
Abstract
MicroRNAs (miRNAs) are short, non-coding RNAs that post-transcriptionally repress translation or induce mRNA degradation of target transcripts through sequence-specific binding. miRNAs target hundreds of transcripts to regulate diverse biological pathways and processes, including aging. Many microRNAs are differentially expressed during aging, generating interest in their use as aging biomarkers and roles as regulators of the aging process. In the invertebrates Caenorhabditis elegans and Drosophila, a number of miRNAs have been found to both positive and negatively modulate longevity through canonical aging pathways. Recent studies have also shown that miRNAs regulate age-associated processes and pathologies in a diverse array of mammalian tissues, including brain, heart, bone, and muscle. The review will present an overview of these studies, highlighting the role of individual miRNAs as biomarkers of aging and regulators of longevity and tissue-specific aging processes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aging is a complex, systemic physiological process characterized by progressive degradation, loss of function, and reduced repair capacity of an animal’s tissues and organ systems. Loss of physical integrity leads to an increased risk of mortality and susceptibility to multiple age-related pathologies, including neurodegenerative disorders, cardiovascular disease, osteoporosis, sarcopenia, and cancer (da Costa et al. 2016). As the average human life expectancy and proportion of elderly individuals in the population continues to increase, there is mounting interest in uncovering the molecular mechanisms underlying aging and related pathologies (Crimmins 2015). A better understanding of these mechanisms may allow for the extension of not only average lifespan but healthy life expectancy, or the number of years spent free from age-related morbidities (Beltrán-Sánchez et al. 2015).
Decades of research in the short-lived invertebrate Caenorhabditis elegans have uncovered multiple genetic pathways that regulate aging and lifespan. Loss-of-function mutations in daf-2 and age-1, components of the insulin/insulin-like growth-factor signaling (IIS) pathway, were the first genetic manipulations shown to dramatically extend lifespan in C. elegans (Friedman and Johnson 1988; Kenyon et al. 1993). Since then, genetic analysis has identified multiple pathways that modulate lifespan, including rapamycin (mTOR) signaling, sirtuins, and mitochondrial respiration (Kenyon 2010). Most lifespan-extending interventions discovered thus far act through the IIS and/or mTOR signaling pathways. Importantly, these pathways and their function in aging are well conserved in mammalian species, including humans.
MicroRNAs (miRNAs) have been increasingly recognized as important regulators of the aging process and modulators of longevity. miRNAs are short (19–22 nucleotides), non-coding RNAs that generally repress translation or induce mRNA degradation of target transcripts through sequence-specific binding to the 3′UTR (Bushati and Cohen 2007). While lin-4, the first miRNA discovered, was found in C. elegans, miRNAs are ubiquitous across plant and animal species (Axtell et al. 2011). At the time of this review, over 1900 human miRNAs have been cataloged in miRbase (http://www.mirbase.org), and nearly 60% of all human transcripts are predicted to be regulated by miRNAs (Lewis et al. 2005; Kozomara and Griffiths-Jones 2014). The miRNA regulatory network is extensive, as each miRNA may target hundreds of different transcripts and each transcript may be targeted by multiple miRNAs. miRNA-mediated regulation is central to a number of biological processes, including developmental timing, cell death, and proliferation; likewise, dysregulation of miRNAs has been implicated in human disease, particularly cancer (Bushati and Cohen 2007).
In this article, we will briefly review miRNAs that are known to regulate longevity and canonical aging pathways (Fig. 1, Table 1). While most knowledge of longevity-regulating miRNAs is derived from work in C. elegans, multiple reviews that extensively cover this topic are available elsewhere (Smith-Vikos and Slack 2012; Kato and Slack 2013; Inukai and Slack 2013). To complement this existing work, here, we will primarily review the functional roles of miRNAs in healthy mammalian aging, with a focus on recent results (2013 to present). Because mammalian aging is characterized by organ-level functional decline that varies greatly between systems (Khan et al. 2017), we discuss the involvement of miRNAs separately in aging of the brain, heart, bone, and muscle (Fig. 2, Table 2). We have chosen to uniquely highlight miRNAs that have been studied in the context of normal aging in these organs rather than in specific age-related pathologies, although there is some degree of overlap. The role of miRNAs in age-related pathologies has been well addressed in a number of recent reviews, including those covering cardiovascular disease (de Lucia et al. 2017; Verjans et al. 2017; Duan et al. 2018), neurodegenerative diseases (Quinlan et al. 2017; Leggio et al. 2017; Wang et al. 2019), and cancer (Peng and Croce 2016; Rupaimoole and Slack 2017). Due to recent interest in the field, we will also discuss the use of circulating miRNAs as biomarkers of aging and longevity in humans. The article will conclude with a perspective on remaining areas of research and the potential for therapeutic applications of miRNAs in human aging.

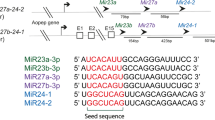
Human orthologs of miRNAs (and miRNA families) that directly regulate lifespan in the model organisms C. elegans, Drosophila, and mouse. Positive and negative modulators of lifespan are shown in blue and orange, respectively. Orthologs that have not yet been demonstrated to directly regulate lifespan in their respective species are shown in black

miRNAs with functional roles in normal aging of the brain, heart, skeletal muscle, and bone. miRNAs that have been experimentally validated (via knockout/knockdown or overexpression studies) to play a beneficial, detrimental, or ambiguous/complex role in the aging process are color-coded in blue, orange, and black, respectively
miRNAs in longevity
A number of miRNAs have been shown to directly influence lifespan through well-described aging pathways, including insulin/insulin-like growth-factor (IGF-1) signaling, target of rapamycin (TOR) and translation signaling, sirtuin deacetylases, mitochondrial/reactive oxygen species (ROS) signaling, and DNA-damage response (Smith-Vikos and Slack 2012). These pathways represent adaptive mechanisms aimed at maintaining organismal homeostasis in response to molecular damage, changes in nutrient availability, and other forms of physiologic stress. Most knowledge about longevity-modulating miRNAs has been derived from work on invertebrates; however, many of the components and functions of miRNA-targeted aging pathways are also conserved in mammalian species. miRNAs that regulate longevity in C. elegans, Drosophila, and mouse are described in detail below and summarized in Fig. 1 and Table 1.
C. elegans
The heterochronic miRNA lin-4 was the first non-coding RNA reported to influence lifespan (Boehm and Slack 2005). Loss of function of lin-4 reduces lifespan, while overexpression of lin-4 or knockdown of its target lin-14 extends lifespan. Lifespan extension via loss of lin-14 is dependent on DAF-16, a downstream transcription factor in the insulin signaling pathway (Boehm and Slack 2005). miR-71, miR-238, miR-246, and miR-239, all of which are upregulated with age, have also been characterized as longevity modulators (de Lencastre et al. 2010). miR-71, miR-238, and miR-246 expression positively correlate with lifespan in isogenic wild-type individuals, while miR-239 expression negatively correlates with future lifespan (Pincus et al. 2011). Furthermore, mir-71, mir-238, and mir-246 loss-of-function mutants are short-lived, and mir-239 mutants are long-lived compared to wild-type; overexpression confers opposite effects for mir-71, mir-246, and mir-239 (de Lencastre et al. 2010). miR-71 and miR-239 have been shown to exert their effects on lifespan through insulin/IGF-1 and DNA-damage checkpoint pathways (de Lencastre et al. 2010; Pincus et al. 2011). miR-71 is also required for lifespan extension induced by either germline loss or dietary restriction (Boulias and Horvitz 2012; Smith-Vikos et al. 2014). Evidence suggests that miR-71 acts in a feedback loop with miR-228, a negative regulator of longevity, and the SKN-1 and PHA-4 transcription factors to mediate lifespan in response to dietary restriction (Smith-Vikos et al. 2014). miR-71 has also been shown to regulate other important aspects of C. elegans biology, including post-starvation survival and development in larval animals, neuronal differentiation, proteostasis, and olfaction (Zhang et al. 2011; Hsieh et al. 2012; Finger et al. 2019). While miR-71 is indispensable for normal lifespan in C. elegans, this miRNA does not appear to be well conserved in other species. Regardless, the multifunctionality of miR-71 demonstrates how microRNAs can act as a common node between different pathways.
Loss-of-function mutations in miR-34, a highly conserved miRNA, greatly extend lifespan through regulation of autophagy genes atg4.1, bec-1, and atg9; atg9 has been confirmed as a direct target of miR-34a, a mammalian member of the miR-34 family (Yang et al. 2013a). Interestingly, upregulation of miR-34 enhances survival under stress conditions and is regulated by DAF-16 in this context; Argonaute-binding data suggest that miR-34 is likely to target daf-16, forming a negative feedback loop (Isik et al. 2016).
In addition to miRNAs, components of the miRNA-guided silencing and processing machinery also regulate lifespan in C. elegans. Adult-specific loss of argonaute-like gene-1 (alg-1), required specifically for miRNA function and processing, results in significantly shortened lifespan (Aalto et al. 2018). Surprisingly, loss of function of alg-2, long considered redundant in function with alg-1, caused lifespan extension. The alg-1 and alg-2 lifespan phenotypes are dependent on the insulin/IGF-1 signaling pathway, with alg-1 positively regulating DAF-16 targets and alg-2 repressing their expression (Aalto et al. 2018).
