
Abstract
Developmental and epileptic encephalopathies (DEEs) are genetically heterogenous conditions, often characterized by early onset, EEG interictal epileptiform abnormalities, polymorphous and drug-resistant seizures, and neurodevelopmental impairments. In this study, we investigated the genetic defects in two siblings who presented with severe DEE, microcephaly, spastic tetraplegia, diffuse brain hypomyelination, cerebellar atrophy, short stature, and kyphoscoliosis. Whole exome next-generation sequencing (WES) identified in both siblings a homozygous non-sense variant in the ACTL6B gene (NM_016188:c.820C>T;p.Gln274*) coding for a subunit of the neuron-specific chromatin remodeling complex nBAF. To further support these findings, a targeted ACTL6B sequencing assay was performed on a cohort of 85 unrelated DEE individuals, leading to the identification of a homozygous missense variant (NM_016188:c.1045G>A;p.Gly349Ser) in a patient. This variant did not segregate in the unaffected siblings in this family and was classified as deleterious by several prediction softwares. Interestingly, in both families, homozygous patients shared a rather homogeneous phenotype. Very few patients with ACTL6B gene variants have been sporadically reported in WES cohort studies of patients with neurodevelopmental disorders and/or congenital brain malformations. However, the limited number of patients with incomplete clinical information yet reported in the literature did not allow to establish a strong gene–disease association. Here, we provide additional genetic and clinical data on three new cases that support the pathogenic role of ACTL6B gene mutation in a syndromic form of DEE.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Developmental and epileptic encephalopathies (DEEs) represent a group of conditions often characterized by onset in infancy, very active EEG interictal epileptiform abnormalities, polymorphous and drug-resistant seizures, and neurological, cognitive, and behavioral deficits (Scheffer et al. 2017). Most of DEEs are genetic in origin and arise from de novo mutations, albeit autosomal recessive and X-linked inheritance patterns have also been reported. Clearly, the identification of the genetic causes in DEEs is extremely important to understand the pathogenetic mechanisms, to guide therapeutic options, and to provide a more accurate genetic counseling. In the last few years, chromosome microarrays and next-generation sequencing (NGS) techniques have allowed the identification of an increasing number of de novo variants in several different genes, confirming the high genetic heterogeneity of DEEs. Up to date, more than 50 genes have demonstrated a definite or possible pathogenic role in specific clinical and EEG phenotypes, while the exact role of other candidate genes needs future validation studies (He et al. 2018; Myers et al. 2019). The increasing knowledge of the genetic background of the disease has allowed designing NGS assays that target panels of associated genes. Although their introduction has improved the diagnostic yield, many patients still remain without a molecular diagnosis, suggesting that many DEE genes have yet to be discovered (Ko et al. 2018).
Here, we report on two non-consanguineous families, in which the affected members shared a phenotype characterized by the early onset drug-resistant epilepsy, severe psychomotor delay, spastic tetraplegia, microcephaly, diffuse cerebral hypomyelination, and brain or cerebellar atrophy. NGS analysis revealed that the affected individuals in each family carried distinct homozygous variants in the ACTL6B gene (also known as BAF53B), coding for a subunit of the neuron-specific chromatin remodeling complex nBAF (BRG1/brm-associated factor) (Olave et al. 2002). So far, very few ACTL6B gene variants have been reported in WES cohort studies of patients with neurodevelopmental disorders and/or congenital brain malformations (Karaca et al. 2015; Maddirevula et al. 2018; Sajan et al. 2017). In the light of the functional roles of ACTL6B, we considered the homozygous changes of this gene as a likely underlying cause of DEE in these families.
Materials and methods
Study subjects
The study was conducted in accordance with the Declaration of Helsinki and national guidelines and has been approved by the local ethics committee “Comitato Etico IRCCS Sicilia—Oasi Maria SS.”, project number: 2018/07/18/CE-IRCCS-OASI/14 as of July 7, 2018. Written informed consent was obtained from each subject or their guardians.
Genetic studies
Genomic DNA was extracted by the standard procedures from peripheral blood samples obtained from family members with informed consent. SNP/CGH array screening was performed using the combined array CGH plus SNP from CytoSure ISCA UPD array 4 × 180 K (Oxford Gene Technology, UK) and analyzed with the Agilent Microarray Scanner, Feature Extraction Software version 11.5, and Agilent Genomic Workbench 7.0 (Agilent Technologies, Santa Clara, CA, USA).
