
Abstract
Through four decades’ development, DNA sequencing has inched into the era of single-molecule sequencing (SMS), or the third-generation sequencing (TGS), as represented by two distinct technical approaches developed independently by Pacific Bioscience (PacBio) and Oxford Nanopore Technologies (ONT). Historically, each generation of sequencing technologies was marked by innovative technological achievements and novel applications. Long reads (LRs) are considered as the most advantageous feature of SMS shared by both PacBio and ONT to distinguish SMS from next-generation sequencing (NGS, or the second-generation sequencing) and Sanger sequencing (the first-generation sequencing). Long reads overcome the limitations of NGS and drastically improves the quality of genome assembly. Besides, ONT also contributes several unique features including ultra-long reads (ULRs) with read length above 300 kb and some close to 1 million bp, direct RNA sequencing and superior portability as made possible by pocket-sized MinION sequencer. Here, we review the history of DNA sequencing technologies and associated applications, with a special focus on the advantages as well as the limitations of ULR sequencing in genome assembly.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
DNA sequencing technologies have undergone massive changes and improvement during last four decades. It can be traced back to 1977 when Sanger partial chain termination method and Maxam–Gilbert chemical degradation method were first reported (Maxam and Gilbert 1977; Sanger and Coulson 1975; Sanger et al. 1977). The maximum sequence length generated by Sanger sequencing is about 1 kb.
Sanger sequencing requires large amount of DNA samples generally produced by cloning target sequences into vectors and amplified by prokaryotic cells such as E. coli. Invention of polymerase chain reaction (PCR) created a tremendous opportunity for later biotechnologies. PCR using oligos immobilized on flow cells enables clonal amplification of templates which is essential for next-generation sequencing (NGS) or the second-generation sequencing.
NGS comprises a number of robust technologies characterized by parallel sequencing of massive DNA molecules. There were four major NGS platforms made commercially available in consecutive years: 454 system by Roche, GA/Solexa system by Illumina, SOLiD system by ABI, and Ion Torrent system by Life Technologies (Goodwin et al. 2016; Liu et al. 2012; Mardis 2013). Over the past decade, Illumina emerged as dominant provider of sequencers due to their lower cost, high speed and high yield.
Illumina has released a series of instruments to fulfill the need of various output demands. Some of these sequencers produce large amount of short reads (billions of reads could be generated from a single run) of ≤ 300 bp for complex eukaryotic genomes or small microbial genomes in relatively a short period of time (Liu et al. 2012). Due to this virtue, Illumina sequencers have gained popularity in their applicability and NGS has been widely deployed to explore various domains of genomics including oncology, microbiology, environmental genomics, metagenomics, etc., for biological, medical, environmental, and agricultural studies (Ashley 2016; Deurenberg et al. 2017; Gardy and Loman 2018; Giampaoli et al. 2018; Hoper et al. 2016; Scheben et al. 2017; Yuan et al. 2017). However, the read length remains a bottleneck for biological studies (Ulahannan et al. 2013).
Limitations of NGS
NGS is advantageous in many aspects such as low cost, high speed and high yield and has been intensively employed in various biological analyses during the past 15 years. Several studies have been conducted to solve same biological question with different NGS and TGS methods. These works, however, have led to the discovery of a number of intrinsic limitations of NGS which may have significant impact on the accuracy of biological studies. Among the bottlenecks, short-read length is the most noticeable drawback for NGS sequencing. This limits the precision of many biological studies, especially genome assembly and transcriptome analysis.
For genome assembly, to deduce the genome sequences from billions of short reads, one has to face computational challenges resulted from genomic complexity, time and hardware limitations. These challenges have become a critical issue for large genome assembly, because short reads often result in highly fragmented assemblies resulted from unsolvable repetitive regions or regions with high GC content (Petersen et al. 2017; Salzberg and Yorke 2005; Schmutz et al. 2004). Using short reads in analysis of segmental duplication, structural variations (SVs) or paralogous regions may result in a significant number of false positives (Guan and Sung 2016; Treangen and Salzberg 2011). This issue was empirically addressed by a number of reports. Despite the advances in sequencing technologies and bioinformatics, de novo assembly of large genomes remains challenging (Chin et al. 2014).
Complexities resulting from heterozygosity, transposable elements, GC-rich regions, tandem repeats and interspersed repetitive regions of 10 kb–10 Mb or more in genome remain unresolved by NGS short reads (Alkan et al. 2011). Sequencing of polymorphic tandem repeats in the genome by NGS is severely impaired by read length (Mousavi et al. 2018), and the read length of 100–250 bases used for determining the TR expansion might be inaccurate (Bahlo et al. 2018). For sequencing complex chromosomal rearrangements and structural variants, previous analytical approaches such as fluorescent in situ hybridization (FISH), array CGH, PCR and NGS are either laborious or imprecise (Pang et al. 2010; Quinlan and Hall 2012). Short paired-end reads, although being able to offer single base precision, are frequently unable to precisely map the repetitive regions (e.g., trinucleotide repeats) (Tattini et al. 2015). On the other hand, SMS long-read length offers a superior alternative for characterization of CGRs (Chaisson et al. 2015; Huddleston et al. 2017; Merker et al. 2018; Spies et al. 2017).
Transcriptome data analysis also encounters similar constrains of short reads as that of in genome assembly. Short reads are often unable to infer the full-length RNA transcripts or to precisely determine specific isoforms (Bayega et al. 2018; Martin and Wang 2011). Because of this problem with short reads, studies were unable to fully address gene regulation, protein-coding potential of genome and phenotypic diversity.
NGS is also limited by its incapability of direct sequencing of RNA and epigenetic/methylation markers. RNA sequencing by NGS requires conversion of RNA molecules to corresponding cDNA molecules and then sequenced as DNA. This procedure is seen in all aspects of biological studies, especially transcription of protein-coding genes and non-coding genes (Costa-Silva et al. 2017). Epigenetic modification plays a vital role in regulation of eukaryotic gene expression. Previous DNA methylation largely relies on 5-methylcytosine (5mC) bisulfite conversion. Although genome-wide 5mC profiling became feasible by NGS, it is still limited by uneven coverage as well as sequencing and mapping artifacts (Nair et al. 2018; Smith et al. 2009; Warnecke et al. 2002).
Moreover, short-read sequencing normally involves the usage of large equipment and laborious experimental procedures and bioinformatics analysis and thus unable to meet the need for fast field testing. The process from transportation of biological samples from one place to another, preparation of sequencing libraries, sequencing and data analysis may take a long time (Quick et al. 2016).
Development of single-molecule sequencing
Shortcomings of NGS fostered the development of single-molecule sequencing (SMS), or the third-generation sequencing (TGS). Evolvement of SMS resulted in an increase of 1–2 orders of magnitude in read length over Sanger sequencing as achieved by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), with a small portion of reads by the latter, may achieve an increase of 3 orders of magnitude (Giordano et al. 2017; Jain et al. 2018; Judge et al. 2015; Weirather et al. 2017).
The development of SMS has been phenomenal, as a number of crucial technologies being developed using cross-disciplinary expertise. Firstly, pores (for Nanopore sequencing) or wells (for PacBio sequencing) at micron scale were made and immobilized on well-designed matrix to harbor single nucleic acid molecules for sequencing (Benner et al. 2007; Eid et al. 2009). Secondly, novel sequencing mechanisms were implemented to detect single fluorescent signals (by PacBio) or to distinguish electrical changes (by Nanopore), which are then converted to nucleotide bases as well as base modifications (Hartel et al. 2019; Rhoads and Au 2015). Thirdly, extremely delicate detection systems were designed independently to detect base-level signals (Benner et al. 2007; Clark et al. 2011). Moreover, a number of software tools were developed for base calling and error correction.
Similar to Sanger sequencing and most NGS platforms, PacBio sequencing also adopts sequencing-by-synthesis mechanism where a DNA strand serves as template for the synthesis of another strand by DNA polymerase. Single-molecule real-time (SMRT) sequencing developed by PacBio, founded in 2004, was the first widely deployed long-read sequencing technology. Reads generated by SMRT sequencing can reach about 200 kb. This feature gives an edge to long-read sequencing technologies in base-to-base comparisons of genomes to identify genetic variations and further understanding of the gene functions and disease association with significantly improved accuracy. RS system, the first SMRT sequencer released in 2009, sequences circular single-stranded DNA templates (i.e., SMRTbell) in real time. SMRTbell is made by ligating hairpin adaptors to both ends of a target double-stranded DNA molecule. Each hairpin loop has a sequencing primer-binding site for primer binding (Ardui et al. 2018; Travers et al. 2010).
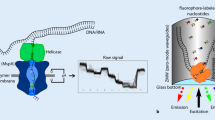
DNA template-polymerase complexes are built and immobilized at the bottom of zero-mode waveguide (ZMW) chambers (Fig. 1). When a fluorescently labeled nucleotide base is incorporated, a light pulse is generated and recorded. Differing from NGS which has fluorescence labeled on the nucleobase moieties, fluorophores of SMRT sequencing are labeled on the terminal phosphate of nucleotides. The formation of phosphodiester bond automatically removes the fluorophore together with the conjugated PPi. Base-linked labeling of fluorophores was found not suitable for SMRT sequencing due to poor incorporation (Ardui et al. 2018; Eid et al. 2009; Rhoads and Au 2015).

