequencing of the non-deleted allele of the FOXF1 enhancer in pt 165.3 identified 59 SNVs. Search for these variants in the core enhancer region in 13 unrelated ACDMPV patients, each of them with heterozygous CNV deletion of the enhancer on maternal chromosome 16, revealed SNV rs150502618-A at chr16:86,257,745 (hg19) (Fig. 2b, Supplemental Fig. S4) that was present in pt 165.3 but absent in ACDMPV patients. This SNV has a low minor allele frequency (MAF) of 0.539% in the general population with an average heterozygosity 0.011 ± 0.072, and is located within the ChIP-seq-determined overlapping binding sites for the transcription factor activator protein 2A and C (TFAP2A, TFAP2C), and CTCF (http://genome.ucsc.edu/ENCODE).
WGS and Sanger sequencing of the enhancer region in pt 179.3 revealed the presence of 42 SNVs, including a variant rs79301423-T of low MAF = 1.258% at chr16:86,257,521 (hg19) with an average heterozygosity 0.025 ± 0.109, located 223 bp centromeric to rs150502618 (Fig. 2b, Supplemental Fig. S5), and, similarly to rs150502618, absent in all 13 ACDMPV patients with heterozygous CNV deletion of the FOXF1 enhancer on maternal chromosome 16. This SNV is located within a ChIP-seq-determined binding site for TFAP2A (http://genome.ucsc.edu/ENCODE).
To explain the unusual genotype–phenotype correlation and segregation of the c.416G>T mutation in family 72, we have Sanger sequenced the core enhancer region in all members of family 72 and identified six SNVs present only in the mother that might have acted in her as potentially protective factors (Supplemental Table S3). Variant A of rs28571077 (chr16:86,254,839, hg19) is particularly interesting since it has low MAF of 2.855% and was not found in pts 165.3, 178.3, 180.3, or any of 13 analyzed unrelated ACDMPV patients with CNV deletion of the FOXF1 enhancer. The ChIP-seq data (Fig. 2b) show that rs28571077 maps 2 kb proximal to rs150502618 and rs79301423 within the overlapping binding sites for TFs MAFK and RAD21 (https://genome.ucsc.edu/ENCODE).
No significant eQTLs were found for any of those non-coding SNVs. The low CADD scores for rs79301423, rs150502618, and rs28571077 equaling 0.779, 1.453, and 2.903, respectively, indicate that these variants are not pathogenic.
SNV rs150502618-A increases activity of the FOXF1 promoter
The functionality of one of the non-coding SNVs, rs150502618, was assessed by testing its ability to modify the activity of the FOXF1 promoter cloned in promoter-less reporter plasmid. We found that, in comparison to the common variant G of rs150502618, variant A increased FOXF1 promoter activity about 2.5-fold (t test P = 0.003) (Fig. 3b). This increase correlated well with the higher than expected level of FOXF1 transcript found in pt 165.3 lung biopsy sample.
Regulation of the FOXF1 expression by TFAP2s
Since both SNPs rs150502618 and rs79301423 map to TFAP2A and TFAP2C binding sites, we asked whether AP2 TFs can regulate FOXF1 expression. To this end, we targeted transcripts of these two TFs with siRNAs in IMR-90 fibroblasts and measured the FOXF1 transcript levels. We found that the depletion of TFAP2C by 80% (t test P = 0.0001) resulted in a 50% decrease (t test P = 0.01) of the FOXF1 transcript level (Fig. 3c, d).
To determine whether variants rs79301423-T and rs150502618-A might affect binding of TFAP2s within the enhancer, we have compared sequences around those SNVs with TFAP2 binding motif using position weight matrix (PWM) (Supplementary Fig. S6). We found that T and A at those particular positions in the binding site occur more often than C and G, respectively, suggesting that TFAP2C binding in the presence of rs79301423-T and rs150502618-A might be increased. In addition, using a QBiC-Pred server (http://qbic.genome.duke.edu; Martin et al. 2019), we have predicted that both variants likely increase binding of TFAP2C (z score = 4.703, P = 0.000003 for rs79301423-T; z score = 3.583, P = 0.0003 for rs150502618-A). Using QBiC-Pre server, we have predicted that also rs28571077-A could potentially increase binding of TFAP2 (z score = 2.338, P = 0.02). The sequences around the described SNPs do not precisely match the TFAP2 consensus binding motif. However, a number of genomic sites, determined experimentally as binding TFAP2s, also differ considerably from this consensus sequence, suggesting that the TFAP2 binding motif represents a promiscuous GC-rich element (Eckert et al. 2005). Moreover, lower affinity non-consensus binding sites are often used in assisted TF recruiting or tethering.