DNA enrichment for whole exome sequencing (WES) was carried out using the Agilent Sure Select Human All Exon V6 + UTRs kit (Agilent Technologies, Santa Clara, CA), and sequenced with the Illumina Novaseq sequencer (Illumina, San Diego, CA). Reads were aligned to the reference human genome (UCSC GRCh37/hg19), using BWA-MEM tool (Li 2012). Base quality score recalibration, indel realignment, duplicate removal, insertions/deletions (indels), and single-nucleotide variants’ (SNVs) detection were performed using GATK (McKenna et al. 2010) according to its “Best Practices” recommendations (Van der Auwera et al. 2013). Variants were annotated using the ANNOVAR software tool (Yang and Wang 2015) and then filtered according to their predicted effects and allele frequencies in the available public databases (dbSNP http://www.ncbi.nlm.nih.gov/projects/SNP/), ExAC (http://exac.broadinstitute.org/), 1000 Genomes (http://www.1000genomes.org/), ESP6500 (http://evs.gs.washington.edu/EVS/), and gnomAD (http://gnomad.broadinstitute.org/). Variants were prioritized either assuming a dominant or recessive pattern of inheritance, and technically verified by Sanger sequencing.
The epilepsy targeted resequencing gene panel (Supplementary Table 1) and the ACTL6B targeted assay were both designed to sequence all the exons and exon–intron flanking regions, using the software Ion AmpliSeq Designer (Thermo Fisher Scientific) and run on an Ion Torrent Personal Genome Machine (Thermo Fisher Scientific).
Results
Clinical findings
Family A
Both siblings (III:3 and III:4, Fig. 1a) were born to healthy non-consanguineous parents with negative family history for neurodevelopmental disorders or late onset neurodegenerative disease.

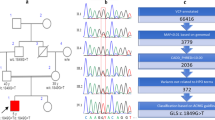
Pedigrees of families under investigation. a Pedigree of family (A) individual I:3 died from stroke at the age of 72. b Pedigree of family (B) affected individuals are represented with filled symbols. ACTL6B genetic testing for each individual is shown, if available (−/−: homozygous for wild-type allele; −/+: heterozygous for the mutated allele; +/+: homozygous for the mutated allele)
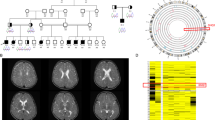
The proband (III:3, Supplementary Fig. 1a), currently 10 years old, was born at gestational age 36 weeks by cesarean section subsequent to morbid cardiotocography (CTG), oligohydramnios, slow fetal movements, and decreased occipitofrontal head circumference (OFC) growth in the last trimester. Apgar score at 1 and 5 min was 7 and 9, birth weight 2780 g (− 0.55 SD), length 45.5 cm (− 1.24 SD), and OFC 31.5 cm (− 1.45 SD). She presented with hypoglycemia (< 10 mg/dL) and neonatal jaundice. In the first week of life, mostly at morning awakening and during wakefulness, she started to present seizures characterized by tonic extension or flexion of the upper limbs, head and eye deviation to the right side, and oral automatisms (chewing and tongue protrusion). At 2 months of life, these episodes occurred approximately three times a day. EEG showed a slow background activity and very frequent slow spike-and-wave complexes synchronous and asynchronous over both hemispheres. For the first months, she suffered from vomiting, lack of appetite, and gastroesophageal reflux disease (GERD). At 2 years and 1 month of age, a gastrostomy tube was placed due to dysphagia and failure to thrive, although oral feeding without aspiration was possible for very small pieces of food. Her seizures did not change in semiology and remain intractable despite numerous medication trials (phenobarbital, valproic acid, corticotropin, clonazepam, nitrazepam, topiramate, carbamazepine, phenytoin, lamotrigine, and levetiracetam) and ketogenic diet. She was severely delayed, non-verbal and non-ambulatory, and unable to sit unassisted. She lacked spontaneous and purposeful hand use. At the last clinical examination at the age of 10 years, weight was 20 kg (− 3.45 SD), height 130 cm (− 2.31 SD), and OFC 45.8 cm (− 5.81 SD). She also presented spastic tetraplegia, microcephaly, short stature, and kyphoscoliosis with severe thoracic deformity. Wakefulness and sleep EEG showed slow background activity, numerous spikes over the occipital regions of both hemispheres, and tonic seizures (Fig. 2a, b). MRI revealed widespread hypomyelination, volume loss in frontal and temporal lobes, and extreme thinning of the corpus callosum (Fig. 3a, b).