Drosophila
In Drosophila, the majority of miRNA loss-of-function mutations appear to have a negative, if any, effect on lifespan; lifespan-extending mutations are generally sex-specific and result in a modest increase in longevity (Chen et al. 2014). miR-125, a well-conserved homolog of lin-4, in addition to the well conserved and co-transcribed (in Drosophila) miRNA let-7, has been shown to modulate lifespan in a sex- and tissue-specific manner (Chawla et al. 2016; Gendron and Pletcher 2017). Loss of mir-125 or let-7 results in shorter lifespan and early onset age-associated neurodegeneration in males. While both directly target transcription factor Chinmo, a regulator of neuronal development, only miR-125 exerts its effects on lifespan through Chinmo repression (Chawla et al. 2016). Neuronal overexpression of let-7 or mir-125 extends or reduces lifespan, respectively, in females; ubiquitous overexpression of either miRNA reduces lifespan. Let-7 expression in neurons confers the opposite effect in males, significantly shortening lifespan (Gendron and Pletcher 2017). The role of the let-7 complex and its members in determination of lifespan is clearly complex, and neuron-specific targets remain to be identified.
Contrary to observations in C. elegans, loss of the conserved miRNA mir-34 reduces lifespan and promotes early onset neurodegeneration (Liu et al. 2012). These phenotypes are rescued by partial loss of miR-34 target Eip74EF, a component of steroid signaling; this pathway is necessary for normal development and has been previously implicated in aging (Simon et al. 2003). Overexpression of mir-34 also extends lifespan; however, whether this extension depends on suppression of Eip74EF is unknown (Liu et al. 2012). Similarly, loss of mir-1000 shortens lifespan and exacerbates age-related neurodegeneration through targeting of the glutamate transporter VGlut (Verma et al. 2015). The role of miR-34 and miR-1000 in age-related neurodegeneration is discussed in more detail in subsequent sections.
For short-lived miRNA mutants, overexpressing the gene product also commonly results in a short-lived or otherwise deleterious phenotype. mir-282 mutants are short-lived and have a reduced hatching rate compared to wild-type animals, while mir-282 overexpression causes larval lethality (Vilmos et al. 2013). Targeting of rutabaga, an adenylate cyclase expressed specifically in the nervous system, may be responsible for these phenotypes; however, it is unknown whether this is the primary function of miR-282. Similarly, loss or overexpression of mir-277, which is down-regulated with age, shortens lifespan (Esslinger et al. 2013; Chen et al. 2014). Overexpression of mir-277 results in upregulation of genes involved in branched chain amino acid (BCAA) catabolism and directly targets two enzymes in the pathway, CG8199 and CG5599. This overexpression also activates TOR kinase in cell culture, possibly through accumulation of BCAA metabolic intermediates; however, an increase in mTOR signaling has not been directly linked to miR-277’s effect on lifespan. One exception to short-lived miRNA mutants is mir-305: knockdown of mir-305, which is normally down-regulated with age, modestly extends lifespan (Ueda et al. 2018). Overexpression of mir-305 reduces lifespan and exacerbates age-related decline in movement and protein homeostasis. Ectopic expression results in down-regulation of genes involved in innate immunity; however, no direct targets have been confirmed.
Mouse
Few lifespan-modulating miRNAs have been described in mammalian species, as genetic manipulations and lifespan studies are much more difficult to execute. To date, miR-17 is the only microRNA that has been reported to directly extend lifespan in a mammalian model. Transgenic mir-17 mice, which ubiquitously overexpress mir-17, are long-lived and exhibit reduced senescence across multiple organ systems (Du et al. 2014). miR-17 directly targets insulin receptor substrate (Irs1) and adenylate cyclase 5 (Adcy5) to inhibit senescence through multiple downstream effectors. Suppression of Adcy5 allows for binding of the RGS2-HIF-1a complex to the promoter of Mkp7, inducing its expression; MKP7 then inhibits mTOR signaling through dephosphorylation of mTOR. The role of miR-17 in promoting longevity is unexpected, since it is generally considered to be an oncogene; however, this may be confounded by the activity of other members of the cluster from which miR-17 is transcribed (Dellago et al. 2017).
Differential regulation of miRNAs in long-lived mouse models may inform as to other miRNAs involved in longevity. The long-lived Ames dwarf mice, which derives its longevity from pituitary defects and growth hormone deficiencies, is also known to have reduced insulin/IGF-1 signaling and increased insulin sensitivity (Bartke and Brown-Borg 2004). Aged Ames mice show increased expression of miR-470, miR-681, and miR-669b in the brain compared to wild-type animals (Liang et al. 2011). These miRNAs all directly target the Igf1r transcript, suppressing expression of Igf1r and the phosphorylated forms of FOXO3 and AKT, two components of the insulin/IGF-1 signaling pathway. Multiple miRNAs are also upregulated in aged Ames mouse liver tissue compared to controls, including miR-27a (Bates et al. 2010). miR-27a directly targets two members of the polyamine synthesis pathway, ornithine decarboxylase (Odc1) and spermidine synthase (Srm). This regulation may contribute to Ames mouse longevity as an anti-tumor mechanism, as polyamine metabolism is required for cell proliferation and is often dysregulated in cancer (Murray-Stewart et al. 2016).
miRNAs in the aging brain
Normal aging in the human brain is characterized by both morphological and functional changes. Brain weight declines steadily with age, total myelination length decreases, and cortical volume is lost as neurons in the front and temporal lobes shrink in size (Sowell et al. 2004). Altered protein homeostasis, characteristic of multiple age-associated neurodegenerative diseases like Alzheimer’s and Parkinson’s, is also common in normal aging. Increased presence of β-amyloid, tau, and α-synuclein aggregates is observed in a significant fraction of aged individuals in the absence of significant cognitive impairment or disease presentation (Elobeid et al. 2016). Reduced brain function and progressive cognitive decline also occur with aging, bringing about impaired learning, memory formation, and processing speed (Hedden and Gabrieli 2004). A number of studies have shown that the miRNA profile of the brain changes with normal aging in mammalian species (Li et al. 2011a; Inukai et al. 2012; Yin et al. 2015; Chen et al. 2019) and is differentially regulated in different regions of the brain (Persengiev et al. 2012). The process of normal brain aging is complex and poorly understood; thus, the role of miRNAs has been most extensively studied in the context of neuropathologies associated with age. Here, we summarize available evidence on the role of miRNAs in normal brain aging.
miR-34, which is highly conserved in C. elegans, Drosophila, mice, and humans, has been identified in a number of brain aging studies. In Drosophila, miR-34 is upregulated with age in the brain and is required for maintaining healthy brain tissue and function (Liu et al. 2012). mir-34 mutants have short lifespans, early onset neurodegeneration, and a faster rate of increase in age-associated transcripts in the brain compared to controls, indicating that miR-34 accelerates the rate of brain aging. Conversely, upregulation of miR-34 extends lifespan and improves neurodegeneration induced by polyglutamine aggregation; modulation of these phenotypes relies on repression of the A isoform of Eip74EF (E74A), a steroid signaling transcription factor. Interestingly, Eip74EF is required for normal development, suggesting that miRNA silencing of developmental genes in adulthood may be necessary for healthy aging (Liu et al. 2012). Recent evidence has shown that miR-34 also modulates brain aging in Drosophila by targeting two components of the polycomb repressive complex 2 (PRC2), Pcl and Su(z)12. Reduction of PRC2 activity resulted in brain transcriptomic profiles associated with youth, including upregulation of chaperone proteins (Kennerdell et al. 2018). The accelerated aging experienced by mir-34 mutants may be caused by failure to repress activity of PRC2, allowing silencing of genes that are associated with healthy brain aging. miR-1000, which is enriched in the central nervous system and decreases progressively with age, also plays a neuroprotective role in aging Drosophila (Verma et al. 2015). mir-1000 mutants have dramatically shorter lifespans, disordered movement, and early onset of neurodegeneration compared to wild-type counterparts. These phenotypes result from de-repression of direct target vesicular glutamate transporter (VGlut), dysregulation of presynaptic glutamate release, and subsequent glutamate excitotoxicity leading to neuronal cell death. Although miR-1000 appears to be Drosophila-specific, the seed sequence (region that binds to the 3′UTR of the target transcript) is largely similar to that of mammalian miR-137. miR-137 has also been demonstrated to target a glutamate transporter (VGluT2) in mouse neurons. Loss of mir-137 leads to increased neuron death, indicating a potentially similar role in regulating neurodegeneration (Verma et al. 2015).
Members of the miR-34 family appear to play a different, less beneficial role in mammalian brain aging compared to their invertebrate counterparts. A mammalian member of the miR-34 family, miR-34c, may actually contribute to cognitive decline associated with normal and pathological brain aging. miR-34c is upregulated in mouse hippocampus in both normal aging and Alzheimer’s disease (AD) models, as well as in humans with AD (Zovoilis et al. 2011). Overexpression of miR-34c in hippocampus of young mice results in memory impairment associated with old age and decreased expression of target sirtuin 1 (Sirt1), a positive regulator of lifespan. Another family member, miR-34a, is upregulated with age in both mouse brain and blood samples, while expression of target Sirt1 declines inversely in both tissues. The correlation between miR-34a/Sirt1 expression in the blood and brain may make them valuable biomarkers for noninvasive detection of brain aging, a tissue which is normally inaccessible (Li et al. 2011b). Interestingly, calorie-restricted mice do not show an age-dependent upregulation of miR-34a in the brain, which is observed in ad libitum controls (Khanna et al. 2011). Conversely, the anti-apoptotic gene Bcl2, a direct target of miR-34a, is upregulated with age in the brain tissue of calorie-restricted mice but down-regulated in controls. In vitro repression of Bcl2 by overexpression of miR-34a caused increased cell death and caspase expression, suggesting that the beneficial effects of calorie restriction on aging and longevity may be mediated through miR-34a. The study showed that two other age-regulated brain miRNAs, miR-181-a-1* and miR-30e, behave similarly to miR-34a (Khanna et al. 2011).