Figure reproduced by copyright permission of AAAS
SMRT sequencing. a Graphic representation of ZMW chamber with DNA polymerase bound single molecule of DNA template. b Association formed between phospholinked nucleotide with the template in the polymerase active site which leads to elevation of the fluorescence signal on respective color channels. Phosphodiester bond formatted further releases dye–linker–pyrophosphate product and diffuses it out of ZMW, and ends the fluorescence pulse. The polymerase shifts to next position, forms bond with nucleotide and generates next pulse. (Eid et al. 2009)
General features of PacBio sequencers are listed in Table 1.
Nanopore sequencing by ONT, founded in 2005, is based on an innovative technology capable of distinguishing minor changes in ionic current when nucleotide bases of single-stranded DNA/RNA molecules pass through protein nanopores immobilized on a saline solution-immersed membrane to which a fixed voltage is applied (Fig. 2).

Nanopore sequencing. DNA or RNA molecules are introduced into nanopores by motor protein and then each causes alterations in electric signal when passing through the pore. These signal fluctuations are further converted into basecalls with special algorithms. (Image adopted from Oxford Nanopore Technologies website.)
Under an applied voltage, the 5′ end of ssDNA is electrophoretically fed into a matrix-embedded protein nanopore, a mutant form of Mycobacterium smegmatis porin A (MspA) designed in such a way that it has a short and narrow constriction (about 1.2 nm wide and 0.6 nm long) to achieve single-nucleotide resolution (Manrao et al. 2012). When a DNA molecule passes through the nanopore, distinct current signals are obtained with respect to each nucleotide. The alterations in ionic current are recorded for each pore and converted to a base sequence previously by Hidden Markov Model-based or currently by Recurrent Neural Network-based basecaller (Boza et al. 2017; Hartel et al. 2019). ONT developed portable sequencer MinION to expedite real-time field sequencing of small amount of DNA samples from biological or environmental samples (Lu et al. 2016). PromethION is ONT’s benchtop instrument that contains 48 flow cells that can be run in parallel and generate up to 100 Gb of data per flow cell per run. General features of ONT sequencers are listed in Table 2. Nanopores can be used to sequence both plus and minus strands of DNA fragments with customized library preparation kits (2D, now 1D2) to increase the sequencing accuracy. Also, ONT’s sequencers allow RNA molecules to be sequenced directly (Keller et al. 2018).
The longest read length achieved by MinION flow cell to this date is more than 2 Mb (Payne et al. 2018). Ultra-long reads (ULRs) are an important feature with a strong potential to facilitate the assembly of large genomes, which was demonstrated by the assembly of a contiguous yeast genome by MinION long reads (LRs) (Istace et al. 2017), followed by human genome assembly by MinION LRs plus ULRs without Illumina short reads (Jain et al. 2018), which will be further discussed later.
Comparison between PacBio and ONT platforms
PacBio and ONT share a common advantage of long-read length and a common disadvantage of high error rate of ~ 5–20% randomly distributed before error correction (Sedlazeck et al. 2018). As such, Illumina short reads are frequently incorporated with long reads for hybrid assembly of large eukaryotic genomes (Antipov et al. 2016; Giordano et al. 2017) or for hybrid sequencing of transcriptomes to characterize transcript isoforms or fusion genes (Deonovic et al. 2017; Weirather et al. 2015).
PacBio and ONT also differ in a number of aspects. To better understand the performances between PacBio and ONT platforms, here we describe three reports separately related to genome assembly (Giordano et al. 2017), transcriptome analysis (Weirather et al. 2017), and structural variant calling (Sedlazeck et al. 2018). Since every sequencing platform endeavors to improve its sequencing quality, the pros and cons of a sequencing platform are expected to shift as new sequencers and bioinformatics continue to evolve. Readers are encouraged to update knowledge through frequent literature search. Here, we summarize some of the reports.
Giordano and colleagues conducted a comprehensive comparison on genome assembly efficiency between PacBio RSII (read length between 5–60 kb, average 12 kb, error rate 13%, accuracy 99.9% after correction, throughput 1 Gb/run), ONT MinION (2D, R7.3/R9 flow cells), and Illumina MiSeq (80 × of 2 × 150 bp) at various read depth using four yeast strains of S. cerevisiae genome (Giordano et al. 2017). They found that RSII performed much better in throughput per run and slightly better in accuracy than MinION. Furthermore, both sequencers performed similarly in error rates and accuracy. Moreover, at 31X coverage, either RSII or MinION reads alone were able to complete the assemblies of the 16 nuclear chromosomes, but not the mitochondrial genome. Moreover, with Illumina short reads, both PacBio and ONT’s datasets could achieve an accuracy above 99.9%. It is worthy to note that, since 2017, however, the output and quality have been significantly improved for MinION flow cell. Read length of MinION can be much longer than that of RSII for both average read length and longest read length.
They also evaluated eight assembly pipelines (namely, PBcR-Self, PBcR-MiSeq, Canu, Falcon, ABruijn, SMARTdenovo, Miniasm and Racon) at various depth and with or without base error correction or consensus construction prior to assembly (Chin et al. 2016; Jayakumar and Sakakibara 2019; Koren et al. 2012, 2017; Li 2016; Lin et al. 2016; Vaser et al. 2017). Miniasm turned out to be the fastest. However, it missed 4–5% of the genome, because of no base error correction. On the other hand, PBcR-MiSeq was the most accurate (> 99.68% reference genome covered with identity above 99.9%), as it included MiSeq short reads to correct long reads produced either by MinION or RSII. These results indicate the importance of error correction and superiority of hybrid assembly over assembly by long reads alone.
A comparison between PacBio RSII and ONT MinION Mk 1B on transcriptomes of human embryonic stem cells (hESCs) was reported by Weirather and colleagues (Weirather et al. 2017). In the study, short reads produced by HiSeq 4000 were used in Hybrid-Seq libraries to evaluate the essentiality of short reads for full-length transcript sequencing and characterization of transcript isoforms. Results indicated that RSII had better quality in terms of error rate (error rate 1.72% in PacBio CCS vs 13.4% in ONT 2D), while ONT has higher yield and throughput/cost ratio, and that both SMS platforms are suitable for full-length transcriptome analysis. In general, Hybrid-Seq strategy performs slightly better in fully utilizing PacBio and ONT reads in transcriptome analysis as both SMS platforms can benefit from short reads for improving accuracy (median errors reduced to 0.05% from 0.13% in ONT-Illumina vs. ONT).
A comparison between PacBio and ONT on accuracy of structural variant calling using a well-studied human genome NA12878 was reported by Sedlazeck and colleagues (Sedlazeck et al. 2018). From 28X ONT data and 55X PacBio data, 26,567 and 15,499 SVs were identified, respectively. Most (94.80%) SVs identified from PacBio data were confirmed by ONT, Illumina or other call sets, while ONT had worse concordance—among the 11,433 SV calls unique to ONT, most (96.01%) were deletions and 92.88% were within homopolymers or other repeats. Contrarily, among the 773 SV calls unique to PacBio, mostly (66.49%) were insertions and only 41.8% were within homopolymers or repeats. The bias in ONT is suspected to result from errors in base calling. However, the results were influenced by coverage. The authors also showed that 15X PacBio reads can call 69.64% of SVs at precision rate about 80% whereas number of calls can be increased to about 80% with precision rate of ~ 85% at coverage of 30X. Subsampled ONT data at 20X coverage were able to call 84.23% of SVs at precision rate of 82.24% which was better than PacBio data at 30X .
Besides the above-mentioned issues, sequencing speed is also a critical factor. For better accuracy, the speed and output of PacBio sequencing are compromised to only a few bp per second, while ONT system can reach above 400 bp per second. Key differences between PacBio and ONT sequencers are summarized in Table 3.
Ultra-long reads as a unique feature of ONT
Using ONT MinION sequencer, Jain et al. (2018) assembled human genome using Canu assembler on 30X coverage composed of ULRs and LRs, without Illumina short reads. In a total of 53 R9.4 flow cells used, they found that ULRs could be better produced from high molecular weight DNA freshly extracted from cells. To the best of our knowledge, ULR has not been demonstrated by PacBio sequencers.
Comparison made by Jain et al. between ONT and PacBio reads also showed that both with read correction and equivalent coverage, genome assembled from ONT reads has lower identity to reference genome GRCh38 than that assembled from PacBio reads (92% vs. > 99%) and the frequency of deletions is also higher for ONT reads—as caused by homopolymers and low complexity regions, suggesting that ONT is more error prone than PacBio. However, as also reported in the comparison, errors in ULRs distribute more or less evenly and do not increase with read length.