EEG of patient III:3 (family A). a Wakefulness EEG of the patient III:3 (family A) at 10 years of age, showing slow background activity, and numerous spikes synchronously and asynchronously localized over the occipital regions of both hemispheres. b EEG recording of a short tonic seizure; the ictal fast activity predominates over the temporal–occipital regions of the right hemisphere, and is followed by rhythmical sharp waves. At the surface EMG of the deltoid muscles (R DELT and L DELT), a bilateral tonic contraction is evident, of higher duration on the right side

MRI of patient III:3 (family A). a T2 axial image of the patient III:3 (family A), at 10 years of age, at level of ventricular atria shows widespread hypomyelination, with normal signal of white matter recognizable only in the internal capsule and callosal splenium. Volume loss in frontal and temporal lobes is associated. b T1 sagittal image showing extreme thinning of the corpus callosum
Her brother (III:4, Supplementary Fig. 1b), currently 4 years old, was born at gestational age 38 weeks by elective cesarean section, performed prior to labor or the rupture of membranes after an uneventful pregnancy. Apgar score at 1 and 5 min was 8 and 10, birth weight 3240 g (− 0.35 SD), length 45 cm (− 2.75 SD), and OFC 34.5 cm (− 0.15 SD). He showed one preauricular tag. At days 3 and 4 of life, tonic and clonic seizures occurred, many times a day. Like his sister, he suffered from vomiting, lack of appetite, and GERD. At 3 months of age, EEG disclosed slow background activity, intermingled with spikes over the occipital regions, more prominent over the left hemisphere.
At 2 years of age, a gastrostomy tube was placed. He was severely delayed, non-verbal, and non-ambulatory, and unable to sit unassisted. Seizures persisted with the same frequency and were characterized by flexion or extension of the limbs, eye and deviation to right side, cyanosis, vocalisms, during wakefulness, and sleep. Seizures resulted resistant to phenobarbital, levetiracetam, clonazepam, and ketogenic diet.
At the last observation, at the age of 4 years, weight was 11.2 kg (− 3.85 SD), height 88 cm (− 4.63 SD), and OFC 44.3 cm (− 5.19 SD). Spastic tetraplegia, microcephaly, short stature, and kyphoscoliosis were also present. Wakefulness and sleep EEG was characterized by slow background activity and numerous spikes localized over the posterior regions of both hemispheres; tonic seizures were also recorded (Fig. 4a, c). MRI showed widespread white-matter atrophy with ventricular enlargement and periventricular signal changes; residual subcortical fibers in anterior frontal and parieto-occipital regions, and hypomyelination of intermediate areas, frontal volume loss, vermian atrophy, cerebral hypomyelination, and callosal thinning (Fig. 5a–c).

EEG of patient III:4 (family A). a Wakefulness EEG of the patient III:4 (family A) at 4 years of age, with slow background activity and numerous spikes, synchronous and asynchronous over the posterior regions, bilaterally. b, c EEG recording of a tonic seizure lasting approximately 15 s, characterized by a fast activity, gradually decreasing in frequency, more prominent over the left posterior regions of both hemispheres. Surface EMG recording of deltoid muscles (E DELT and L DELT) reveals a tonic contraction

MRI of patient III:4 (family A). a T2 axial image of the patient III:4 (family A), at 4 years of age, at the level of lateral ventricles shows widespread white-matter atrophy with ventricular enlargement and periventricular signal changes; residual subcortical fibers in anterior frontal and parieto-occipital regions, and hypomyelination of intermediate areas can be seen. Frontal volume loss is associated. b T2 coronal image showing cerebellar atrophy and cerebral hypomyelination. c T1 sagittal image shows callosal thinning and vermian atrophy
Family B
Healthy parents were non-consanguineous, and there was no family history of neurodevelopmental disorders or late onset neurodegenerative diseases. Healthy grandparents were not available for genetic testing.
The proband (II:5, Supplementary Fig. 1c), a 4-year-old female, was born at term by cesarean section, after a normal pregnancy. Birth weight was 3100 g (− 0.55 SD); birth length and OFC were not reported. Neonatal period was uneventful and no dysmorphic features were present. At 2 months of age, she started to present clusters of epileptic spasms, occurring many per day. EEG was reported as characterized by slow background activity, with spikes over the right temporal regions. She was treated initially with phenobarbital, but seizures persisted with an unmodified frequency, presenting as epileptic spasms and tonic seizures. Subsequently, levetiracetam and nitrazepam were added, resulting in a weekly occurrence of seizures.