Aggregation of beta amyloid (Aβ) in the brain, a hallmark of normal and pathological brain aging, results from abnormal processing of amyloid precursor protein (APP) by the enzyme BACE1. Evidence suggests that there may be a number of Bace1-targeting miRNAs that are dysregulated in the brain with age (Che et al. 2014). However, most of these miRNAs have been studied specifically in the context of Alzheimer’s disease, and their role in normal aging is unclear. One exception is miR-186, the expression of which decreases with age in the mouse brain cortex (Kim et al. 2016). In vitro overexpression of miR-186 suppresses Bace1 to reduce Aβ formation; it follows that age-induced down-regulation of miR-186 and resulting upregulation of Bace1 may set the stage for Aβ accumulation and development of disease.
Age-associated upregulation of the miR-29 family in mouse brain, particularly miR-29a and miR-29b, results in dysregulation of microglia and increased neuroinflammation, a hallmark of brain aging. Igf1 and fractalkine ligand Cx3cl1, known inhibitors of microglia activity, are directly suppressed by miR-29a/b, and both targets are reduced in the brain with age (Fenn et al. 2013). Under inflammatory conditions, miR-29b expression is induced and specifically suppresses Igf1 in the microglia, suggesting that the age-associated increase in miR-29 expression may be part of a detrimental feedback response to increasing inflammation in the aged brain. miR-29 is also induced in the brain neurons of killifish as part of an adaptive response to age-associated iron accumulation, a process ubiquitous in model invertebrate and mammalian organisms (Ripa et al. 2017). miR-29 regulates iron homeostasis through targeting Ireb2, which codes for an RNA-binding protein that promotes intracellular transport of iron in the brain. It appears the upregulation of miR-29 with age may be part of a broad response to counteract age-associated phenotypes in the brain. Surprisingly, knockout of mir-29 in the brain does not significantly affect lifespan in mice, although knockouts trended towards shorter lifespan (Takeda and Tanabe 2016).
miRNAs in aging muscle
Sarcopenia, a progressive, age-related loss of skeletal muscle mass and function, is highly prevalent in older adults and is associated with frailty, impaired balance, falls, and reduced quality of life. Loss of normal muscle movement and function leads to a reduction in physical activity, exacerbating muscle loss and increasing elderly individuals’ susceptibility to permanent disability, metabolic disorders, and other aging pathologies (Fuggle et al. 2017). The pathophysiology of sarcopenia is multifactorial and not fully understood, although reduced IGF-1 signaling, chronic inflammation, muscle fiber denervation, and increased myostatin/SMAD signaling have all been implicated in disease development (Marty et al. 2017). Expression profiling in mouse (Hamrick et al. 2010; Kim et al. 2014; Soriano-Arroquia et al. 2016), primate (Mercken et al. 2013), porcine (Redshaw et al. 2014), and human (Drummond et al. 2011; Zacharewicz et al. 2014; Rivas et al. 2014) skeletal muscle has revealed a number of miRNAs dysregulated with age. A subset of these miRNAs, including miR-181a, miR-434-3p, miR-431, miR-29, and miR-126, have been found to play a role in the underlying pathology of sarcopenia, modulating processes like apoptosis, senescence, and IGF-1 signaling in aging muscle cells.
miR-181a is down-regulated in muscle from aged mice (Hamrick et al. 2010; Soriano-Arroquia et al. 2016) and rhesus monkeys (Mercken et al. 2013). Protein levels of Sirt1, a direct target of miR-181a, increase with age in the skeletal muscle; in vitro, miR-181a negatively regulates myotube diameter through repression of Sirt1 (Soriano-Arroquia et al. 2016). Down-regulation of miR-181a with age may be part of a compensatory mechanism to counteract muscle loss; interestingly, this down-regulation is mitigated by calorie restriction in primates, possibly reflecting delayed muscle aging (Mercken et al. 2013). Regardless, a functional role for miR-181a specifically in the context of aged skeletal muscle remains to be described.
miR-434-3p is also down-regulated with age in mouse skeletal muscle (Hamrick et al. 2010; Pardo et al. 2017; Jung et al. 2017). In vitro, miR-434-3p protects myotubes from TPEN-induced apoptosis and reduces caspase activation through target eukaryotic translation initiation factor 5A1 (Eif5a1), a pro-apoptotic gene. Increased protein levels of eIF5A1 are also observed in aged skeletal muscle, suggesting that dysregulation of miR-434-3p may contribute to muscle loss by promoting apoptosis through de-repression of Eif5a1 (Pardo et al. 2017). Age-related reduction in muscle expression of miR-434-3p is also reflected in serum of old mice, highlighting its potential use as a circulating biomarker for muscle aging (Jung et al. 2017). Another potentially protective miRNA, miR-431, is down-regulated in myoblasts from old mice compared to younger individuals (Lee et al. 2015). Exogenous expression of miR-431 restores myogenesis in old myoblasts through suppression of Smad4, a component of the SMAD transcriptional complex known to inhibit the regenerative capacity of muscle cells. Both overexpression of miR-431 and knockdown of Smad4 in vivo improved muscle regeneration in aged mice, highlighting the therapeutic potential of miR-431 in muscle aging.
Unlike the other muscle-expressed miRNAs described here, miR-29 is upregulated with age in rodents (Hu et al. 2014) and a progeria mouse model (Ugalde et al. 2011). In vivo overexpression of miR-29 induces senescence and inhibits muscle cell proliferation in young skeletal muscle through suppression of targets Igf1, P85, and Bmyb (Hu et al. 2014). Down-regulation of these mediators of muscle growth and proliferation by miR-29 may contribute to muscle loss. Evidence from Ugalde et al. suggests that the age-related induction of miR-29 family expression is caused by accumulation of DNA damage. miR-29 is upregulated as part of the DNA-damage response and represses Ppm1d phosphatase, a negative regulator of p53 activity (Ugalde et al. 2011). Thus, miR-29 may play a pivotal role in muscle cell survival and proliferation with age and serve as a promising therapeutic target for sarcopenia.
Recent evidence from Rivas et al. showed that a reduction in muscle hypertrophic response to resistance exercise, a possible contributor to sarcopenia, is also regulated by miRNAs. Interestingly, changes from baseline in the miRNA expression profile post-exercise were observed in young but not old human skeletal muscle, suggesting that elderly individuals may have a reduced transcriptional response to activity (Rivas et al. 2014). One such dysregulated miRNA, miR-126, was significantly lower in older individuals at baseline and down-regulated in response to exercise in young but not old men. Inhibition of miR-126 significantly increased Akt and FOXO1 activation in myotubes upon IGF-1 anabolic stimulation, demonstrating a possible role for miR-126 in muscle growth suppression and the age-dysregulated hypertrophic response.
miRNAs in aging bone
Progressive bone loss, a consequence of normal aging in humans, typically begins after age 30 and is accompanied by significant structural and compositional changes that predispose elderly individuals to osteoporosis (Boskey and Imbert 2017). Osteoporosis is a chronic disease in which bone resorption exceeds formation, causing pathologic changes to bone microarchitecture, mass, and strength that dramatically increase lifetime fracture risk. Osteoporosis-related fractures commonly occur in the hip or spine, negatively impacting individual mobility and quality of life while significantly increasing mortality (Rachner et al. 2011). Age-related deregulation in bone remodeling, the cycle of bone removal by osteoclasts and bone deposition by osteoblasts, is a key process underlying disease (Demontiero et al. 2012). miRNAs are increasingly recognized as playing a role in this complex process, presenting new therapeutic targets to prevent bone disease and resulting frailty in the elderly (Feng et al. 2018).
Recent work suggests that miR-21 plays a role in mammalian osteocyte apoptosis (Davis et al. 2017b), a process that increases with age and contributes to bone loss by recruiting and promoting osteoclast activity (Plotkin and Bellido 2016). miR-21 expression is significantly decreased in aged mouse bone, and genetic knockdown of miR-21 induces osteocyte death through increased expression of target phosphatase and tensin homolog (Pten), a pro-apoptotic gene (Davis et al. 2017b). Evidence supports a model where miR-21 lies downstream of connexin43 (Cx43), which codes for a gap junction protein that also decreases with age and protects against osteocyte apoptosis; however, the exact relationship between Cx43 and miR-21 has yet to be characterized (Davis et al. 2017b). miR-21 is also down-regulated in bone marrow mesenchymal stem cells from mice and humans with estrogen deficiency/ovariectomy-induced osteoporosis, and promotes osteoblast differentiation and bone formation in vivo (Yang et al. 2013b). In this disease context (and possibly normal aging), TNF-α was found to suppress miR-21 expression and increase expression of direct target Spry1, a negative regulator of FGF and ERK-MAPK signaling, ultimately impairing osteogenesis and bone formation. miR-21 may also modulate osteoporosis through targeting Reck, a negative regulator of matrix metalloproteinases involved in osteogenesis; Reck knockdown improves bone loss in ovariectomy-induced osteoporotic mice (Zhao et al. 2015b). Surprisingly, another study has shown that miR-21 knockout protects against bone loss and resorption in both normal aging and ovariectomy-induced osteoporosis; these effects are attributed to reduced osteoclast differentiation mediated by direct target Pdcd4 (Hu et al. 2017). The role of miR-21 in maintaining bone homeostasis in normal and pathological aging is clearly complex and merits further study.