To further demonstrate the ULRs mentioned in the report by Jain et al., here we display ONT data that were used in their study (https://github.com/nanopore-wgs-consortium/NA12878/blob/master/Genome.md), together with several other PacBio and ONT datasets from recent studies (Fig. 3) (Supplementary Tables 1–3). Based on previous reports and to have a reasonable separation, we temporarily define ULRs as those with read length ≥ 300 kb.

Comparison of read length between ONT and PacBio datasets. Recent datasets generated by ONT or PacBio were analyzed by NanoPack tool (De Coster et al. 2018) (https://github.com/wdecoster/nanopack). (ONT datasets: JAIN (MinION), ONT1 (MinION), ONT2 (MinION), ONT3 (PromethION), ONT4 (PromethION); PacBio datasets: PB0 (RS II), PB1 (Sequel), PB2 (Sequel II)). Numbers written below the blue line are number of reads with length ≥ 100,000 bp per million of total reads. Numbers written above the red line are number of reads with length ≥ 300,000 bp per million of total reads
Figure 3 shows the result from comparison of ONT and PacBio datasets (Supplementary Tables 1–3). We observed that although the median read length of ONT data is comparable to that of PacBio, a small portion of ONT reads was above 300 kb in length. At the same time, PacBio data do not contain any reads above 150 kb, but the N50 of these datasets varies depending on the sequencing protocol used. Size selection and sequencing kits are few of many factors.
It would be interesting to further understand how protocol influences the production of ULRs. We thus compare ONTs and the method used by Jain et al. for making more ULRs. Figure 4 shows the results generated from three protocols: one with standard ligation kit (SQK-LSK108 1D ligation kit), second with standard rapid kit (SQK-RAD002 genomic DNA kit) and third with a modified protocol using rapid kit (SQK-RAD002 genomic DNA Rapid kit) for input DNA extracted by modified Sambrook and Russell’s protocol, followed by MinION sequencing. Mean read length from modified protocol was about 3.5 and 1.5 times higher than that from standard ligation kit and standard rapid kit, respectively, while the N50 from the modified protocol was more than 8.5 and 2.5 times higher than that from standard ligation kit and standard rapid kit, respectively. ULRs (i.e., read length ≥ 300 kb) totaled around 2 Gb, 472 Mb and 84 Mb for modified protocol and standard ligation kit and standard rapid kit, respectively. Modified protocol is about 700-fold more efficient than ligation kit in producing ULRs (7000 ULR pm vs. 12 ULR pm).

Comparison of read lengths between libraries produced from different protocols with a focus on ultra-long reads. Reads produced from ligation kit, rapid kit and modified protocol (Ultra) were compared to highlight ultra-long reads. Figures are generated with NanoPack tools (https://github.com/wdecoster/nanopack). Numbers written below the blue line are number of reads with length ≥ 100,000 bp per million of total reads. Numbers written above the red line are number of reads with length ≥ 300,000 bp per million of total reads
The advantage of ULRs in genome assembly is evident as shown in the report. Interestingly, additional incorporation of 5X coverage of ULRs was able to increase NG50 by at least twofold (from ~ 3 Mb of long reads to ~ 6.4 Mb). Assembly contiguity also increased, while yield per flow cell was not compromised by ULRs and sequencing accuracy does not decrease along with the read lengths of ULRs.
Applications leveraging on single-molecule sequencing technologies De novo genome assembly
Long-read length benefits a lot to genome assembly by increasing N50 and contiguity, while short reads result in highly fragmented assemblies. Since long reads span through regions of high GC, low complexity and repetitive regions, they resolve the bubble formation in de bruijn graph-based assembly and also determine the lengths of microsatellites and tandem repeat regions.
Giordano et al. (2017) showed that yeast genome could be assembled with 31X of PacBio or ONT reads with accuracies about 99% and 98%, respectively. De novo assembly of GM12878 human genome with 30X Nanopore reads can produce an assembly with NG50 around 3 Mb, whereas adding 5X ULRs can increase the NG50 to ~ 6.4 Mb (Jain et al. 2018). Thus, SMS platforms offer a better solution for large genome assembly (Seo et al. 2016; Shi et al. 2016). In particular, ONT method that can provide reads up to 2 Mb would be extremely helpful for achieving high contiguity and resolving repetitive regions.
Sequencing tandem repeats in human diseases
Tandem repeats (TR) regions are short genomic regions ranging up to thousand base pairs that are repeated multiple times in human genome (de Koning et al. 2011). Although widely spread in non-coding regions, some tandem repeats can still be found in coding regions (Usdin 2008). It has been reported that up to 9% gene have tandem repeats within their coding region and about 12% of genes have tandem repeats in their promoter regions. These regions are hyper-mutable and are often used in forensics, population genetics and are associated with several genetic diseases. Compared to other genomic elements, the mutation rates of tandem repeats are 10 to 10,000 fold higher (Ameur et al. 2019; Duitama et al. 2014).
The read lengths offered by SMS can subtly profile tandem repeat regions and various diseases caused by tandem repeat expansions have been studied with both PacBio and ONT (Ameur et al. 2019; Loomis et al. 2013; Mitsuhashi et al. 2017).
McGinty et al. (2017) demonstrated the potential of nanopore sequencing in characterization of the role played by tandem repeats in chromosomal rearrangement and sequencing time is much shorter than PacBio sequencing. Using nanopore sequencing of 11 individuals (6 patients of Alzheimer’s), De Roeck et al. (2018, 2019) showed that variable number tandem repeat (VNTR) expansion results in increased risk of Alzheimer’s disease. They used NanoSatellite software for resolving tandem repeats from PromethION data. When compared together, both PacBio and ONT were able to sequence through the SCA36 ‘GGCCTG’ and the C9orf72 G4C2 repeat expansions. Both regions were cloned into plasmids and then sequenced with PacBio and ONT MinION. While median read length was found to be similar by both these platforms, MinION had a higher percentage of reads that spanned through these repeat regions (Ebbert et al. 2018).
Although both PacBio and ONT suffer high error rates. Ability of direct detection of nucleotides without DNA synthesis and, hence no GC bias, makes ONT sequencing lucrative to analyze tandem repeats (Bahlo et al. 2018). By employing the ONT sequencing, identification of novel tandem repeats associated with disease can be done in a cost-effective manner (Gießelmann et al. 2018).
Detecting complex chromosomal rearrangements and structural variants
Complex genomic rearrangements (CGRs) refer to insertions, deletions, inversions, duplications, and translocations of variable genomic sequences (Sudmant et al. 2015). These genomic sequences, which are frequently repetitive in sequence and polymorphic in structure and length, contribute to the etiology of a number of diseases, including cancer (de Koning et al. 2011; Macintyre et al. 2016; Tubio 2015), early-onset Alzheimer’s disease (Rovelet-Lecrux et al. 2006), and autism (Hedges et al. 2012).
McGinty et al. (2017) demonstrated the potential of nanopore sequencing to characterize the DNA repair pathways involved in (GAA)n-induced CGRs. In the study, they showed that ONT long reads can detect the CGR breakpoints with single base pair resolution. The intricacies of CGR would not have been discovered without long reads.
SVs characterization of genomic SVs by PE short reads often results in false negative or false positive, and long reads are more likely to span across questionable repetitive regions or the breakpoints of SVs. To facilitate alignment of SVs with long reads, open-source methods NGMLR and Sniffles were introduced by Sedlazeck and colleagues (Sedlazeck et al. 2018). A comparison between PacBio and ONT data using the mentioned methods was discussed above in “Comparisons between PacBio and ONT platforms”. In genomic analysis of two chromothripsis patients, comparisons between short-reads and long reads were made in identification of complex structural variants. They showed that 32% more SVs could be identified using NanoSV with long reads from ONT’s MinION as compared to short reads (Cretu Stancu et al. 2017).
Haplotype phasing of variants and dissecting the complexities of highly polymorphic regions MHC/HLA
A diploid human genome generally has 4–5 million sites that differ from a reference genome. Most genomic variations are heterozygous in nature and their density across the genome varies with ethnic, geographic background of parents (Eberle et al. 2017). This haplotype information of parental alleles affects the analysis of allele-specific expression, DNA-binding sites, de novo mutations and other genomic features. Due to the limitations of current methods, there is great interest to acquire haplotype information directly from the reads (Raymond et al. 2005; Tewhey et al. 2011). In simple words, phasing of variants can be achieved if they are present on the same read. Longer read can cover maximum variants to be phased, but read length, sequencing errors and fluctuating coverage could be major limiting factors which may induce false positive and true negatives (Cretu Stancu et al. 2017; Delaneau et al. 2013; Laver et al. 2016).
The hyperpolymorphic major histocompatibility complex (MHC) or human leukocyte antigen (HLA) encodes for proteins of antigen presentation pathway. The variations in 3.6 Mb genomic region of MHC located on chromosome 6p21 are associated with immune response. These genes define susceptibility or resistance for various infections as well as confer hypersensitivities to specific drugs (Traherne 2008; Trowsdale 1993).