Contribution of SNV rs150502618-A to CTCF binding
To further test how the identified SNVs might have altered affinity of transcriptional regulators to the FOXF1 enhancer, we have explored the binding of CTCF to rs150502618-A or G-containing ChIP-seq-determined CTCF binding site at chr16:86,257,713-86,258,147 (hg19) (a region that partially overlaps with TFAP2 binding site). We have previously shown that CTCF contributes to regulation of chromatin architecture around the FOXF1 locus (Szafranski et al. 2016a). According to ChIP-seq data, CTCF is the strongest binding regulator at the site where one of the identified SNVs, rs150502618-A, is located. We have used shorter and longer DNA fragments bearing this SNV as probes. Binding of CTCF to the shorter DNA probe resulted in the appearance of a slower migrating DNA-CTCF band, whereas CTCF binding to the longer probe apparently caused change in DNA topology that increased the migration of the complex band in comparison to the free probe (Supplemental Fig. S7). In both cases, the presence of the low frequency variant increased binding of CTCF by 15 ± 2% and 8 ± 1%, respectively. Thus the tested SNV, rs150502618-A, could be of functional relevance also in the context of CTCF binding to the enhancer.
Discussion
To explain the statistically significant parental bias of ACDMPV-causative CNV deletions at the FOXF1 locus, we have proposed a model of FOXF1 regulation involving a stronger distant lung-specific enhancer located on the chromosome 16 inherited from father and a weaker one on the maternal chromosome 16 (Szafranski et al. 2016a). Here, we hypothesize that the described phenotypic differences in the presentation of ACDMPV, resulting from FOXF1 enhancer heterozygous deletions, might be also caused by the presence of the hypermorphic non-coding SNVs within the non-deleted allele of the enhancer. Compound inheritance of rare coding pathogenic variants and rare or common non-coding variants has been reported for thrombocytopenia absent radius syndrome with recurrent 1q21 deletion (Albers et al. 2012), congenital scoliosis with recurrent 16p11.2 deletion (Wu et al. 2015), and lethal developmental lung diseases caused by pathogenic SNVs and CNVs involving TBX4 and FGF10 (Karolak et al. 2019). We have previously shown that none of the other genes deleted at 16q23.3q24.1 together with the FOXF1 enhancer, contribute to ACDMPV phenotype, as point mutations involving the FOXF1 gene manifest fully expressed ACDMPV, similar to the upstream deletions that included the FOXF1 enhancer but not its target gene (Stankiewicz et al. 2009; Szafranski et al. 2013; 2016a). Moreover, four 16q23.3q24.1 deletions (pts 60.4, 165.3, DR2015, 179.3) removed the same genes in addition to the FOXF1 enhancer, but resulted in either severe or mitigated phenotypes.
Sequencing of the remaining allele of the FOXF1 enhancer in pt 165.3 revealed a rare SNV rs150502618-A, located within partially overlapping binding sites for TFAP2s and CTCF. Luciferase reporter assay showed that, in comparison with the common variant G of this SNP, variant A significantly increased the activity of the FOXF1 promoter. Thus, we propose that variant A contributed to an increase of the FOXF1 expression from pt 165.3 paternal allele, resulting in a milder disease phenotype. Similarly, in pt 179.3 a putatively hypermorphic SNV rs79301423-T located close to rs150502618 within the binding site for TFAP2 might have contributed to the increased FOXF1 expression from the maternal allele, leading to neonatal lethal ACDMPV rather than the expected prenatal lethality. In support of this notion, our RNAi-based knock-down data indicated that TFAP2C likely contributes to regulation of FOXF1 expression. Consistent with our model of parental inheritance, in pt 180.3 in whom no rare SNV was found residing within the enhancer core, enhancer deletion on paternal chromosome 16 resulted in early development of severe phenotype and pregnancy termination.