The miRNA content of bone marrow-secreted microvesicles changes with age and may contribute to bone’s reduced capacity for growth and repair. miR-183-5p, a member of the miR-183 cluster, is highly expressed in aged mouse bone marrow-derived extracellular vesicles. When these vesicles are absorbed by young bone marrow stromal cells (BMSCs), mir-183-5p inhibits BMSC proliferation and bone formation (Davis et al. 2017a). miR-183 also induces osteoclast differentiation in bone marrow-derived macrophages through target heme oxygenase-1 (Ho1), suggesting that miR-183 plays a role in age-related bone loss by affecting both adsorption and resorption processes (Ke et al. 2015). miR-31, a miRNA found in senescent endothelial cell-secreted vesicles, is elevated with age in human plasma and upregulated in osteoporotic men compared to healthy controls. Vesicular miR-31 inhibits osteogenic differentiation through target Fzd3, a Wnt5a receptor (Weilner et al. 2016).
Another age-related miRNA, miR-214, is upregulated with age in bone from osteoporotic humans and mice. miR-214 inhibits osteoblast activity and mineralization through target Atf-4, and in vivo overexpression of miR-214 in osteoblasts significantly decreases bone formation (Wang et al. 2013). miR-214 is also essential for osteoclastogenesis and promotes this process through target Pten and subsequent activation of the PI3 K/Akt pathway (Zhao et al. 2015a). Inhibition of miR-214 in osteoblasts of aged ovariectomy-induced osteoporotic mice improves bone mass, architecture, and formation of new bone (Wang et al. 2013). Conversely, overexpression of miR-214 in osteoclasts in vivo leads to increased osteoclast activity and bone loss (Zhao et al. 2015a).
Recently, osteoclast-derived exosomal miR-214 has been found to play a significant role in inhibition of osteoblast activity and bone formation (Sun et al. 2016; Li et al. 2016). miR-214 is secreted from osteoclasts in miRNA-enriched exosomes, which are internalized by osteoblasts to inhibit their activity both in vitro and in vivo. Furthermore, blocking exosome release or inhibiting miR-214 in osteoclasts increases osteoblast activity and bone formation in aging ovariectomized mice (Sun et al. 2016; Li et al. 2016). Levels of miR-214 in serum exosomes of elderly women with fractures not only reflect expression levels in bone tissue, but negatively correlate with bone formation, making miR-214 both a potential biomarker of bone loss and a therapeutic target (Li et al. 2016).
With age, BMSCs preferentially differentiate into adipocytes rather than osteoblasts, leading to increased fat accumulation in bone marrow and reduced bone growth (Moerman et al. 2004). The switch from osteogenic to adipogenic differentiation is promoted by miR-188, which is elevated with age in both mouse and human BMSCs (Li et al. 2015). miR-188 targets Rictor and Hdac9, important bone metabolism genes, in BMSCs, activating the adipocyte differentiation transcription factor PPARy and increasing BMSC differentiation into adipocytes rather than osteoblasts (Li et al. 2015). As such, aged miR-188 knockout mice show increased osteoblastic bone formation, reduced bone loss, and less fat accumulation in bone marrow compared to wild type. Interestingly, no difference was observed between the two backgrounds in young animals, suggesting that miR-188 acts specifically during aging. Furthermore, BMSC-specific injection of miR-188 inhibitor was also able to increase bone formation in aged mouse (Li et al. 2015). Similar to miR-188, miR-218 is also elevated in aged mouse BMSCs and a direct regulator of Rictor; however, miR-218 promotes bone loss by inhibiting osteoblast adhesion and survival, preventing formation of new bone rather than affecting differentiation (Lai et al. 2016). Due to their negative effects on bone formation, both miR-218 and mir-188 are potential therapeutic targets for age-related bone loss.
miRNAs in the aging heart
Age is the prevailing risk factor for the development of cardiovascular disease (CVD), a leading cause of mortality in the 65 and older population (North and Sinclair 2012). Part of the age-associated risk for CVD stems from morphological and functional changes that occur in the heart during the normal aging process. In the human heart, the number of myocytes progressively declines, while the presence of fibrotic tissue and lipid deposition increases with advancing age. Functionally, cardiac output and maximum heart rate generally decrease with age, and impairment of diastolic function and ventricular loading can be observed (Epstein and Wei 1992). These changes, along with a decline in cardiac repair mechanisms and capacity for self-renewal, set the stage for the development of cardiac pathology in elderly individuals (Strait and Lakatta 2012). Cardiovascular disease commonly presents with the occurrence of a myocardial infarction (MI); aged individuals are at much higher risk for experiencing an MI and are less likely to survive than their younger counterparts. The elderly are also at higher risk to develop chronic heart failure as a result of the MI compared to younger patients (Shih et al. 2011). The expression of miR-21, miR-22, miR-34a, and the miR-17–92 cluster is significantly altered in normal cardiac aging and age-related cardiac pathologies and important functional roles these for miRNAs in both processes have been described.
miR-21 is highly expressed in the heart tissue of aged mouse compared to young animals (Zhang et al. 2012) and is strongly induced in the post-MI infarction zone by TGF-β1 (Yuan et al. 2017; Zhou et al. 2018). Through repression of its targets Jagged1 and Smad7, miR-21 mediates the cardiac fibroblast-to-myofibroblast transition and subsequent formation of fibrotic tissue in the heart, a key pathology in post-MI cardiac remodeling (Yuan et al. 2017; Zhou et al. 2018). Gene therapy targeting miR-21 in cardiac nonmyocytes reduced pathogenic remodeling and functional impairment in a mouse cardiac disease model, making inhibition of miR-21 an attractive therapeutic strategy for improving disease outcomes in humans post-MI (Ramanujam et al. 2016). Circulating levels of miR-21 have also been used to successfully identify patients experiencing aortic stenosis-induced fibrosis (Villar et al. 2013) and acute MI (Zhang et al. 2016), demonstrating the potential utility of miR-21 as a diagnostic biomarker for cardiac fibrosis and disease.
miR-22 expression progressively increases with age in mouse and human cardiac tissue (Jazbutyte et al. 2013; Gupta et al. 2016). In mice, miR-22 regulates senescence in cardiac fibroblasts through mimecan (osteoglycin/Ogn), a direct target of miR-22 that decreases with age in the myocardium (Jazbutyte et al. 2013). Interestingly, mimecan knockout mice show significantly higher mortality post-MI compared to wild type, as well as increased cardiac dysfunction and collagen abnormalities (Van Aelst et al. 2015). Cardiomyocyte-specific overexpression of miR-22 causes hypertrophy and impaired diastolic function, mediated through repression of Ppar, Sirt1, and Pgc1 (direct interaction has been demonstrated for Ppar and Sirt1) (Gurha et al. 2013). Conversely, inhibition of miR-22 in aged rat cardiomyocytes reduces hypertrophy and activates myocardial autophagy, a critical process for maintaining cardiac function with age (Gupta et al. 2016). Pharmacological inhibition in aged mouse post-MI also improves cardiac function and pathological remodeling (Gupta et al. 2016). miR-22 may be a promising therapeutic target for both normal aging and MI-related cardiac pathologies.
miR-34a expression is induced in the aging mouse heart and promotes age-associated cardiomyocyte death and decline in cardiac contractile function (Boon et al. 2013). miR-34a is also induced post-MI, and miR-34a inhibition reduces cell death, fibrosis, and functional decline associated with heart attack (Boon et al. 2013; Huang et al. 2014; Yang et al. 2015). These effects are in part mediated through targeting of the protein phosphatase Pnuts, an apoptosis inhibitor and regulator of telomere maintenance, and DNA-damage response (Boon et al. 2013). Cardiomyocyte survival post-MI is regulated through targets Bcl2, Ccnd1, and Sirt1, which are known to play roles in aging and cell cycle activity (Yang et al. 2015). Interestingly, miR-34a expression is protective against ischemia-reperfusion injury post-MI, suggesting that the therapeutic role for this miRNA is complex (Shao et al. 2018).
The miR-17-92 cluster, which consists of six individually transcribed miRNAs (miR-17, mir-18a, miR-19a, mir-19b, miR-20a, and miR-92a-1), has been implicated in a number of age-related processes and diseases (Dellago et al. 2017). Expression of all cluster members is down-regulated with age in heart failure-prone (but not heart failure-resistant) mice, and miR-18a, miR-19a, and miR-19b are specifically depleted with age in failed but not healthy human heart tissue (van Almen et al. 2011). miR-18a and mir-19a/b regulate pro-fibrotic genes Ctgf and Tsp1 in aged cardiomyocytes, affecting collagen synthesis and fibrotic remodeling (van Almen et al. 2011). miR-17 has been shown to suppress senescence and apoptosis in mouse myocardium and cardiac fibroblasts through targeting of Par4, a pro-apoptotic protein (Du et al. 2015). miR-17 also targets Timp1 and Timp2, tissue inhibitors of metalloproteinases, to prevent excessive proteolysis and cardiac remodeling; inhibiting miR-17 in vivo prevents this remodeling and improves cardiac function post-MI (Li et al. 2013).
Circulatory miRNAs as biomarkers of aging and longevity
miRNAs are secreted from cells into peripheral bodily fluids through microvesicles and exosomes, either in association with RNA-binding proteins like Argonaute 2, or bound to high-density lipoproteins (Jung and Suh 2014). These ‘circulatory’ miRNAs have been observed in at least 12 different human bodily fluids, including blood, saliva, cerebrospinal fluid, amniotic fluid, breast milk, and urine (Weber et al. 2010). Circulatory miRNAs are highly abundant, stable, and have been demonstrated to change with chronological age and development of age-related pathologies, sparking interest in their use as noninvasive “biomarkers of aging”, i.e., parameters that predict longevity better than chronological age itself (Kumar et al. 2017). An accurate, noninvasive biomarker of aging would have a variety of potential diagnostic and therapeutic applications, providing the ability to identify and intervene for individuals who are experiencing accelerated aging and measure the efficacy of therapeutic interventions aimed at reducing the rate of aging. Recent studies (2013 to present) have measured differential miRNA expression between young and aged individuals in several different peripheral fluids, including serum (Hooten et al. 2013; Sawada et al. 2014; Zhang et al. 2015), plasma (Olivieri et al. 2014; Ameling et al. 2015), and saliva (Machida et al. 2015). Efforts to link circulating miRNA profiles directly to lifespan in humans have also been reported (Smith-Vikos et al. 2016; Wu et al. 2016). These studies are summarized in Table 3 and described in brief below.