In clinical practice, precise HLA genotyping is imperative before allogeneic hematopoietic stem cell transplants or organ transplants for ascertaining the compatibility of HLA between donor and recipient (Sasazuki et al. 2016). However, unambiguous HLA genotyping is technically challenging due to high polymorphism, high sequence similarity and extreme linkage disequilibrium (Profaizer et al. 2016). Till date, 24,093 allelic variants are identified in human genomes (http://www.ebi.ac.uk/ipd/imgt/hla/stats.html).
The advent of NGS greatly benefited HLA genotyping techniques as it offered higher throughput and better resolution than previous technologies (Erlich et al. 2011; Ozaki et al. 2015; Wang et al. 2012). Although the NGS methods provide good resolution, time taken in sequencing, phasing of HLA genes and associated regulatory regions remains a challenge (Hosomichi et al. 2015). The TGS techniques offers longer read lengths and provide sequence information of HLA regions which are hard to assemble with short reads facilitating identification of novel alleles, phasing and haplotype identification(Ambardar and Gowda 2018; Lang et al. 2018; Turner et al. 2018). Despite the high error rate, TGS technologies provided 100% accuracy in class I HLA typing at two-field resolution (Liu et al. 2018). To construe HLA architecture, combination of both NGS and TGS can ameliorate clinical practices.
RNA sequencing to detect alternative splicing/transcripts or RNA isoforms
On the other hand, long-read RNA sequencing is a superior approach over NGS short-read RNA-Seq in detecting alternative splicing transcripts or transcript isoforms, due to the fact that short reads are unable to span fully across gene transcripts and uneven coverages of inter-exonic or intra-exonic regions may fluctuate severely, making it difficult to be interpreted by bioinformatics means (Bayega et al. 2018; Steijger et al. 2013). SMS long-read sequencing has been found particularly useful in comprehensive characterization of RNA isoforms at various levels including single cell transcriptome analysis (Boldogkoi et al. 2019).
In a study of using RNA-seq approach together with Nanopore MinION long-read sequencing to investigate isoform diversity in individual B cells, Byrne et al. (2017) showed that complex isoforms can be precisely quantified at the single cell level. Their approach can be very useful for the study of immune response. In another analysis of alternative splicing transripts in Amborella trichopoda plant, Liu and colleagues demonstrated the feasibility of using PacBio Iso-Seq long reads to identify alternative splicing events without a reference genome (Liu et al. 2017).
Direct sequencing of RNA
RNA is recognized as a crucial component to interrogate biological phenomena and direct RNA sequencing is gaining a new momentum through direct sequencing using ONT platform.
For PacBio SMRT sequencing method, cDNA prepared from RNA can be used as input without undergoing fragmentation step, thereby enabling full-length information of RNA. Since chemistry of ONT sequencing involves the usage of nanopores through which either DNA or RNA molecule can pass. ONT permits direct RNA sequencing. A recent study of herpes simplex virus type 1 transcriptome by direct RNA-seq by Depledge et al. (2019) demonstrated the superior capability of direct RNA-seq in redefining transcriptional complexity, as novel class of chimeric transcripts was detected. They also stated that high error rate can be partially overcome by error correction using Illumina short reads. A direct sequencing of influenza RNA genome was reported by Keller et al.(2018), who designed an adapter to conserved termini of the viral genome and thus to direct the (-) sense RNA into MinION protein nanopore for direct sequencing. Taken together, nanopore direct RNA sequencing possesses ample advantages and is expected to benefit the understanding of host–pathogen interactions.
Direct sequencing of epigenetic/methylation markers
Both PacBio and ONT methods are proven to be much advantageous than current bisulfite method as they provide direct identification of various nucleotide methylation not just limited to 5mC (Clarke et al. 2009; McIntyre et al. 2019; Rand et al. 2017; Simpson et al. 2017; Xiao et al. 2018).
Euskirchen and colleagues screened glioma samples to identify copy number variations and methylation profiles in IDH1, IDH2, H3F3A, TP53 and TERT promoters using deep amplicon sequencing by Nanopore MinION Mk 1B/R9 or R9.4 flow cells (Euskirchen et al. 2017). Study was designed to achieve same-day detection of structural variants, point mutations, and methylation profiling using a nanopore device. A significant correlation was observed in outcomes of nanopore sequencing and data generated from short-read exome sequencing, Sanger sequencing, SNP array, and/or genome-wide methylation microarray. Nanopore sequencing method outperformed hybridization-based methods and current sequencing technologies in time consumed in diagnosis and laboratory equipment and expertise required. Overall, ONT method can be applied for precision medicine development for cancer patients in limited resources, within short duration and cost-effective manner.
Fast sequencing in PGS for decision making
Preimplantation genetic screening (PGS) is the process of screening of all 23 pairs of chromosomes to identify genetic defects within embryo prior to implantation. Success of in vitro fertilization (IVF) depends on the selection of viable embryo, which was previously based on morphological, developmental characteristics and chromosomal status (Lee et al. 2015b). The reliability of these criteria was found very low and new methods for detailed assessment of genetic defects and aneuploidy are desired (Fragouli et al. 2010; Lee et al. 2015a). NGS-based methods are advantageous over CGH-based methods in cost, detection of partial or segmental aneuploidy and mosaicism in multicellular samples, and automation (Fragouli et al. 2011; Yang et al. 2015). Friedenthal et al. (2018) compared CGH with NGS in single thawed euploid embryo transfer (STEET) and revealed that implantation rate and ongoing pregnancy/live birth rate were higher in NGS-based PGS.
There is very little information available about the usage of PacBio sequencing in PGS, while Nanopore MinION is suggested by Wei et al. (2018) to be a faster sequencing tool for PGS. They showed that the whole procedure of embryo selection can be completed within a day, making it a protocol faster than any NGS-based method. This study showed that results obtained with Nanopore sequencing methods were comparable with those of other NGS-based methods. Given that traditional NGS-based methods could be laborious, longer and more costly. Nanopore MinION is less laborious, faster and cheaper for PGS decision making and thus is able to facilitate fresh embryo transfer and reduce stress and cost for patients.
A fast and portable sequencing method in investigating outbreak of human infectious diseases
In case of an outbreak of human infectious disease, the first and foremost task is to sequence the genome of the infectious agent. Analysis of the genomic sequence can help inferring viral evolution and facilitate the identification of genetic sequences crucial for its survival and transmission, and thus expedite diagnosis and treatment.
PacBio long-read sequencing is able to overcome some of the limitations such as repetitive sequences and high GC content. However, it also requires laborious laboratory setup, long runtime and high cost, causing limited usage in case of pathogenic outbreaks.
In a scenario of disease outbreak, portable and rapidly deployable setup is desired to lower the cost of transport and to accelerate the diagnostic process. ONT’s MinION has demonstrated its strong efficacy in handling Ebola outbreak in western Africa (Hoenen et al. 2016; Quick et al. 2016), and Lassa virus (LASV) outbreak during 2018 in Nigeria (Kafetzopoulou et al. 2019). A comparison between Illumina and ONT methods in metagenomic sequencing was conducted. Through recovering whole-genome sequences of Dengue virus and chikungunya virus directly from serum and plasma of patients, they demonstrated the feasibility of nanopore metagenomic sequencing at a lower requirement of resources (Kafetzopoulou et al. 2018). Nanopore devices can work in less favorable locations and conditions and do not need a sophisticated laboratory setup (Greninger et al. 2015; Hansen et al. 2018). These devices can also reduce risk and avoid expensive logistics in terms of cost and time incurred in carrying samples from place to place.
Summary
Many biological questions can be answered with various sequencing technologies available till date. Choice of any sequencing method to be employed in human genetics project is generally context dependent. Additionally, one can consider cost, accuracy, running time and technical biases of these methods. Improved read length in TGS/SMS technologies is a milestone in the field of human genetics. Both PacBio and ONT have been continuously putting efforts in upgrading their sequencing solutions toward increased read length, reduced error rate and cost of sequencing. Oxford Nanopore’s ability to generate ultra-long reads and to differentiate modified nucleotides at high speed are few of its advantages over PacBio methods. Longer reads can greatly benefit genome assemblies of complex organisms, resolving tandem repeats and complex structural rearrangements in human diseases, phasing of haplotypes, deciphering the MHC sequence and identifying correct isoforms of RNA. Direct detection of RNA molecules or epigenetic modification can overcome the need of reverse transcription in case of RNA sequencing and bisulfite treatment to decipher methylation. Sequencing solutions that reduce the analysis time would improve the decision making in PGS and also in case of disease outbreaks by pathogens.
Despite their advantages, long reads produced by ONT sequencing methods suffer a high error rate, which might hamper the accuracy of genome sequencing projects. This error rate is expected to reduce/diminish to certain extent by improving the signal detection systems of these sequencers. Many researchers have shown that although long-reads technologies have been developed, short reads have not lost their relevance yet. High accuracy rates of short reads and longer length of long reads can be combined to achieve better accuracy. Another limitation faced by projects involving long reads is the computational requirement of analysis. During genome assembly, as number of reads increases, the number of overlaps computed between the reads also increases exponentially. Nonetheless, with methods that leverage the power of ultra-long reads, SMS will be a revolution in genomic studies and will create new possibilities.