TFAP2 proteins have not been previously linked to regulation of FOXF1 expression. However, TFAP2A was shown to be involved in maintaining the integrity of the airway epithelium (Xiang et al. 2012) and overexpression of TFAP2C has been associated with lung tumorigenesis (Kim et al. 2016). In general, AP2 factors control the balance between cell proliferation and differentiation during embryogenesis (Eckert et al. 2005), and some of their effects on cell proliferation and migration mimic those exhibited by FOXF1 in the lungs (Gialmanidis et al. 2013; Kalinichenko et al. 2004; Kalin et al. 2008; Saito et al. 2010; Wei et al. 2014) and in other tissues (Huang et al. 2018; Lo et al. 2010; Wang et al. 2018; Saito et al. 2010; Tamura et al. 2014). Interestingly, a non-coding variant in the TFAP2A binding site in the enhancer of the LINC00339 gene on chromosome 1 was recently shown to increase the expression of this long non-coding RNA (Chen et al. 2018).
The proposed functional role for the discussed non-coding SNVs seems to be also supported by our EMSA experiment, showing that the tested variant rs150502618-A slightly increased in vitro the affinity of CTCF for its binding site that partially overlaps with an AP2 binding site within the FOXF1 enhancer. This might have affected local chromatin folding, resulting in increased FOXF1 transcription. Based on the Hi-C data, we proposed previously that CTCF-mediated chromatin looping does involve FOXF1 enhancer region (Szafranski et al. 2016a), and disruptions of TAD domains has been shown to have pathogenic effect by altering the expression of disease-implicated genes (Spielmann and Mundlos 2016).
Re-analysis of the reported unusual transmission of the missense variant c.416G>T (p.Arg139Leu) from a healthy mother to her affected children (Sen et al. 2013a) prompted us to propose that a non-coding SNV rs28571077-A within the FOXF1 enhancer core might have acted also as a hypermorph in cis with the wild-type allele of FOXF1 during lung development. This SNV is particularly interesting as it is located only 2 kb from the two regulatory SNVs discussed above, has a low MAF of 2.855% and like the two other putatively hypermorphic SNVs, was absent in 13 tested unrelated ACDMPV patients with the FOXF1 enhancer deletion. This variant might have altered the affinity of MAFK, RAD21, or even TFAP2 to their binding sites, increasing the expression of FOXF1 in the mother during her lung development. MAFK is a lung-expressed transcriptional repressor (Onodera et al. 1999; Sakurai et al. 2016), whereas RAD21 (Hu et al. 2018) is a subunit of the CTCF-associated cohesion complex essential for maintaining chromatin architecture.
In sum, our studies suggest that rare non-coding SNVs present within a regulatory region of a disease-implicated FOXF1 might modify the expressivity and/or the penetrance of the lethal ACDMPV phenotype. They also pinpoint the underappreciated role of non-coding variants in congenital disorders. Finally, they continue to strengthen the observation that almost 90% of disease-associated SNVs identified in genome-wide association studies do not localize to protein-coding sequences (Welter et al. 2014).
References
Abdallah HI, Karmazin N, Marks LA (1993) Late presentation of misalignment of lung vessels with alveolar capillary dysplasia. Crit Care Med 21:628–630
Ahmed S, Ackerman V, Faught P, Langston C (2008) Profound hypoxemia and pulmonary hypertension in a 7-month-old infant: late presentation of alveolar capillary dysplasia. Pediatr Crit Care Med 9:e43–e46
Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P et al (2012) Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 44:435–439
Astorga J, Carlsson P (2007) Hedgehog induction of murine vasculogenesis is mediated by Foxf1 and Bmp4. Development 134:3753–3761
Bishop NB, Stankiewicz P, Steinhorn RH (2011) Alveolar capillary dysplasia. Am J Respir Crit Care Med 184:172–179