Hooten et al. profiled circulating miRNAs in serum of old (mean age of 64.6 years) and young individuals (mean age of 30.5 years), identifying miR-181a-5p, miR-1248, and miR-151a-3p to be significantly down-regulated with age (Hooten et al. 2013). miR-1248 and miR-151-5p were also found to be down-regulated in serum of aged rhesus monkeys, demonstrating similarities of age-related changes in miRNA expression across aging models. Target prediction and pathway analysis of miR-181a-5p, miR-1248, and miR-151a-3p showed overlapping involvement in inflammatory pathways, consistent with the relationship between chronic inflammation and aging (Hooten et al. 2013). Another study profiling miRNAs in serum from adults of various ages found miR-29b, miR-106b, miR-130b, miR-142-5p, and miR-340 to be down-regulated with age, while miR-92a, miR-222, and miR-375 increased in expression (Zhang et al. 2015). Bioinformatic analysis indicated that cancer, an age-related pathology, was the most common disease predicted to be affected by differential expression of these miRNAs; similarly, the most common functions of predicted age-dependent miRNA targets were roles involving the cell cycle, proliferation, and transcription. A large, population-based study comparing plasma between 374 young and old human individuals found miR-126-3p, miR-30c-5p, miR-30b-5p, miR-210, miR-142-3p, and let-7a-5p to increase with age, and miR-93-5p to decrease with age (Ameling et al. 2015). In a rare example of overlap, an independent study also found mir-126-3p to be upregulated in aged human plasma (Olivieri et al. 2014).
The ability to link circulatory miRNA expression with not only age but future longevity would be of great prognostic value; however, few of these studies have been conducted due to the rarity of longitudinal human data sets that include lifespan. One such study used serum samples collected longitudinally from short-lived (58–75 years) and long-lived (76–92 years) participants of the Baltimore Longitudinal Study of Aging (Smith-Vikos et al. 2016). Twenty-four miRNAs were found to be significantly upregulated and seventy-three significantly down-regulated in long-lived individuals; six of these miRNAs, miR-211-5p, miR-5095, miR-1225-3p, miR-374a-5p, miR-340-3p, and miR-376c-3p, were both differentially expressed and correlated with individual lifespan. Of the validated targets of this subset of miRNAs, 24 encode gene products associated with aging processes, including a DNA repair protein (PARP1) and insulin signaling pathway receptors (IGF1R, IGF2R). Another report aiming to link circulating miRNAs with longevity used plasma samples from the National Heart, Lung, and Blood Institute Twin Study (Wu et al. 2016). Five monozygotic twin pairs with a difference in lifespan of at least 5 years were analyzed, using a plasma sample collected at middle age (average of 46 years) and archived. The study found that miR-3615 and miR-619 were significantly higher in short-lived twins compared to their long-lived co-twin; however, these results were not significant once multiple hypothesis correction was applied.
Multiple studies have demonstrated an association between circulatory miRNA expression and age or longevity in humans. However, there is little overlap between studies, even among those utilizing the same sample type, making it difficult to develop a ‘gold-standard’ miRNA biomarker of aging. Some of the discrepancies may be explained by differences in study design, participant demographics, and miRNA profiling methods. Regardless, circulating miRNAs remain a promising opportunity as noninvasive biomarkers of aging.
Conclusions and perspectives
miRNAs play important regulatory roles in the complex process of aging, targeting a diverse array of transcripts and pathways—in some cases organism-wide and others in a tissue-specific manner. Multiple miRNAs that directly modulate longevity in the invertebrates C. elegans and Drosophila have been identified, including those that extend lifespan upon overexpression. Some of these longevity-promoting miRNAs, including lin-4, let-7, and miR-34 are well-conserved in humans, presenting the possibility that modulating expression of such miRNAs could extend human lifespan. However, only one lifespan-modulating miRNA, miR-17, has thus far been reported in a mammalian species. It remains to be seen whether other longevity-promoting miRNAs will be discovered in mammals and whether they will be relevant to human longevity. Regardless of their conservation in higher order species, studies of lifespan-modulating miRNAs in model organisms have greatly contributed to our understanding of the molecular mechanisms and pathways involved in aging.
miRNAs appear to play both beneficial and detrimental roles in the aging of different organs and tissues in mammalian systems (Fig. 2). As our understanding of these roles continues to expand, so does the therapeutic potential of modulating specific miRNAs to change the course of the aging process and associated diseases. miRNA mimics—synthetic RNA molecules designed to mimic endogenous miRNAs—may be used to replenish protective miRNAs that are normally down-regulated with age. Likewise, miRNA inhibitors (antimiRs) and/or sponges—synthetic miRNA targets that titrate away active miRNAs—could be used to reduce the impact of miRNAs that promote aging. Over the past decade, a number of promising early phase clinical trials utilizing miRNA therapeutics have been conducted for the treatment of Hepatitis C, diabetes, and various cancers (Rupaimoole and Slack 2017). However, miRNA therapeutics have yet to demonstrate efficacy in late-stage clinical trials or gain FDA approval (Hanna et al. 2019). In designing miRNA therapeutics for aging and age-related pathologies, a number of challenges such as tissue-specific targeting, delivery efficiency, and off-target effects will need to be addressed (Li and Rana 2014). While targeting miRNAs is a promising treatment strategy for aging, it is likely that fully realized therapeutics remain many years away.
Many miRNAs are differentially expressed with aging across multiple organs and tissues in mammals, and some have been demonstrated to regulate processes associated with age-related decline, including sarcopenia, bone loss, cognitive decline, and cardiac dysfunction. However, it is important to note that significant up- or down-regulation of a miRNA with age only suggests rather than proves an important role in the aging process. Additional genetic studies examining the effects of miRNA knockdown or overexpression will be critical to demonstrate direct regulatory roles for miRNAs in aging. Such studies are often performed in disease models of age-related pathologies, which provide valuable insights into aging dysfunction. However, examining the function of miRNAs in the context of natural aging, which may be mediated by processes that are distinct from age-related pathologies, will also be of significant value. These studies will also be necessary to determine whether direct modulation of miRNA expression is feasible and effective for therapeutic applications. The tissue specificity of miRNA expression and action, particularly in mammalian species, also presents additional complexity to understanding their role in aging. A miRNA may have different targets, functionality, and effects on aging and lifespan depending on the tissue in which it is expressed. Thus, tissue-specific knockdown or overexpression of miRNAs will be highly relevant to understanding an miRNA’s effect on aging in mammalian systems.
Many studies have shown that differential expression of miRNAs in circulating biological fluids (circulatory miRNAs) is correlated to aging and lifespan in humans. These findings, along with the ease of obtaining samples from human patients, have generated significant interest in their use as noninvasive biomarkers of aging and gauges of individual decline. Such a biomarker would have significant diagnostic and therapeutic potential, providing the ability to identify cases of accelerated aging and measure the efficacy of therapeutic interventions aimed at addressing aging. However, there is little concordance between published findings (even those using the same sample type), most likely due to inter-individual variation and differences in study design and technique. As such, a ‘gold-standard’ circulatory miRNA biomarker of aging does not yet exist. More studies on diverse populations and analytical standardization may serve to address this issue.
There are many aspects of miRNA involvement in longevity and aging that remain to be understood. While target genes (and computationally predicted targets) have been identified for most of the miRNAs discussed in this review, it is likely that many of the targets for any given miRNA have yet to be discovered. More studies verifying direct binding between a miRNA and predicted targets, with demonstrated functional consequences in the aging process, will substantially advance the field. Because miRNAs show marked changes in expression during aging, these changes are expected to significantly influence the expression of many target genes, with broad and significant impacts on health and functionality of the aging individual. Continuing to identify genes that are regulated by aging-associated miRNAs will provide further insight into the pathways and processes that underly aging. While much has been done to identify regulatory targets for age-associated miRNAs, regulation of the miRNAs themselves is almost entirely unexplored. Studies identifying the upstream factors, particularly transcription factors and RNA-binding proteins, that mediate differential miRNA expression will be critical in understanding how and why the aging process is set into motion.