References
Alkan C, Sajjadian S, Eichler EE (2011) Limitations of next-generation genome sequence assembly. Nat Methods 8:61–65. https://doi.org/10.1038/nmeth.1527
Ambardar S, Gowda M (2018) High-resolution full-length HLA typing method using third generation (Pac-Bio SMRT) sequencing technology. Methods Mol Biol 1802:135–153. https://doi.org/10.1007/978-1-4939-8546-3_9
Ameur A, Kloosterman WP, Hestand MS (2019) Single-molecule sequencing: towards clinical applications. Trends Biotechnol 37:72–85. https://doi.org/10.1016/j.tibtech.2018.07.013
Antipov D, Korobeynikov A, McLean JS, Pevzner PA (2016) hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics 32:1009–1015. https://doi.org/10.1093/bioinformatics/btv688
Ardui S, Ameur A, Vermeesch JR, Hestand MS (2018) Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res 46:2159–2168. https://doi.org/10.1093/nar/gky066
Ashley EA (2016) Towards precision medicine. Nat Rev Genet 17:507–522. https://doi.org/10.1038/nrg.2016.86
Bahlo M, Bennett MF, Degorski P, Tankard RM, Delatycki MB, Lockhart PJ (2018) Recent advances in the detection of repeat expansions with short-read next-generation sequencing. F1000Res 7(F1000 Faculty Rev):736. https://doi.org/10.12688/f1000research.13980.1
Bayega A, Wang YC, Oikonomopoulos S, Djambazian H, Fahiminiya S, Ragoussis J (2018) Transcript profiling using long-read sequencing technologies. Methods Mol Biol 1783:121–147. https://doi.org/10.1007/978-1-4939-7834-2_6
Benner S, Chen RJ, Wilson NA, Abu-Shumays R, Hurt N, Lieberman KR, Deamer DW, Dunbar WB, Akeson M (2007) Sequence-specific detection of individual DNA polymerase complexes in real time using a nanopore. Nat Nanotechnol 2:718–724. https://doi.org/10.1038/nnano.2007.344
Boldogkoi Z, Moldovan N, Balazs Z, Snyder M, Tombacz D (2019) Long-read sequencing—a powerful tool in viral transcriptome research. Trends Microbiol 27:578–592. https://doi.org/10.1016/j.tim.2019.01.010
Boza V, Brejova B, Vinar T (2017) DeepNano: deep recurrent neural networks for base calling in MinION nanopore reads. PLoS One 12:e0178751. https://doi.org/10.1371/journal.pone.0178751
Byrne A, Beaudin AE, Olsen HE, Jain M, Cole C, Palmer T, DuBois RM, Forsberg EC, Akeson M, Vollmers C (2017) Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat Commun 8:16027. https://doi.org/10.1038/ncomms16027
Chaisson MJ, Wilson RK, Eichler EE (2015) Genetic variation and the de novo assembly of human genomes. Nat Rev Genet 16:627–640. https://doi.org/10.1038/nrg3933
Chin FY, Leung HC, Yiu SM (2014) Sequence assembly using next generation sequencing data–challenges and solutions. Sci China Life Sci 57:1140–1148. https://doi.org/10.1007/s11427-014-4752-9
Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O’Malley R, Figueroa-Balderas R, Morales-Cruz A, Cramer GR, Delledonne M, Luo C, Ecker JR, Cantu D, Rank DR, Schatz MC (2016) Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13:1050–1054. https://doi.org/10.1038/nmeth.4035
Clark TA, Spittle KE, Turner SW, Korlach J (2011) Direct detection and sequencing of damaged DNA bases. Genome Integr 2:10. https://doi.org/10.1186/2041-9414-2-10
Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H (2009) Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol 4:265–270. https://doi.org/10.1038/nnano.2009.12
Costa-Silva J, Domingues D, Lopes FM (2017) RNA-Seq differential expression analysis: an extended review and a software tool. PLoS One 12:e0190152. https://doi.org/10.1371/journal.pone.0190152
Cretu Stancu M, van Roosmalen MJ, Renkens I, Nieboer MM, Middelkamp S, de Ligt J, Pregno G, Giachino D, Mandrile G, Espejo Valle-Inclan J, Korzelius J, de Bruijn E, Cuppen E, Talkowski ME, Marschall T, de Ridder J, Kloosterman WP (2017) Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat Commun 8:1326. https://doi.org/10.1038/s41467-017-01343-4
De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C (2018) NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34:2666–2669. https://doi.org/10.1093/bioinformatics/bty149
de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD (2011) Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 7:e1002384. https://doi.org/10.1371/journal.pgen.1002384
De Roeck A, De Coster W, Bossaerts L, Cacace R, De Pooter T, Van Dongen J, D’Hert S, De Rijk P, Strazisar M, Van Broeckhoven C, Sleegers K (2018) Accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. BioRxiv. https://doi.org/10.1101/439026
De Roeck A, Van Broeckhoven C, Sleegers K (2019) The role of ABCA7 in Alzheimer’s disease: evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. https://doi.org/10.1007/s00401-019-01994-1
Delaneau O, Howie B, Cox AJ, Zagury JF, Marchini J (2013) Haplotype estimation using sequencing reads. Am J Hum Genet 93:687–696. https://doi.org/10.1016/j.ajhg.2013.09.002
Deonovic B, Wang Y, Weirather J, Wang XJ, Au KF (2017) IDP-ASE: haplotyping and quantifying allele-specific expression at the gene and gene isoform level by hybrid sequencing. Nucleic Acids Res 45:e32. https://doi.org/10.1093/nar/gkw1076
Depledge DP, Srinivas KP, Sadaoka T, Bready D, Mori Y, Placantonakis DG, Mohr I, Wilson AC (2019) Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat Commun 10:754. https://doi.org/10.1038/s41467-019-08734-9
Deurenberg RH, Bathoorn E, Chlebowicz MA, Couto N, Ferdous M, Garcia-Cobos S, Kooistra-Smid AM, Raangs EC, Rosema S, Veloo AC, Zhou K, Friedrich AW, Rossen JW (2017) Application of next generation sequencing in clinical microbiology and infection prevention. J Biotechnol 243:16–24. https://doi.org/10.1016/j.jbiotec.2016.12.022
Duitama J, Zablotskaya A, Gemayel R, Jansen A, Belet S, Vermeesch JR, Verstrepen KJ, Froyen G (2014) Large-scale analysis of tandem repeat variability in the human genome. Nucleic Acids Res 42:5728–5741. https://doi.org/10.1093/nar/gku212
Ebbert MTW, Farrugia SL, Sens JP, Jansen-West K, Gendron TF, Prudencio M, McLaughlin IJ, Bowman B, Seetin M, DeJesus-Hernandez M, Jackson J, Brown PH, Dickson DW, van Blitterswijk M, Rademakers R, Petrucelli L, Fryer JD (2018) Long-read sequencing across the C9orf72 ‘GGGGCC’ repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol Neurodegener 13:46. https://doi.org/10.1186/s13024-018-0274-4
Eberle MA, Fritzilas E, Krusche P, Kallberg M, Moore BL, Bekritsky MA, Iqbal Z, Chuang HY, Humphray SJ, Halpern AL, Kruglyak S, Margulies EH, McVean G, Bentley DR (2017) A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res 27:157–164. https://doi.org/10.1101/gr.210500.116
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S (2009) Real-time DNA sequencing from single polymerase molecules. Science 323:133–138. https://doi.org/10.1126/science.1162986
Erlich RL, Jia X, Anderson S, Banks E, Gao X, Carrington M, Gupta N, DePristo MA, Henn MR, Lennon NJ, de Bakker PI (2011) Next-generation sequencing for HLA typing of class I loci. BMC Genomics 12:42. https://doi.org/10.1186/1471-2164-12-42
Euskirchen P, Bielle F, Labreche K, Kloosterman WP, Rosenberg S, Daniau M, Schmitt C, Masliah-Planchon J, Bourdeaut F, Dehais C, Marie Y, Delattre JY, Idbaih A (2017) Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol 134:691–703. https://doi.org/10.1007/s00401-017-1743-5
Fragouli E, Katz-Jaffe M, Alfarawati S, Stevens J, Colls P, Goodall NN, Tormasi S, Gutierrez-Mateo C, Prates R, Schoolcraft WB, Munne S, Wells D (2010) Comprehensive chromosome screening of polar bodies and blastocysts from couples experiencing repeated implantation failure. Fertil Steril 94:875–887. https://doi.org/10.1016/j.fertnstert.2009.04.053
Fragouli E, Alfarawati S, Daphnis DD, Goodall NN, Mania A, Griffiths T, Gordon A, Wells D (2011) Cytogenetic analysis of human blastocysts with the use of FISH, CGH and aCGH: scientific data and technical evaluation. Hum Reprod 26:480–490. https://doi.org/10.1093/humrep/deq344