Neurological examination showed spastic tetraplegia, profound psychomotor delay, no motor achievements, and absence of language. At the age of 3 years, she underwent gastrostomy for severe dysphagia. At 4 years of age, weight was 11.5 kg (− 2.99 SD), height 86 cm (− 3.98 SD), and OFC 42.7 cm (− 6.39 SD). Wakefulness EEG revealed a slow background activity, and numerous spikes, synchronous and asynchronous over the middle–posterior regions of both hemispheres; during sleep, the paroxysmal abnormalities often appeared diffuse, with also a suppression-burst pattern. Short tonic seizures were recorded (Fig. 6a–c). MRI disclosed delayed myelination, global volume loss, vermian atrophy, and callosal thinning (Fig. 7a–c).

EEG of patient II:5 (family B). a Wakefulness EEG recording of patient II:5 (family B), characterized by slow background activity and synchronous and asynchronous sharp waves over the posterior regions. b During sleep EEG recording, irregular spike-and-wave complexes appear diffuse, and a burst-suppression pattern is also evident. c EEG recording of a short tonic seizure, correlated with the appearance of low-voltage diffuse fast activity lasting approximately 5 s. R DELT right deltoid muscle, L DELT left deltoid muscle

MRI of patient II:5 (family B). a T2 axial image of the patient II:5 (family B) at 4 years of age shows delayed myelination and global volume loss. b T2 coronal image shows cerebellar atrophy; cerebral hypomyelination and volume loss are visible again. c T1 sagittal image shows callosal thinning and vermian atrophy
The proband had twin monozygotic brothers (II:1, II:2; Fig. 1b) who deceased before their DNA could be collected. They were born at gestational age 36 weeks, by cesarean section. Birth weight of both was 2200 g. They presented neonatal tonic seizures, and subsequently multiple daily epileptic spasms which were resistant to vigabatrin. An EEG report of both twins, at 3 years of age, described frequent spikes over the posterior regions of both hemispheres, with diffusion during sleep. At the same age, an MRI report pointed out a mild periventricular hypomyelination, a slight dilation of the lateral ventricles and cerebral subarachnoid spaces, and thinning of corpus callosum. Both twins presented severe psychomotor delay and tetraplegia and suffered from vomiting. They died from pneumonia, respectively, at 4 and 5 years of age.
Genetic findings
Family A
SNP/CGH array analysis did not reveal any previously unreported copy-number variant (CNV) that might have clinical significance but identified in both probands chromosomal regions with runs of homozygosity (ROH). These regions were not shared by the affected children except for a ~ 3 Mb-long genomic segment located at chromosome 7q22.2 (98,128,828–101,041,758 bp; hg19). The total length of ROHs in individual III:3 and individual III:4 was 17 Mb and 29 Mb, respectively. These findings suggested that these regions were identical by descent (IBD), reflecting common ancestral haplotypes in parents.
WES analysis unraveled a homozygous non-sense variant in the ACTL6B gene (NM_016188: c. 820C>T;p.Gln274*) in patient III:3 and her brother III:4 (Supplementary Fig. 2a). No further recessive candidate variants shared by both siblings were identified. Parental germline mosaicism for dominant mutation was excluded as we did not find any de novo variant common to both probands. The homozygous truncating mutation was inherited from heterozygous parents and located in the ROH region at chromosome 7q22.2, previously identified in both siblings. Segregation analysis of the ACTL6B variant in the available family members is shown in Fig. 1a. Although we do not have experimental evidence as ACTL6B is almost exclusively expressed in neurons, the mutant mRNA most likely undergoes non-sense-mediated decay.
Family B
To further investigate the role of ACTL6B in DEEs, we decided to screen a cohort of 85 unrelated DEE Sicilian patients using a targeted ACTL6B NGS assay. One individual among them carried a significant change in the ACTL6B gene, consisting in a homozygous missense variant (NM_016188:c.1045G>A;p.Gly349Ser). Segregation analysis by Sanger sequencing in this family demonstrated that the homozygous variant was inherited from heterozygous parents (Supplementary Fig. 2b) and that healthy brothers II:3 and II:4 were homozygous for the wild-type allele. Unluckily, we were not able to genotype the affected dead twins (II:1; II:2) due the unavailability of their DNAs.
This variant was not reported in the public available databases, and was predicted to be deleterious by several prediction softwares including MutationTaster (score 1), CADD (score 23.2), PolyPhen-2 (score 0.999), SIFT (score 0), and Provean (score − 5.77). Phylogenetic analysis demonstrated that the glycine at position 349 was highly conserved, even in the evolutionarily distant species such as D. rerio, D. melanogaster, and C. elegans (Supplementary Fig. 3). Moreover, the DNA change was not identified in a group of 850 ethnically matched normal controls. Altogether, these findings indicate that this variant is likely causative in our patient.
To exclude that any other genomic rearrangement or genetic variant elsewhere in the genome might underlie the disease in the proband, SNP/CGH array and WES analysis were carried out in the family.