References
Aalto AP, Nicastro IA, Broughton JP et al (2018) Opposing roles of microRNA Argonautes during Caenorhabditis elegans aging. PLoS Genet 14:e1007379. https://doi.org/10.1371/journal.pgen.1007379
Ameling S, Kacprowski T, Chilukoti RK et al (2015) Associations of circulating plasma microRNAs with age, body mass index and sex in a population-based study. BMC Med Genomics 8:61. https://doi.org/10.1186/s12920-015-0136-7
Axtell MJ, Westholm JO, Lai EC (2011) Vive la différence: biogenesis and evolution of microRNAs in plants and animals. Genome Biol 12:221. https://doi.org/10.1186/gb-2011-12-4-221
Bartke A, Brown-Borg H (2004) Life extension in the dwarf mouse. Curr Top Dev Biol 63:189–225. https://doi.org/10.1016/S0070-2153(04)63006-7
Bates DJ, Li N, Liang R et al (2010) MicroRNA regulation in Ames dwarf mouse liver may contribute to delayed aging. Aging Cell 9:1–18. https://doi.org/10.1111/j.1474-9726.2009.00529.x
Beltrán-Sánchez H, Soneji S, Crimmins EM (2015) Past, present, and future of healthy life expectancy: figure 1. Cold Spring Harb Perspect Med 5:a025957. https://doi.org/10.1101/cshperspect.a025957
Boehm M, Slack F (2005) A developmental timing microRNA and its target regulate life span in C. elegans. Science 310:1954–1957. https://doi.org/10.1126/science.1115596
Boon RA, Iekushi K, Lechner S et al (2013) MicroRNA-34a regulates cardiac ageing and function. Nature 495:107–110. https://doi.org/10.1038/nature11919
Boskey AL, Imbert L (2017) Bone quality changes associated with aging and disease: a review. Ann N Y Acad Sci 1410:93–106. https://doi.org/10.1111/nyas.13572
Boulias K, Horvitz HR (2012) The C. elegans microRNA mir-71 acts in neurons to promote germline-mediated longevity through regulation of DAF-16/FOXO. Cell Metab 15:439–450. https://doi.org/10.1016/J.CMET.2012.02.014
Bushati N, Cohen SM (2007) MicroRNA functions. Annu Rev Cell Dev Biol 23:175–205. https://doi.org/10.1146/annurev.cellbio.23.090506.123406
Chawla G, Deosthale P, Childress S et al (2016) A let-7-to-miR-125 MicroRNA switch regulates neuronal integrity and lifespan in Drosophila. PLoS Genet 12:e1006247. https://doi.org/10.1371/journal.pgen.1006247
Che H, Sun L-H, Guo F et al (2014) Expression of amyloid-associated miRNAs in both the forebrain cortex and hippocampus of middle-aged rat. Cell Physiol Biochem 33:11–22. https://doi.org/10.1159/000356646
Chen Y-W, Song S, Weng R et al (2014) Systematic study of Drosophila microRNA functions using a collection of targeted knockout mutations. Dev Cell 31:784–800. https://doi.org/10.1016/j.devcel.2014.11.029
Chen J, Zou Q, Lv D et al (2019) Comprehensive transcriptional profiling of porcine brain aging. Gene 693:1–9. https://doi.org/10.1016/J.GENE.2019.01.019
Crimmins EM (2015) Lifespan and healthspan: past, present, and promise. Gerontologist 55:901–911. https://doi.org/10.1093/geront/gnv130
da Costa JP, Vitorino R, Silva GM et al (2016) A synopsis on aging-theories, mechanisms and future prospects. Ageing Res Rev 29:90–112. https://doi.org/10.1016/j.arr.2016.06.005
Davis C, Dukes A, Drewry M et al (2017a) MicroRNA-183-5p increases with age in bone-derived extracellular vesicles, suppresses bone marrow stromal (stem) cell proliferation, and induces stem cell senescence. Tissue Eng Part A 23:1231–1240. https://doi.org/10.1089/ten.TEA.2016.0525
Davis HM, Pacheco-Costa R, Atkinson EG et al (2017b) Disruption of the Cx43/miR21 pathway leads to osteocyte apoptosis and increased osteoclastogenesis with aging. Aging Cell 16:551–563. https://doi.org/10.1111/acel.12586
de Lencastre A, Pincus Z, Zhou K et al (2010) MicroRNAs both promote and antagonize longevity in C. elegans. Curr Biol 20:2159–2168. https://doi.org/10.1016/j.cub.2010.11.015
de Lucia C, Komici K, Borghetti G et al (2017) MicroRNA in cardiovascular aging and age-related cardiovascular diseases. Front Med 4:74. https://doi.org/10.3389/fmed.2017.00074
Dellago H, Bobbili MR, Grillari J (2017) MicroRNA-17-5p: at the crossroads of cancer and aging—a mini-review. Gerontology 63:20–28. https://doi.org/10.1159/000447773
Demontiero O, Vidal C, Duque G (2012) Aging and bone loss: new insights for the clinician. Ther Adv Musculoskelet Dis 4:61–76. https://doi.org/10.1177/1759720X11430858
Drummond MJ, McCarthy JJ, Sinha M et al (2011) Aging and microRNA expression in human skeletal muscle: a microarray and bioinformatics analysis. Physiol Genomics 43:595–603. https://doi.org/10.1152/physiolgenomics.00148.2010
Du WW, Yang W, Fang L et al (2014) miR-17 extends mouse lifespan by inhibiting senescence signaling mediated by MKP7. Cell Death Dis 5:e1355. https://doi.org/10.1038/cddis.2014.305
Du WW, Li X, Li T et al (2015) The microRNA miR-17-3p inhibits mouse cardiac fibroblast senescence by targeting Par4. J Cell Sci 128:293–304. https://doi.org/10.1242/jcs.158360
Duan L, Liu C, Hu J et al (2018) Epigenetic mechanisms in coronary artery disease: the current state and prospects. Trends Cardiovasc Med 28:311–319. https://doi.org/10.1016/J.TCM.2017.12.012
Elobeid A, Libard S, Leino M et al (2016) Altered Proteins in the Aging Brain. J Neuropathol Exp Neurol 75:316–325. https://doi.org/10.1093/jnen/nlw002
Epstein FH, Wei JY (1992) Age and the cardiovascular system. N Engl J Med 327:1735–1739. https://doi.org/10.1056/NEJM199212103272408
Esslinger SM, Schwalb B, Helfer S et al (2013) Drosophila miR-277 controls branched-chain amino acid catabolism and affects lifespan. RNA Biol 10:1042–1056. https://doi.org/10.4161/rna.24810
Feng Q, Zheng S, Zheng J (2018) The emerging role of microRNAs in bone remodeling and its therapeutic implications for osteoporosis. Biosci Rep. https://doi.org/10.1042/bsr20180453
Fenn AM, Smith KM, Lovett-Racke AE et al (2013) Increased micro-RNA 29b in the aged brain correlates with the reduction of insulin-like growth factor-1 and fractalkine ligand. Neurobiol Aging 34:2748–2758. https://doi.org/10.1016/j.neurobiolaging.2013.06.007
Finger F, Ottens F, Springhorn A et al (2019) Olfaction regulates organismal proteostasis and longevity via microRNA-dependent signalling. Nat Metab 1:350–359. https://doi.org/10.1038/s42255-019-0033-z
Friedman DB, Johnson TE (1988) A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 118:75–86
Fuggle N, Shaw S, Dennison E, Cooper C (2017) Sarcopenia. Best Pract Res Clin Rheumatol 31:218–242. https://doi.org/10.1016/j.berh.2017.11.007
Gendron CM, Pletcher SD (2017) MicroRNAs mir-184 and let-7 alter Drosophila metabolism and longevity. Aging Cell 16:1434–1438. https://doi.org/10.1111/acel.12673
Gupta SK, Foinquinos A, Thum S et al (2016) Preclinical development of a microRNA-based therapy for elderly patients with myocardial infarction. J Am Coll Cardiol 68:1557–1571. https://doi.org/10.1016/j.jacc.2016.07.739
Gurha P, Wang T, Larimore AH et al (2013) MicroRNA-22 promotes heart failure through coordinate suppression of PPAR/ERR-nuclear hormone receptor transcription. PLoS One 8:e75882. https://doi.org/10.1371/journal.pone.0075882
Hamrick MW, Herberg S, Arounleut P et al (2010) The adipokine leptin increases skeletal muscle mass and significantly alters skeletal muscle miRNA expression profile in aged mice. Biochem Biophys Res Commun 400:379–383. https://doi.org/10.1016/j.bbrc.2010.08.079
Hanna J, Hossain GS, Kocerha J (2019) The potential for microRNA therapeutics and clinical research. Front Genet 10:478. https://doi.org/10.3389/fgene.2019.00478
Hedden T, Gabrieli JDE (2004) Insights into the ageing mind: a view from cognitive neuroscience. Nat Rev Neurosci 5:87–96. https://doi.org/10.1038/nrn1323
Hooten NN, Fitzpatrick M, Wood WH et al (2013) Age-related changes in microRNA levels in serum. Aging (Albany NY) 5:725–740. https://doi.org/10.18632/aging.100603
Hsieh Y-W, Chang C, Chuang C-F (2012) The microRNA mir-71 inhibits calcium signaling by targeting the TIR-1/Sarm1 adaptor protein to control stochastic L/R neuronal asymmetry in C. elegans. PLoS Genet 8:e1002864. https://doi.org/10.1371/journal.pgen.1002864
Hu Z, Klein JD, Mitch WE et al (2014) MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging (Albany NY) 6:160–175. https://doi.org/10.18632/aging.100643
Hu C-H, Sui B-D, Du F-Y et al (2017) miR-21 deficiency inhibits osteoclast function and prevents bone loss in mice. Sci Rep 7:43191. https://doi.org/10.1038/srep43191
Huang Y, Qi Y, Du J-Q, Zhang D (2014) MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin Ther Targets 18:1–11. https://doi.org/10.1517/14728222.2014.961424