Friedenthal J, Maxwell SM, Munne S, Kramer Y, McCulloh DH, McCaffrey C, Grifo JA (2018) Next generation sequencing for preimplantation genetic screening improves pregnancy outcomes compared with array comparative genomic hybridization in single thawed euploid embryo transfer cycles. Fertil Steril 109:627–632. https://doi.org/10.1016/j.fertnstert.2017.12.017
Gardy JL, Loman NJ (2018) Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet 19:9–20. https://doi.org/10.1038/nrg.2017.88
Giampaoli S, Alessandrini F, Frajese GV, Guglielmi G, Tagliabracci A, Berti A (2018) Environmental microbiology: perspectives for legal and occupational medicine. Leg Med (Tokyo) 35:34–43. https://doi.org/10.1016/j.legalmed.2018.09.014
Gießelmann P, Brändl B, Raimondeau E, Bowen R, Rohrandt C, Tandon R, Kretzmer H, Assum G, Galonska C, Siebert R, Ammerpohl O, Heron A, Schneider SA, Ladewig J, Koch P, Schuldt BM, Graham JE, Meissner A, Müller F-J (2018) Repeat expansion and methylation state analysis with nanopore sequencing. BioRxiv. https://doi.org/10.1101/480285
Giordano F, Aigrain L, Quail MA, Coupland P, Bonfield JK, Davies RM, Tischler G, Jackson DK, Keane TM, Li J, Yue JX, Liti G, Durbin R, Ning Z (2017) De novo yeast genome assemblies from MinION, PacBio and MiSeq platforms. Sci Rep 7:3935. https://doi.org/10.1038/s41598-017-03996-z
Goodwin S, McPherson JD, McCombie WR (2016) Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 17:333–351. https://doi.org/10.1038/nrg.2016.49
Greninger AL, Naccache SN, Federman S, Yu G, Mbala P, Bres V, Stryke D, Bouquet J, Somasekar S, Linnen JM, Dodd R, Mulembakani P, Schneider BS, Muyembe-Tamfum JJ, Stramer SL, Chiu CY (2015) Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med 7:99. https://doi.org/10.1186/s13073-015-0220-9
Guan P, Sung WK (2016) Structural variation detection using next-generation sequencing data: a comparative technical review. Methods 102:36–49. https://doi.org/10.1016/j.ymeth.2016.01.020
Hansen S, Faye O, Sanabani SS, Faye M, Bohlken-Fascher S, Faye O, Sall AA, Bekaert M, Weidmann M, Czerny CP, Abd El Wahed A (2018) Combination random isothermal amplification and nanopore sequencing for rapid identification of the causative agent of an outbreak. J Clin Virol 106:23–27. https://doi.org/10.1016/j.jcv.2018.07.001
Hartel AJW, Shekar S, Ong P, Schroeder I, Thiel G, Shepard KL (2019) High bandwidth approaches in nanopore and ion channel recordings—a tutorial review. Anal Chim Acta 1061:13–27. https://doi.org/10.1016/j.aca.2019.01.034
Hedges DJ, Hamilton-Nelson KL, Sacharow SJ, Nations L, Beecham GW, Kozhekbaeva ZM, Butler BL, Cukier HN, Whitehead PL, Ma D, Jaworski JM, Nathanson L, Lee JM, Hauser SL, Oksenberg JR, Cuccaro ML, Haines JL, Gilbert JR, Pericak-Vance MA (2012) Evidence of novel fine-scale structural variation at autism spectrum disorder candidate loci. Mol Autism 3:2. https://doi.org/10.1186/2040-2392-3-2
Hoenen T, Groseth A, Rosenke K, Fischer RJ, Hoenen A, Judson SD, Martellaro C, Falzarano D, Marzi A, Squires RB, Wollenberg KR, de Wit E, Prescott J, Safronetz D, van Doremalen N, Bushmaker T, Feldmann F, McNally K, Bolay FK, Fields B, Sealy T, Rayfield M, Nichol ST, Zoon KC, Massaquoi M, Munster VJ, Feldmann H (2016) Nanopore sequencing as a rapidly deployable ebola outbreak tool. Emerg Infect Dis 22:331–334. https://doi.org/10.3201/eid2202.151796
Hoper D, Mettenleiter TC, Beer M (2016) Metagenomic approaches to identifying infectious agents. Rev Sci Tech 35:83–93. https://doi.org/10.20506/rst.35.1.2419
Hosomichi K, Shiina T, Tajima A, Inoue I (2015) The impact of next-generation sequencing technologies on HLA research. J Hum Genet 60:665–673. https://doi.org/10.1038/jhg.2015.102
Huddleston J, Chaisson MJP, Steinberg KM, Warren W, Hoekzema K, Gordon D, Graves-Lindsay TA, Munson KM, Kronenberg ZN, Vives L, Peluso P, Boitano M, Chin CS, Korlach J, Wilson RK, Eichler EE (2017) Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res 27:677–685. https://doi.org/10.1101/gr.214007.116
Istace B, Friedrich A, d’Agata L, Faye S, Payen E, Beluche O, Caradec C, Davidas S, Cruaud C, Liti G, Lemainque A, Engelen S, Wincker P, Schacherer J, Aury JM (2017) de novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. Gigascience 6:1–13. https://doi.org/10.1093/gigascience/giw018
Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S, Marriott H, Nieto T, O’Grady J, Olsen HE, Pedersen BS, Rhie A, Richardson H, Quinlan AR, Snutch TP, Tee L, Paten B, Phillippy AM, Simpson JT, Loman NJ, Loose M (2018) Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36:338–345. https://doi.org/10.1038/nbt.4060
Jayakumar V, Sakakibara Y (2019) Comprehensive evaluation of non-hybrid genome assembly tools for third-generation PacBio long-read sequence data. Brief Bioinform 20:866–876. https://doi.org/10.1093/bib/bbx147
Judge K, Harris SR, Reuter S, Parkhill J, Peacock SJ (2015) Early insights into the potential of the Oxford Nanopore MinION for the detection of antimicrobial resistance genes. J Antimicrob Chemother 70:2775–2778. https://doi.org/10.1093/jac/dkv206
Kafetzopoulou LE, Efthymiadis K, Lewandowski K, Crook A, Carter D, Osborne J, Aarons E, Hewson R, Hiscox JA, Carroll MW, Vipond R, Pullan ST (2018) Assessment of metagenomic Nanopore and Illumina sequencing for recovering whole genome sequences of chikungunya and dengue viruses directly from clinical samples. Euro Surveill. https://doi.org/10.2807/1560-7917.es.2018.23.50.1800228
Kafetzopoulou LE, Pullan ST, Lemey P, Suchard MA, Ehichioya DU, Pahlmann M, Thielebein A, Hinzmann J, Oestereich L, Wozniak DM, Efthymiadis K, Schachten D, Koenig F, Matjeschk J, Lorenzen S, Lumley S, Ighodalo Y, Adomeh DI, Olokor T, Omomoh E, Omiunu R, Agbukor J, Ebo B, Aiyepada J, Ebhodaghe P, Osiemi B, Ehikhametalor S, Akhilomen P, Airende M, Esumeh R, Muoebonam E, Giwa R, Ekanem A, Igenegbale G, Odigie G, Okonofua G, Enigbe R, Oyakhilome J, Yerumoh EO, Odia I, Aire C, Okonofua M, Atafo R, Tobin E, Asogun D, Akpede N, Okokhere PO, Rafiu MO, Iraoyah KO, Iruolagbe CO, Akhideno P, Erameh C, Akpede G, Isibor E, Naidoo D, Hewson R, Hiscox JA, Vipond R, Carroll MW, Ihekweazu C, Formenty P, Okogbenin S, Ogbaini-Emovon E, Gunther S, Duraffour S (2019) Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science 363:74–77. https://doi.org/10.1126/science.aau9343
Keller MW, Rambo-Martin BL, Wilson MM, Ridenour CA, Shepard SS, Stark TJ, Neuhaus EB, Dugan VG, Wentworth DE, Barnes JR (2018) Direct RNA sequencing of the coding complete influenza a virus genome. Sci Rep 8:14408. https://doi.org/10.1038/s41598-018-32615-8
Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, Adam MP (2012) Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 30:693–700. https://doi.org/10.1038/nbt.2280
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM (2017) Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27:722–736. https://doi.org/10.1101/gr.215087.116
Lang K, Surendranath V, Quenzel P, Schofl G, Schmidt AH, Lange V (2018) Full-length HLA class I genotyping with the MinION nanopore sequencer. Methods Mol Biol 1802:155–162. https://doi.org/10.1007/978-1-4939-8546-3_10
Laver TW, Caswell RC, Moore KA, Poschmann J, Johnson MB, Owens MM, Ellard S, Paszkiewicz KH, Weedon MN (2016) Pitfalls of haplotype phasing from amplicon-based long-read sequencing. Sci Rep 6:21746. https://doi.org/10.1038/srep21746
Lee E, Illingworth P, Wilton L, Chambers GM (2015a) The clinical effectiveness of preimplantation genetic diagnosis for aneuploidy in all 24 chromosomes (PGD-A): systematic review. Hum Reprod 30:473–483. https://doi.org/10.1093/humrep/deu303