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Campbell IM, Yuan B, Robberecht C, Pfundt R, Szafranski P, McEntagart ME, Nagamani SC, Erez A, Bartnik M, Wiśniowiecka-Kowalnik B et al (2014) Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet 95:173–182
Challis D, Yu J, Evani US, Jackson AR, Paithankar S, Coarfa C, Milosavljevic A, Gibbs RA, Yu F (2012) An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinform 13:8
Chen XF, Zhu DL, Yang M, Hu WX, Duan YY, Lu BJ, Rong Y, Dong SS, Hao RH, Chen JB et al (2018) An osteoporosis risk SNP at 1p36.12 acts as an allele-specific enhancer to modulate LINC00339 expression via long-range loop formation. Am J Hum Genet 102:776–793
Dello Russo P, Franzoni A, Baldan F, Puppin C, De Maglio G, Pittini C, Cattarossi L, Pizzolitto S, Damante G (2015) A 16q deletion involving FOXF1 enhancer is associated to pulmonary capillary hemangiomatosis. BMC Med Genet 16:94
Eckert D, Buhl S, Weber S, Jäger R, Schorle H (2005) The AP-2 family of transcription factors. Genome Biol 6:246
Edwards JJ, Murali C, Pogoriler J, Frank DB, Handler SS, Deardorff MA, Hopper RK (2019) Histopathologic and genetic features of alveolar capillary dysplasia with atypical late presentation and prolonged survival. J Pediatr 210:214–219
Fernandes-Silva H, Correia-Pinto J, Moura RS (2017) Canonical sonic hedgehog signaling in early lung development. J Dev Biol 5:E3
Galambos C, Sims-Lucas S, Ali N, Gien J, Dishop MK, Abman SH (2015) Intrapulmonary vascular shunt pathways in alveolar capillary dysplasia with misalignment of pulmonary veins. Thorax 70:84–85
Gialmanidis IP, Bravou V, Petrou I, Kourea H, Mathioudakis A, Lilis I, Papadaki H (2013) Expression of Bmi1, FoxF1, Nanog, and γ-catenin in relation to hedgehog signaling pathway in human non-small-cell lung cancer. Lung 191:511–521
Ho UY, Wainwright BJ (2017) Patched1 patterns fibroblast growth factor 10 and Forkhead box F1 expression during pulmonary branch formation. Mech Dev 147:37–48
Hu W, Pei W, Zhu L, Nie J, Pei H, Zhang J, Li B, Hei TK, Zhou G (2018) Microarray profiling of TGF-β1-induced long non-coding RNA expression patterns in human lung bronchial epithelial BEAS-2B cells. Cell Physiol Biochem 50:2071–2085
Huang L, Chen M, Pan J, Yu W (2018) Circular RNA circNASP modulates the malignant behaviors in osteosarcoma via miR-1253/FOXF1 pathway. Biochem Biophys Res Commun 500:511–517
Ito Y, Akimoto T, Cho K, Yamada M, Tanino M, Dobata T, Kitaichi M, Kumaki S, Kinugawa Y (2015) A late presenter and long-term survivor of alveolar capillary dysplasia with misalignment of the pulmonary veins. Eur J Pediatr 174:1123–1126
Janney CG, Askin FB, Kuhn C (1981) Congenital alveolar capillary dysplasia–an unusual cause of respiratory distress in the newborn. Am J Clin Pathol 76:722–727
Kalin TV, Meliton L, Meliton AY, Zhu X, Whitsett JA, Kalinichenko VV (2008) Pulmonary mastocytosis and enhanced lung inflammation in mice heterozygous null for the Foxf1 gene. Am J Respir Cell Mol Biol 39:390–399
Kalinichenko VV, Lim L, Stolz DB, Shin B, Rausa FM, Clark J, Whitsett JA, Watkins SC, Costa RH (2001) Defects in pulmonary vasculature and perinatal lung hemorrhage in mice heterozygous null for the Forkhead Box f1 transcription factor. Dev Biol 235:489–506
Kalinichenko VV, Gusarova GA, Kim IM, Shin B, Yoder HM, Clark J, Sapozhnikov AM, Whitsett JA, Costa RH (2004) Foxf1 haploinsufficiency reduces Notch-2 signaling during mouse lung development. Am J Physiol Lung Cell Mol Physiol 286:L521–L530
Karolak JA, Vincent M, Deutsch G, Gambin T, Cogné B, Pichon O, Vetrini F, Mefford HC, Dines JN, Golden-Grant K et al (2019) Complex compound inheritance of lethal lung developmental disorders due to disruption of the TBX-FGF pathway. Am J Hum Genet 104:213–228
Kim W, Kim E, Lee S, Kim D, Chun J, Park KH, Youn H, Youn B (2016) TFAP2C-mediated upregulation of TGFBR1 promotes lung tumorigenesis and epithelial-mesenchymal transition. Exp Mol Med 48:e273