Ibáñez-Ventoso C, Vora M, Driscoll M (2008) Sequence relationships among C. elegans, D. melanogaster and human microRNAs highlight the extensive conservation of microRNAs in biology. PLoS One 3:e2818. https://doi.org/10.1371/journal.pone.0002818
Inukai S, Slack F (2013) MicroRNAs and the genetic network in aging. J Mol Biol 425:3601–3608. https://doi.org/10.1016/j.jmb.2013.01.023
Inukai S, de Lencastre A, Turner M, Slack F (2012) Novel microRNAs differentially expressed during aging in the mouse brain. PLoS One 7:e40028. https://doi.org/10.1371/journal.pone.0040028
Isik M, Blackwell TK, Berezikov E et al (2016) MicroRNA mir-34 provides robustness to environmental stress response via the DAF-16 network in C. elegans. Sci Rep 6:36766. https://doi.org/10.1038/srep36766
Jazbutyte V, Fiedler J, Kneitz S et al (2013) MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr) 35:747–762. https://doi.org/10.1007/s11357-012-9407-9
Jung HJ, Suh Y (2014) Circulating miRNAs in ageing and ageing-related diseases. J Genet Genomics 41(9):465–472. https://doi.org/10.1016/j.jgg.2014.07.003
Jung HJ, Lee K-P, Milholland B et al (2017) Comprehensive miRNA profiling of skeletal muscle and serum in induced and normal mouse muscle atrophy during aging. J Gerontol A Biol Sci Med Sci 72:1483–1491. https://doi.org/10.1093/gerona/glx025
Kato M, Slack FJ (2013) Ageing and the small, non-coding RNA world. Ageing Res Rev 12:429–435. https://doi.org/10.1016/j.arr.2012.03.012
Ke K, Sul O-J, Rajasekaran M, Choi H-S (2015) MicroRNA-183 increases osteoclastogenesis by repressing heme oxygenase-1. Bone 81:237–246. https://doi.org/10.1016/J.BONE.2015.07.006
Kennerdell JR, Liu N, Bonini NM (2018) MiR-34 inhibits polycomb repressive complex 2 to modulate chaperone expression and promote healthy brain aging. Nat Commun 9:4188. https://doi.org/10.1038/s41467-018-06592-5
Kenyon CJ (2010) The genetics of ageing. Nature 464:504–512. https://doi.org/10.1038/nature08980
Kenyon C, Chang J, Gensch E et al (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366:461–464. https://doi.org/10.1038/366461a0
Khan SS, Singer BD, Vaughan DE (2017) Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 16:624–633. https://doi.org/10.1111/acel.12601
Khanna A, Muthusamy S, Liang R et al (2011) Gain of survival signaling by down-regulation of three key miRNAs in brain of calorie-restricted mice. Aging (Albany NY) 3:223–236. https://doi.org/10.18632/aging.100276
Kiezun A, Artzi S, Modai S et al (2012) miRviewer: a multispecies microRNA homologous viewer. BMC Res Notes 5:92. https://doi.org/10.1186/1756-0500-5-92
Kim JY, Park Y-K, Lee K-P et al (2014) Genome-wide profiling of the microRNA-mRNA regulatory network in skeletal muscle with aging. Aging (Albany NY) 6:524–544. https://doi.org/10.18632/aging.100677
Kim J, Yoon H, Chung D-E et al (2016) miR-186 is decreased in aged brain and suppresses BACE1 expression. J Neurochem 137:436–445. https://doi.org/10.1111/jnc.13507
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73. https://doi.org/10.1093/nar/gkt1181
Kumar S, Vijayan M, Bhatti JS, Reddy PH (2017) MicroRNAs as peripheral biomarkers in aging and age-related diseases. Prog Mol Biol Transl Sci 146:47–94. https://doi.org/10.1016/BS.PMBTS.2016.12.013
Lai P, Song Q, Yang C et al (2016) Loss of Rictor with aging in osteoblasts promotes age-related bone loss. Cell Death Dis 7:e2408. https://doi.org/10.1038/cddis.2016.249
Lee K-P, Shin YJ, Panda AC et al (2015) miR-431 promotes differentiation and regeneration of old skeletal muscle by targeting Smad4. Genes Dev 29:1605–1617. https://doi.org/10.1101/gad.263574.115
Leggio L, Vivarelli S, L’Episcopo F et al (2017) microRNAs in parkinson’s disease: from pathogenesis to novel diagnostic and therapeutic approaches. Int J Mol Sci. https://doi.org/10.3390/ijms18122698
Lewis BP, Burge CB, Bartel DP (2005) Conserved seed pairing, often flanked by Adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20. https://doi.org/10.1016/J.CELL.2004.12.035
Li Z, Rana TM (2014) Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov 13:622–638. https://doi.org/10.1038/nrd4359
Li N, Bates DJ, An J et al (2011a) Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol Aging 32:944–955. https://doi.org/10.1016/J.NEUROBIOLAGING.2009.04.020
Li X, Khanna A, Li N, Wang E (2011b) Circulatory miR34a as an RNA-based, noninvasive biomarker for brain aging. Aging (Albany NY) 3:985–1002. https://doi.org/10.18632/aging.100371
Li S-H, Guo J, Wu J et al (2013) miR-17 targets tissue inhibitor of metalloproteinase 1 and 2 to modulate cardiac matrix remodeling. FASEB J 27:4254–4265. https://doi.org/10.1096/fj.13-231688
Li C-J, Cheng P, Liang M-K et al (2015) MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J Clin Invest 125:1509–1522. https://doi.org/10.1172/JCI77716
Li D, Liu J, Guo B et al (2016) Osteoclast-derived exosomal miR-214-3p inhibits osteoblastic bone formation. Nat Commun 7:10872. https://doi.org/10.1038/ncomms10872
Liang R, Khanna A, Muthusamy S et al (2011) Post-transcriptional regulation of IGF1R by key microRNAs in long-lived mutant mice. Aging Cell 10:1080–1088. https://doi.org/10.1111/j.1474-9726.2011.00751.x
Liu N, Landreh M, Cao K et al (2012) The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482:519–523. https://doi.org/10.1038/nature10810
Machida T, Tomofuji T, Ekuni D et al (2015) MicroRNAs in salivary exosome as potential biomarkers of aging. Int J Mol Sci 16:21294–21309. https://doi.org/10.3390/ijms160921294
Marty E, Liu Y, Samuel A et al (2017) A review of sarcopenia: enhancing awareness of an increasingly prevalent disease. Bone 105:276–286. https://doi.org/10.1016/J.BONE.2017.09.008
Meder B, Backes C, Haas J et al (2014) Influence of the confounding factors age and sex on microRNA profiles from peripheral blood. Clin Chem 60:1200–1208. https://doi.org/10.1373/clinchem.2014.224238
Mercken EM, Majounie E, Ding J et al (2013) Age-associated miRNA alterations in skeletal muscle from rhesus monkeys reversed by caloric restriction. Aging (Albany NY) 5:692–703. https://doi.org/10.18632/aging.100598
Moerman EJ, Teng K, Lipschitz DA, Lecka-Czernik B (2004) Aging activates adipogenic and suppresses osteogenic programs in mesenchymal marrow stroma/stem cells: the role of PPAR-γ2 transcription factor and TGF-β/BMP signaling pathways. Aging Cell 3:379–389. https://doi.org/10.1111/j.1474-9728.2004.00127.x
Murray-Stewart TR, Woster PM, Casero RA (2016) Targeting polyamine metabolism for cancer therapy and prevention. Biochem J 473:2937–2953. https://doi.org/10.1042/BCJ20160383
North BJ, Sinclair DA (2012) The intersection between aging and cardiovascular disease. Circ Res 110:1097–1108. https://doi.org/10.1161/CIRCRESAHA.111.246876
Olivieri F, Bonafè M, Spazzafumo L et al (2014) Age- and glycemia-related miR-126-3p levels in plasma and endothelial cells. Aging (Albany NY) 6:771–786. https://doi.org/10.18632/aging.100693
Pardo PS, Hajira A, Boriek AM, Mohamed JS (2017) MicroRNA-434-3p regulates age-related apoptosis through eIF5A1 in the skeletal muscle. Aging (Albany NY) 9:1012–1029. https://doi.org/10.18632/aging.101207
Peng Y, Croce CM (2016) The role of microRNAs in human cancer. Signal Transduct Target Ther 1:15004. https://doi.org/10.1038/sigtrans.2015.4
Persengiev SP, Kondova II, Bontrop RE (2012) The impact of MicroRNAs on brain aging and neurodegeneration. Curr Gerontol Geriatr Res 2012:359369. https://doi.org/10.1155/2012/359369
Pincus Z, Smith-Vikos T, Slack FJ (2011) MicroRNA predictors of longevity in Caenorhabditis elegans. PLoS Genet 7:e1002306. https://doi.org/10.1371/journal.pgen.1002306
Plotkin LI, Bellido T (2016) Osteocytic signalling pathways as therapeutic targets for bone fragility. Nat Rev Endocrinol 12:593–605. https://doi.org/10.1038/nrendo.2016.71
Quinlan S, Kenny A, Medina M et al (2017) MicroRNAs in neurodegenerative diseases. Int Rev Cell Mol Biol 334:309–343. https://doi.org/10.1016/BS.IRCMB.2017.04.002
Rachner TD, Khosla S, Hofbauer LC (2011) Osteoporosis: now and the future. Lancet (Lond, Engl) 377:1276–1287. https://doi.org/10.1016/S0140-6736(10)62349-5
Ramanujam D, Sassi Y, Laggerbauer B, Engelhardt S (2016) Viral vector-based targeting of miR-21 in cardiac nonmyocyte cells reduces pathologic remodeling of the heart. Mol Ther 24:1939–1948. https://doi.org/10.1038/mt.2016.166
Redshaw Z, Sweetman D, Loughna PT (2014) The effects of age upon the expression of three miRNAs in muscle stem cells isolated from two different porcine skeletal muscles. Differentiation 88:117–123. https://doi.org/10.1016/J.DIFF.2014.12.001