Lee HL, McCulloh DH, Hodes-Wertz B, Adler A, McCaffrey C, Grifo JA (2015b) In vitro fertilization with preimplantation genetic screening improves implantation and live birth in women age 40 through 43. J Assist Reprod Genet 32:435–444. https://doi.org/10.1007/s10815-014-0417-7
Li H (2016) Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32:2103–2110. https://doi.org/10.1093/bioinformatics/btw152
Lin Y, Yuan J, Kolmogorov M, Shen MW, Chaisson M, Pevzner PA (2016) Assembly of long error-prone reads using de Bruijn graphs. Proc Natl Acad Sci USA 113:E8396–E8405. https://doi.org/10.1073/pnas.1604560113
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M (2012) Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012:251364. https://doi.org/10.1155/2012/251364
Liu X, Mei W, Soltis PS, Soltis DE, Barbazuk WB (2017) Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol Ecol Resour 17:1243–1256. https://doi.org/10.1111/1755-0998.12670
Liu C, Xiao F, Hoisington-Lopez J, Lang K, Quenzel P, Duffy B, Mitra RD (2018) Accurate typing of human leukocyte antigen class i genes by oxford nanopore sequencing. J Mol Diagn 20:428–435. https://doi.org/10.1016/j.jmoldx.2018.02.006
Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, McCalmon S, Hagerman RJ, Tassone F, Hagerman PJ (2013) Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res 23:121–128. https://doi.org/10.1101/gr.141705.112
Lu H, Giordano F, Ning Z (2016) Oxford nanopore MinION sequencing and genome assembly. Genomics Proteomics Bioinformatics 14:265–279. https://doi.org/10.1016/j.gpb.2016.05.004
Macintyre G, Ylstra B, Brenton JD (2016) Sequencing structural variants in cancer for precision therapeutics. Trends Genet 32:530–542. https://doi.org/10.1016/j.tig.2016.07.002
Manrao EA, Derrington IM, Laszlo AH, Langford KW, Hopper MK, Gillgren N, Pavlenok M, Niederweis M, Gundlach JH (2012) Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat Biotechnol 30:349–353. https://doi.org/10.1038/nbt.2171
Mardis ER (2013) Next-generation sequencing platforms. Annu Rev Anal Chem (Palo Alto Calif) 6:287–303. https://doi.org/10.1146/annurev-anchem-062012-092628
Martin JA, Wang Z (2011) Next-generation transcriptome assembly. Nat Rev Genet 12:671–682. https://doi.org/10.1038/nrg3068
Maxam AM, Gilbert W (1977) A new method for sequencing DNA. Proc Natl Acad Sci USA 74:560–564
McGinty RJ, Rubinstein RG, Neil AJ, Dominska M, Kiktev D, Petes TD, Mirkin SM (2017) Nanopore sequencing of complex genomic rearrangements in yeast reveals mechanisms of repeat-mediated double-strand break repair. Genome Res 27:2072–2082. https://doi.org/10.1101/gr.228148.117
McIntyre ABR, Alexander N, Grigorev K, Bezdan D, Sichtig H, Chiu CY, Mason CE (2019) Single-molecule sequencing detection of N6-methyladenine in microbial reference materials. Nat Commun 10:579. https://doi.org/10.1038/s41467-019-08289-9
Merker JD, Wenger AM, Sneddon T, Grove M, Zappala Z, Fresard L, Waggott D, Utiramerur S, Hou Y, Smith KS, Montgomery SB, Wheeler M, Buchan JG, Lambert CC, Eng KS, Hickey L, Korlach J, Ford J, Ashley EA (2018) Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet Med 20:159–163. https://doi.org/10.1038/gim.2017.86
Mitsuhashi S, Nakagawa S, Takahashi Ueda M, Imanishi T, Frith MC, Mitsuhashi H (2017) Nanopore-based single molecule sequencing of the D4Z4 array responsible for facioscapulohumeral muscular dystrophy. Sci Rep 7:14789. https://doi.org/10.1038/s41598-017-13712-6
Mousavi N, Shleizer-Burko S, Gymrek M (2018) Profiling the genome-wide landscape of tandem repeat expansions. BioRxiv. https://doi.org/10.1101/361162
Nair SS, Luu PL, Qu W, Maddugoda M, Huschtscha L, Reddel R, Chenevix-Trench G, Toso M, Kench JG, Horvath LG, Hayes VM, Stricker PD, Hughes TP, White DL, Rasko JEJ, Wong JJ, Clark SJ (2018) Guidelines for whole genome bisulphite sequencing of intact and FFPET DNA on the Illumina HiSeq X Ten. Epigenetics Chromatin 11:24. https://doi.org/10.1186/s13072-018-0194-0
Ozaki Y, Suzuki S, Kashiwase K, Shigenari A, Okudaira Y, Ito S, Masuya A, Azuma F, Yabe T, Morishima S, Mitsunaga S, Satake M, Ota M, Morishima Y, Kulski JK, Saito K, Inoko H, Shiina T (2015) Cost-efficient multiplex PCR for routine genotyping of up to nine classical HLA loci in a single analytical run of multiple samples by next generation sequencing. BMC Genomics 16:318. https://doi.org/10.1186/s12864-015-1514-4
Pang AW, MacDonald JR, Pinto D, Wei J, Rafiq MA, Conrad DF, Park H, Hurles ME, Lee C, Venter JC, Kirkness EF, Levy S, Feuk L, Scherer SW (2010) Towards a comprehensive structural variation map of an individual human genome. Genome Biol 11:R52. https://doi.org/10.1186/gb-2010-11-5-r52
Payne A, Holmes N, Rakyan V, Loose M (2018) BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics. https://doi.org/10.1093/bioinformatics/bty841
Petersen BS, Fredrich B, Hoeppner MP, Ellinghaus D, Franke A (2017) Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet 18:14. https://doi.org/10.1186/s12863-017-0479-5
Profaizer T, Lazar-Molnar E, Close DW, Delgado JC, Kumanovics A (2016) HLA genotyping in the clinical laboratory: comparison of next-generation sequencing methods. HLA 88:14–24. https://doi.org/10.1111/tan.12850
Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, Bore JA, Koundouno R, Dudas G, Mikhail A, Ouedraogo N, Afrough B, Bah A, Baum JH, Becker-Ziaja B, Boettcher JP, Cabeza-Cabrerizo M, Camino-Sanchez A, Carter LL, Doerrbecker J, Enkirch T, Dorival IGG, Hetzelt N, Hinzmann J, Holm T, Kafetzopoulou LE, Koropogui M, Kosgey A, Kuisma E, Logue CH, Mazzarelli A, Meisel S, Mertens M, Michel J, Ngabo D, Nitzsche K, Pallash E, Patrono LV, Portmann J, Repits JG, Rickett NY, Sachse A, Singethan K, Vitoriano I, Yemanaberhan RL, Zekeng EG, Trina R, Bello A, Sall AA, Faye O, Faye O, Magassouba N, Williams CV, Amburgey V, Winona L, Davis E, Gerlach J, Washington F, Monteil V, Jourdain M, Bererd M, Camara A, Somlare H, Camara A, Gerard M, Bado G, Baillet B, Delaune D, Nebie KY, Diarra A, Savane Y, Pallawo RB, Gutierrez GJ, Milhano N, Roger I, Williams CJ, Yattara F, Lewandowski K, Taylor J, Rachwal P, Turner D, Pollakis G, Hiscox JA, Matthews DA, O’Shea MK, Johnston AM, Wilson D, Hutley E, Smit E, Di Caro A, Woelfel R, Stoecker K, Fleischmann E, Gabriel M, Weller SA, Koivogui L, Diallo B, Keita S, Rambaut A, Formenty P et al (2016) Real-time, portable genome sequencing for Ebola surveillance. Nature 530:228–232. https://doi.org/10.1038/nature16996
Quinlan AR, Hall IM (2012) Characterizing complex structural variation in germline and somatic genomes. Trends Genet 28:43–53. https://doi.org/10.1016/j.tig.2011.10.002
Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, Paten B (2017) Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods 14:411–413. https://doi.org/10.1038/nmeth.4189
Raymond CK, Subramanian S, Paddock M, Qiu R, Deodato C, Palmieri A, Chang J, Radke T, Haugen E, Kas A, Waring D, Bovee D, Stacy R, Kaul R, Olson MV (2005) Targeted, haplotype-resolved resequencing of long segments of the human genome. Genomics 86:759–766. https://doi.org/10.1016/j.ygeno.2005.08.013
Rhoads A, Au KF (2015) pacbio sequencing and its applications. Genomics Proteomics Bioinformatics 13:278–289. https://doi.org/10.1016/j.gpb.2015.08.002
Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38:24–26. https://doi.org/10.1038/ng1718
Salzberg SL, Yorke JA (2005) Beware of mis-assembled genomes. Bioinformatics 21:4320–4321. https://doi.org/10.1093/bioinformatics/bti769
Sanger F, Coulson AR (1975) A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 94:441–448