Kodama Y, Tao K, Ishida F, Kawakami T, Tsuchiya K, Ishida K, Takemura T, Nakazawa A, Matsuoka K, Yoda H (2012) Long survival of congenital alveolar capillary dysplasia patient with NO inhalation and epoprostenol: effect of sildenafil, beraprost and bosentan. Pediatr Int 54:923–926
Langston C (1991) Misalignment of pulmonary veins and alveolar capillary dysplasia. Pediatr Pathol 11:163–170
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760
Lim L, Kalinichenko VV, Whitsett JA, Costa RH (2002) Fusion of lung lobes and vessels in mouse embryos heterozygous for the forkhead box f1 targeted allele. Am J Physiol Lung Cell Mol Physiol 282:L1012–L1022
Lo PK, Lee JS, Liang X, Han L, Mori T, Fackler MJ, Sadik H, Argani P, Pandita TK, Sukumar S (2010) Epigenetic inactivation of the potential tumor suppressor gene FOXF1 in breast cancer. Cancer Res 70:6047–6058
Mahlapuu M, Pelto-Huikko M, Aitola M, Enerbäck S, Carlsson P (1998) FREAC-1 contains a cell-type-specific transcriptional activation domain and is expressed in epithelial-mesenchymal interfaces. Dev Biol 202:183–195
Mahlapuu M, Enerbäck S, Carlsson P (2001) Haploinsufficiency of the forkhead gene Foxf1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 128:2397–2406
Martin V, Zhao J, Afek A, Mielko Z, Gordân R (2019) QBiC-Pred: quantitative predictions of transcription factor binding changes due to sequence variants. Nucleic Acids Res 47:W127–W135
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Michalsky MP, Arca MJ, Groenman F, Hammond S, Tibboel D, Caniano DA (2005) Alveolar capillary dysplasia: a logical approach to a fatal disease. J Pediatr Surg 40:1100–1105
Murphy DB, Wiese S, Burfeind P, Schmundt D, Mattei MG, Schulz-Schaeffer W, Thies U (1994) Human brain factor 1, a new member of the fork head gene family. Genomics 21:551–557
Onodera K, Shavit JA, Motohashi H, Katsuoka F, Akasaka JE, Engel JD, Yamamoto M (1999) Characterization of the murine mafF gene. J Biol Chem 274:21162–21169
Pierrou S, Hellqvist M, Samuelsson L, Enerbäck S, Carlsson P (1994) Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J 13:5002–5012
Saito RA, Micke P, Paulsson J, Augsten M, Peña C, Jönsson P, Botling J, Edlund K, Johansson L, Carlsson P et al (2010) Forkhead box F1 regulates tumor-promoting properties of cancer-associated fibroblasts in lung cancer. Cancer Res 70:2644–2654
Sakurai T, Isogaya K, Sakai S, Morikawa M, Morishita Y, Ehata S, Miyazono K, Koinuma D (2016) RNA-binding motif protein 47 inhibits Nrf2 activity to suppress tumor growth in lung adenocarcinoma. Oncogene 35:5000–5009
Sen P, Thakur N, Stockton DW, Langston C, Bejjani BA (2004) Expanding the phenotype of alveolar capillary dysplasia (ACD). J Pediatr 145:646–651
Sen P, Gerychova R, Janku P, Jezova M, Valaskova I, Navarro C, Silva I, Langston C, Welty S, Belmont J, Stankiewicz P (2013a) A familial case of alveolar capillary dysplasia with misalignment of pulmonary veins supports paternal imprinting of FOXF1 in human. Eur J Hum Genet 21:474–477
Sen P, Yang Y, Navarro C, Silva I, Szafranski P, Kolodziejska KE, Dharmadhikari AV, Mostafa H, Kozakewich H, Kearney D et al (2013b) Novel FOXF1 mutations in sporadic and familial cases of alveolar capillary dysplasia with misaligned pulmonary veins imply a role for its DNA binding domain. Hum Mutat 34:801–811
Sen P, Dharmadhikari AV, Majewski T, Mohammad MA, Kalin TV, Zabielska J, Ren X, Bray M, Brown HM, Welty S et al (2014) Comparative analyses of lung transcriptomes in patients with alveolar capillary dysplasia with misalignment of pulmonary veins and in Foxf1 heterozygous knockout mice. PLoS One 9:e94390
Seo H, Kim J, Park GH, Kim Y, Cho SW (2016) Long-range enhancers modulate Foxf1 transcription in blood vessels of pulmonary vascular network. Histochem Cell Biol 146:289–300
Shankar V, Haque A, Johnson J, Pietsch J (2006) Late presentation of alveolar capillary dysplasia in an infant. Pediatr Crit Care Med 7:177–179