Ripa R, Dolfi L, Terrigno M et al (2017) MicroRNA miR-29 controls a compensatory response to limit neuronal iron accumulation during adult life and aging. BMC Biol 15:9. https://doi.org/10.1186/s12915-017-0354-x
Rivas DA, Lessard SJ, Rice NP et al (2014) Diminished skeletal muscle microRNA expression with aging is associated with attenuated muscle plasticity and inhibition of IGF-1 signaling. FASEB J 28:4133–4147. https://doi.org/10.1096/fj.14-254490
Rupaimoole R, Slack FJ (2017) MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 16:203–222. https://doi.org/10.1038/nrd.2016.246
Sawada S, Akimoto T, Takahashi M et al (2014) Effect of aging and sex on circulating microRNAs in humans. Adv Aging Res 03:152–159. https://doi.org/10.4236/aar.2014.32023
Shao H, Yang L, Wang L et al (2018) MicroRNA-34a protects myocardial cells against ischemia–reperfusion injury through inhibiting autophagy via regulating TNFα expression. Biochem Cell Biol 96:349–354. https://doi.org/10.1139/bcb-2016-0158
Shih H, Lee B, Lee RJ, Boyle AJ (2011) The aging heart and post-infarction left ventricular remodeling. J Am Coll Cardiol 57:9–17. https://doi.org/10.1016/J.JACC.2010.08.623
Simon AF, Shih C, Mack A, Benzer S (2003) Steroid control of longevity in Drosophila melanogaster. Science (80−) 299:1407–1410. https://doi.org/10.1126/science.1080539
Smith-Vikos T, Slack FJ (2012) MicroRNAs and their roles in aging. J Cell Sci 125:7–17. https://doi.org/10.1242/jcs.099200
Smith-Vikos T, de Lencastre A, Inukai S et al (2014) MicroRNAs mediate dietary-restriction-induced longevity through PHA-4/FOXA and SKN-1/Nrf transcription factors. Curr Biol 24:2238–2246. https://doi.org/10.1016/j.cub.2014.08.013
Smith-Vikos T, Liu Z, Parsons C et al (2016) A serum miRNA profile of human longevity: findings from the Baltimore Longitudinal Study of Aging (BLSA). Aging (Albany NY) 8:2971–2987. https://doi.org/10.18632/aging.101106
Soriano-Arroquia A, House L, Tregilgas L et al (2016) The functional consequences of age-related changes in microRNA expression in skeletal muscle. Biogerontology 17:641–654. https://doi.org/10.1007/s10522-016-9638-8
Sowell ER, Thompson PM, Toga AW (2004) Mapping changes in the human cortex throughout the span of life. Neurosci 10:372–392. https://doi.org/10.1177/1073858404263960
Strait JB, Lakatta EG (2012) Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail Clin 8:143–164. https://doi.org/10.1016/j.hfc.2011.08.011
Sun W, Zhao C, Li Y et al (2016) Osteoclast-derived microRNA-containing exosomes selectively inhibit osteoblast activity. Cell Discov 2:16015. https://doi.org/10.1038/celldisc.2016.15
Takeda T, Tanabe H (2016) Lifespan and reproduction in brain-specific miR-29-knockdown mouse. Biochem Biophys Res Commun 471:454–458. https://doi.org/10.1016/j.bbrc.2016.02.055
Ueda M, Sato T, Ohkawa Y, Inoue YH (2018) Identification of miR-305, a microRNA that promotes aging, and its target mRNAs in Drosophila. Genes Cells 23:80–93. https://doi.org/10.1111/gtc.12555
Ugalde AP, Ramsay AJ, de la Rosa J et al (2011) Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J 30:2219–2232. https://doi.org/10.1038/emboj.2011.124
Van Aelst LNL, Voss S, Carai P et al (2015) Osteoglycin prevents cardiac dilatation and dysfunction after myocardial infarction through infarct collagen strengthening. Circ Res 116:425–436. https://doi.org/10.1161/CIRCRESAHA.116.304599
van Almen GC, Verhesen W, van Leeuwen REW et al (2011) MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell 10:769–779. https://doi.org/10.1111/j.1474-9726.2011.00714.x
Verjans R, van Bilsen M, Schroen B (2017) MiRNA deregulation in cardiac aging and associated disorders. Int Rev Cell Mol Biol 334:207–263. https://doi.org/10.1016/BS.IRCMB.2017.03.004
Verma P, Augustine GJ, Ammar M-R et al (2015) A neuroprotective role for microRNA miR-1000 mediated by limiting glutamate excitotoxicity. Nat Neurosci 18:379–385. https://doi.org/10.1038/nn.3935
Villar AV, García R, Merino D et al (2013) Myocardial and circulating levels of microRNA-21 reflect left ventricular fibrosis in aortic stenosis patients. Int J Cardiol 167:2875–2881. https://doi.org/10.1016/J.IJCARD.2012.07.021
Vilmos P, Bujna A, Szuperák M et al (2013) Viability, longevity, and egg production of Drosophila melanogaster are regulated by the miR-282 microRNA. Genetics 195:469–480. https://doi.org/10.1534/genetics.113.153585
Wang X, Guo B, Li Q et al (2013) miR-214 targets ATF4 to inhibit bone formation. Nat Med 19:93–100. https://doi.org/10.1038/nm.3026
Wang M, Qin L, Tang B (2019) MicroRNAs in Alzheimer’s disease. Front Genet 10:153. https://doi.org/10.3389/fgene.2019.00153
Weber JA, Baxter DH, Zhang S et al (2010) The microRNA spectrum in 12 body fluids. Clin Chem 56:1733–1741. https://doi.org/10.1373/clinchem.2010.147405
Weilner S, Schraml E, Wieser M et al (2016) Secreted microvesicular miR-31 inhibits osteogenic differentiation of mesenchymal stem cells. Aging Cell 15:744–754. https://doi.org/10.1111/acel.12484
Wu S, Kim T-K, Wu X et al (2016) Circulating microRNAs and life expectancy among identical twins. Ann Hum Genet 80:247–256. https://doi.org/10.1111/ahg.12160
Yang J, Chen D, He Y et al (2013a) MiR-34 modulates Caenorhabditis elegans lifespan via repressing the autophagy gene atg9. Age (Omaha) 35:11–22. https://doi.org/10.1007/s11357-011-9324-3
Yang N, Wang G, Hu C et al (2013b) Tumor necrosis factor α suppresses the mesenchymal stem cell osteogenesis promoter miR-21 in estrogen deficiency–induced osteoporosis. J Bone Miner Res 28:559–573. https://doi.org/10.1002/jbmr.1798
Yang Y, Cheng H-W, Qiu Y et al (2015) MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ Res 117:450–459. https://doi.org/10.1161/CIRCRESAHA.117.305962
Yin L, Sun Y, Wu J et al (2015) Discovering novel microRNAs and age-related nonlinear changes in rat brains using deep sequencing. Neurobiol Aging 36:1037–1044. https://doi.org/10.1016/J.NEUROBIOLAGING.2014.11.001
Yuan J, Chen H, Ge D et al (2017) Mir-21 promotes cardiac fibrosis after myocardial infarction via targeting Smad7. Cell Physiol Biochem 42:2207–2219. https://doi.org/10.1159/000479995
Zacharewicz E, Della Gatta P, Reynolds J et al (2014) Identification of microRNAs linked to regulators of muscle protein synthesis and regeneration in young and old skeletal muscle. PLoS One 9:e114009. https://doi.org/10.1371/journal.pone.0114009
Zhang X, Zabinsky R, Teng Y et al (2011) MicroRNAs play critical roles in the survival and recovery of Caenorhabditis elegans from starvation-induced L1 diapause. Proc Natl Acad Sci USA 108:17997–18002. https://doi.org/10.1073/pnas.1105982108
Zhang X, Azhar G, Wei JY (2012) The expression of microRNA and microRNA clusters in the aging heart. PLoS One 7:e34688. https://doi.org/10.1371/journal.pone.0034688
Zhang H, Yang H, Zhang C et al (2015) Investigation of microRNA expression in human serum during the aging process. J Gerontol Ser A 70:102–109. https://doi.org/10.1093/gerona/glu145
Zhang Y, Liu Y-J, Liu T et al (2016) Plasma microRNA-21 is a potential diagnostic biomarker of acute myocardial infarction. Eur Rev Med Pharmacol Sci 20:323–329
Zhao C, Sun W, Zhang P et al (2015a) miR-214 promotes osteoclastogenesis by targeting Pten/PI3 k/Akt pathway. RNA Biol 12:343–353. https://doi.org/10.1080/15476286.2015.1017205
Zhao W, Dong Y, Wu C et al (2015b) MiR-21 overexpression improves osteoporosis by targeting RECK. Mol Cell Biochem 405:125–133. https://doi.org/10.1007/s11010-015-2404-4
Zhou X, Xu H, Liu Z et al (2018) miR-21 promotes cardiac fibroblast-to-myofibroblast transformation and myocardial fibrosis by targeting Jagged1. J Cell Mol Med 22:3816–3824. https://doi.org/10.1111/jcmm.13654
Zovoilis A, Agbemenyah HY, Agis-Balboa RC et al (2011) microRNA-34c is a novel target to treat dementias. EMBO J 30:4299–4308. https://doi.org/10.1038/emboj.2011.327
Acknowledgements
This work was supported by National Human Genome Institute Grant T32HG000045, National Institutes of Health Grant 1R01AG057748, and a Beckman Young Investigator award from the Arnold and Mabel Beckman Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all the authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kinser, H.E., Pincus, Z. MicroRNAs as modulators of longevity and the aging process. Hum Genet 139, 291–308 (2020). https://doi.org/10.1007/s00439-019-02046-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-019-02046-0