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Sasazuki T, Inoko H, Morishima S, Morishima Y (2016) Gene map of the HLA region, graves’ disease and hashimoto thyroiditis, and hematopoietic stem cell transplantation. Adv Immunol 129:175–249. https://doi.org/10.1016/bs.ai.2015.08.003
Scheben A, Batley J, Edwards D (2017) Genotyping-by-sequencing approaches to characterize crop genomes: choosing the right tool for the right application. Plant Biotechnol J 15:149–161. https://doi.org/10.1111/pbi.12645
Schmutz J, Wheeler J, Grimwood J, Dickson M, Yang J, Caoile C, Bajorek E, Black S, Chan YM, Denys M, Escobar J, Flowers D, Fotopulos D, Garcia C, Gomez M, Gonzales E, Haydu L, Lopez F, Ramirez L, Retterer J, Rodriguez A, Rogers S, Salazar A, Tsai M, Myers RM (2004) Quality assessment of the human genome sequence. Nature 429:365–368. https://doi.org/10.1038/nature02390
Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, Schatz MC (2018) Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods 15:461–468. https://doi.org/10.1038/s41592-018-0001-7
Seo JS, Rhie A, Kim J, Lee S, Sohn MH, Kim CU, Hastie A, Cao H, Yun JY, Kim J, Kuk J, Park GH, Kim J, Ryu H, Kim J, Roh M, Baek J, Hunkapiller MW, Korlach J, Shin JY, Kim C (2016) De novo assembly and phasing of a Korean human genome. Nature 538:243–247. https://doi.org/10.1038/nature20098
Shi L, Guo Y, Dong C, Huddleston J, Yang H, Han X, Fu A, Li Q, Li N, Gong S, Lintner KE, Ding Q, Wang Z, Hu J, Wang D, Wang F, Wang L, Lyon GJ, Guan Y, Shen Y, Evgrafov OV, Knowles JA, Thibaud-Nissen F, Schneider V, Yu CY, Zhou L, Eichler EE, So KF, Wang K (2016) Long-read sequencing and de novo assembly of a Chinese genome. Nat Commun 7:12065. https://doi.org/10.1038/ncomms12065
Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W (2017) Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods 14:407–410. https://doi.org/10.1038/nmeth.4184
Smith ZD, Gu H, Bock C, Gnirke A, Meissner A (2009) High-throughput bisulfite sequencing in mammalian genomes. Methods 48:226–232. https://doi.org/10.1016/j.ymeth.2009.05.003
Spies N, Weng Z, Bishara A, McDaniel J, Catoe D, Zook JM, Salit M, West RB, Batzoglou S, Sidow A (2017) Genome-wide reconstruction of complex structural variants using read clouds. Nat Methods 14:915–920. https://doi.org/10.1038/nmeth.4366
Steijger T, Abril JF, Engstrom PG, Kokocinski F, Consortium R, Hubbard TJ, Guigo R, Harrow J, Bertone P (2013) Assessment of transcript reconstruction methods for RNA-seq. Nat Methods 10:1177–1184. https://doi.org/10.1038/nmeth.2714
Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH, Konkel MK, Malhotra A, Stutz AM, Shi X, Casale FP, Chen J, Hormozdiari F, Dayama G, Chen K, Malig M, Chaisson MJP, Walter K, Meiers S, Kashin S, Garrison E, Auton A, Lam HYK, Mu XJ, Alkan C, Antaki D, Bae T, Cerveira E, Chines P, Chong Z, Clarke L, Dal E, Ding L, Emery S, Fan X, Gujral M, Kahveci F, Kidd JM, Kong Y, Lameijer EW, McCarthy S, Flicek P, Gibbs RA, Marth G, Mason CE, Menelaou A, Muzny DM, Nelson BJ, Noor A, Parrish NF, Pendleton M, Quitadamo A, Raeder B, Schadt EE, Romanovitch M, Schlattl A, Sebra R, Shabalin AA, Untergasser A, Walker JA, Wang M, Yu F, Zhang C, Zhang J, Zheng-Bradley X, Zhou W, Zichner T, Sebat J, Batzer MA, McCarroll SA, Genomes Project C, Mills RE, Gerstein MB, Bashir A, Stegle O, Devine SE, Lee C, Eichler EE, Korbel JO (2015) An integrated map of structural variation in 2,504 human genomes. Nature 526:75–81. https://doi.org/10.1038/nature15394
Tattini L, D’Aurizio R, Magi A (2015) Detection of genomic structural variants from next-generation sequencing data. Front Bioeng Biotechnol 3:92. https://doi.org/10.3389/fbioe.2015.00092
Tewhey R, Bansal V, Torkamani A, Topol EJ, Schork NJ (2011) The importance of phase information for human genomics. Nat Rev Genet 12:215–223. https://doi.org/10.1038/nrg2950
Traherne JA (2008) Human MHC architecture and evolution: implications for disease association studies. Int J Immunogenet 35:179–192. https://doi.org/10.1111/j.1744-313X.2008.00765.x
Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW (2010) A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res 38:e159. https://doi.org/10.1093/nar/gkq543
Treangen TJ, Salzberg SL (2011) Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13:36–46. https://doi.org/10.1038/nrg3117
Trowsdale J (1993) Genomic structure and function in the MHC. Trends Genet 9:117–122
Tubio JM (2015) Somatic structural variation and cancer. Brief Funct Genomics 14:339–351. https://doi.org/10.1093/bfgp/elv016
Turner TR, Hayhurst JD, Hayward DR, Bultitude WP, Barker DJ, Robinson J, Madrigal JA, Mayor NP, Marsh SGE (2018) Single molecule real-time DNA sequencing of HLA genes at ultra-high resolution from 126 international HLA and immunogenetics workshop cell lines. HLA 91:88–101. https://doi.org/10.1111/tan.13184
Ulahannan D, Kovac MB, Mulholland PJ, Cazier JB, Tomlinson I (2013) Technical and implementation issues in using next-generation sequencing of cancers in clinical practice. Br J Cancer 109:827–835. https://doi.org/10.1038/bjc.2013.416
Usdin K (2008) The biological effects of simple tandem repeats: lessons from the repeat expansion diseases. Genome Res 18:1011–1019. https://doi.org/10.1101/gr.070409.107
Vaser R, Sovic I, Nagarajan N, Sikic M (2017) Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res 27:737–746. https://doi.org/10.1101/gr.214270.116
Wang C, Krishnakumar S, Wilhelmy J, Babrzadeh F, Stepanyan L, Su LF, Levinson D, Fernandez-Vina MA, Davis RW, Davis MM, Mindrinos M (2012) High-throughput, high-fidelity HLA genotyping with deep sequencing. Proc Natl Acad Sci USA 109:8676–8681. https://doi.org/10.1073/pnas.1206614109
Warnecke PM, Stirzaker C, Song J, Grunau C, Melki JR, Clark SJ (2002) Identification and resolution of artifacts in bisulfite sequencing. Methods 27:101–107
Wei S, Weiss ZR, Gaur P, Forman E, Williams Z (2018) Rapid preimplantation genetic screening using a handheld, nanopore-based DNA sequencer. Fertil Steril 110(910–916):e2. https://doi.org/10.1016/j.fertnstert.2018.06.014
Weirather JL, Afshar PT, Clark TA, Tseng E, Powers LS, Underwood JG, Zabner J, Korlach J, Wong WH, Au KF (2015) Characterization of fusion genes and the significantly expressed fusion isoforms in breast cancer by hybrid sequencing. Nucleic Acids Res 43:e116. https://doi.org/10.1093/nar/gkv562
Weirather JL, de Cesare M, Wang Y, Piazza P, Sebastiano V, Wang XJ, Buck D, Au KF (2017) Comprehensive comparison of pacific biosciences and oxford nanopore technologies and their applications to transcriptome analysis. F1000Res 6:100. https://doi.org/10.12688/f1000research.10571.2
Xiao CL, Zhu S, He M, Chen Zhang Q, Chen Y, Yu G, Liu J, Xie SQ, Luo F, Liang Z, Wang DP, Bo XC, Gu XF, Wang K, Yan GR (2018) N(6)-methyladenine DNA modification in the human genome. Mol Cell 71(306–318):e7. https://doi.org/10.1016/j.molcel.2018.06.015
Yang Z, Lin J, Zhang J, Fong WI, Li P, Zhao R, Liu X, Podevin W, Kuang Y, Liu J (2015) Randomized comparison of next-generation sequencing and array comparative genomic hybridization for preimplantation genetic screening: a pilot study. BMC Med Genomics 8:30. https://doi.org/10.1186/s12920-015-0110-4
Yuan Y, Bayer PE, Batley J, Edwards D (2017) Improvements in genomic technologies: application to crop genomics. Trends Biotechnol 35:547–558. https://doi.org/10.1016/j.tibtech.2017.02.009
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No conflict of interest declared by MKM and KPC. MW works in a company which provides Nanopore services.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Midha, M.K., Wu, M. & Chiu, KP. Long-read sequencing in deciphering human genetics to a greater depth. Hum Genet 138, 1201–1215 (2019). https://doi.org/10.1007/s00439-019-02064-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-019-02064-y