Slot E, Edel G, Cutz E, van Heijst A, Post M, Schnater M, Wijnen R, Tibboel D, Rottier R, de Klein A (2018) Alveolar capillary dysplasia with misalignment of the pulmonary veins: clinical, histological, and genetic aspects. Pulm Circ 8:2045894018795143
Spielmann M, Mundlos S (2016) Looking beyond the genes: the role of non-coding variants in human disease. Hum Mol Genet 25:R157–R165
Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA, Ou Z, Wiszniewska J, Driscoll DJ, Maisenbacher MK et al (2009) Genomicand genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet 84:780–791
Szafranski P, Dharmadhikari AV, Brosens E, Gurha P, Kolodziejska KE, Zhishuo O, Dittwald P, Majewski T, Mohan KN, Chen B et al (2013) Small noncoding differentially methylated copy-number variants, including lncRNA genes, cause a lethal lung developmental disorder. Genome Res 23:23–33
Szafranski P, Dharmadhikari AV, Wambach JA, Towe CT, White FV, Grady RM, Eghtesady P, Cole FS, Deutsch G, Sen P, Stankiewicz P (2014) Two deletions overlapping a distant FOXF1 enhancer unravel the role of lncRNA LINC01081 in etiology of alveolar capillary dysplasia with misalignment of pulmonary veins. Am J Med Genet A 164A:2013–2019
Szafranski P, Gambin T, Dharmadhikari AV, Akdemir KC, Jhangiani SN, Schuette J, Godiwala N, Yatsenko SA, Sebastian J, Madan-Khetarpal S (2016a) Pathogenetics of alveolar capillary dysplasia with misalignment of pulmonary veins. Hum Genet 135:569–586
Szafranski P, Herrera C, Proe LA, Coffman B, Kearney DL, Popek E, Stankiewicz P (2016b) Narrowing the FOXF1 distant enhancer region on 16q24.1 critical for ACDMPV. Clin Epigenet 8:112
Szafranski P, Kośmider E, Liu Q, Karolak JA, Currie L, Parkash S, Kahler SG, Roeder E, Littlejohn RO, DeNapoli TS et al (2018) LINE- and Alu-containing genomic instability hotspot at 16q24.1 associated with recurrent and nonrecurrent CNV deletions causative for ACDMPV. Hum Mutat 39:1916–1925
Tamura M, Sasaki Y, Koyama R, Takeda K, Idogawa M, Tokino T (2014) Forkhead transcription factor FOXF1 is a novel target gene of the p53 family and regulates cancer cell migration and invasiveness. Oncogene 33:4837–4846
Towe CT, White FV, Grady RM, Sweet SC, Eghtesady P, Wegner DJ, Sen P, Szafranski P, Stankiewicz P, Hamvas A et al (2018) Infants with a typical presentations of alveolar capillary dysplasia with misalignment of the pulmonary veins who underwent bilateral lung transplantation. J Pediatr 194:158–164
Wang S, Yan S, Zhu S, Zhao Y, Yan J, Xiao Z, Bi J, Qiu J, Zhang D, Hong Z et al (2018) FOXF1 induces epithelial-mesenchymal transition in colorectal cancer metastasis by transcriptionally activating SNAI1. Neoplasia 20:996–1007
Wei HJ, Nickoloff JA, Chen WH, Liu HY, Lo WC, Chang YT, Yang PC, Wu CW, Williams DF, Gelovani JG, Deng WP (2014) FOXF1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget 5:9514–9529
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H (2014) The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42:D1001–D1006
Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, Shen Y, Yu G, Liu J, Xie H et al (2015) TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med 372:341–350
Xiang Y, Qin XQ, Liu HJ, Tan YR, Liu C, Liu CX (2012) Identification of transcription factors regulating CTNNAL1 expression in human bronchial epithelial cells. PLoS One 7:e31158
Acknowledgements
This work was supported by grants awarded by National Heart, Lung, and Blood Institute (NHLBI) Grant R01HL137203 to PSt and National Organization for Rare Disorders (NORD) Grant 16001 to PSz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Szafranski, P., Liu, Q., Karolak, J.A. et al. Association of rare non-coding SNVs in the lung-specific FOXF1 enhancer with a mitigation of the lethal ACDMPV phenotype. Hum Genet 138, 1301–1311 (2019). https://doi.org/10.1007/s00439-019-02073-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-019-02073-x