SNP/CGH array analysis revealed in the proband II:5 a single 7 MB-long ROH at chromosome 12q12, while CNV analysis did not identify any potentially pathogenic rearrangement. WES analysis, carried out on proband and her parents (I:1 and I:2), did not unveil any likely pathogenic sequence change in other genes. Interestingly, using H3M2 software (Magi et al. 2014) on WES data from proband, we detected an ROH of ~ 5 Mb which included the missense ACTL6B variant and that escaped from the previous SNP/CGH analysis due to the lack of statistical significance. As in the case of family A, these data suggest that her parents shared a common ancestor.
Discussion
Recent technological advances in copy-number identification and high-throughput sequencing have allowed the discovery of novel DDE-related genes, involving different molecular pathways in the etiology of the disease, including transcriptional and mTOR regulation. Mutations in different chromatin remodeling genes, playing a crucial role for appropriate programming of developmental gene expression, have long been associated with neurodevelopmental disorders, including intellectual disability and autism (Goodwin and Picketts 2018; Lopez and Wood 2015). More recently, mutations in CHD2, a member of chromodomain helicase DNA-binding (CHD) family of proteins, have been identified in several DEE individuals, thus supporting the role of chromatin remodeling genes in DEEs (Carvill et al. 2013).
Here, we report on two DEE families in which affected individuals had homozygous variants in the ACTL6B gene, coding for a subunit of the post-mitotic neurons complex nBAF, a neuron-specific chromatin remodeling complex which is critical for neuronal differentiation, dendritic extension, synaptic function, and long-term memory processes (Vogel-Ciernia et al. 2013; Wu et al. 2007). In mammals, the neuron-specific chromatin regulator complexes npBAF and nBAF act sequentially during the mitotic and post-mitotic stages of neuronal development and maturation (Sokpor et al. 2017; Staahl and Crabtree 2013). npBAF and nBAF have 15 subunits each, 12 of them common to both complexes. Interestingly, dominant causative variants in several genes coding for BAF-associated subunits have been identified in subjects with different forms of neurodevelopmental disorders (Bögershausen and Wollnik 2018; Vogel-Ciernia et al. 2017). Mutations in SMARCA4 (MIM: 603254), SMARCE1 (MIM: 603111), ARID2 (MIM: 609539), ARID1B (MIM: 614556), and ARID1A (MIM: 603024) have been found in individuals with a clinical diagnosis of Coffin–Siris syndrome (CSS; MIMs 135900, 614607, 614608, 614609, 616938, and 617808), while SMARCA2 (MIM: 600014) heterozygous variants have been identified in individuals with Nicolaides–Baraitser syndrome (MIM: 601358). More recently, dominant mutations in the ACTL6A gene, the homologous alternative of ACTL6B in the npBAF complex, have been associated with varying degree of intellectual disability dysmorphic features, and genitourinary and skeletal defects (Marom et al. 2017). Interestingly, de novo mutations in SS18L1 (also known as CREST) that, along with ACTLB6, encodes for a dedicated subunit of the nBAF complex have been detected in patients with amyotrophic lateral sclerosis (Chesi et al. 2013), suggesting a role for the nBAF complex in neurodegenerative disorders. In our study, however, neither parents nor available grandparents had the clinical signs of neurodegeneration.
Our patients presented with a rather homogeneous life-threatening, phenotype characterized by severe psychomotor delay, spastic tetraplegia, microcephaly, drug-resistant DEE with onset in the first months of life, brain diffuse hypomyelination, and vermian atrophy. Seizures were in the form of epileptic spasms and tonic seizures, and interictal EEG showed slow background activity, multifocal interictal epileptiform abnormalities which were in all cases more prominent over the posterior regions, and also a burst-suppression during sleep in one subject.
Knockout mice for ACTL6B have elevated perinatal lethality, impaired dendritic outgrowth, and defective axonal myelination but, in agreement with the restricted pattern of ACTL6B expression, they do not show any nonneural phenotype (Wu et al. 2007). These data suggest that biallelic ACTL6B gene mutations might determine this early neurological manifestation through dysregulation of genes essential for a proper dendritic outgrowth and axonal development in post-mitotic neuron. In fact, gene ontology and pathway enrichment analyses of pathogenic variant-harboring genes have demonstrated that axonal, dendritic, and synaptic regions preferentially are associated with the early life epilepsy other than West syndrome (Berg et al. 2018).
Variants of the ACTL6B gene have been sporadically reported up to now, in WES cohort studies of patients with neurodevelopmental disorders and/or congenital brain malformations (Table 1). Only two siblings, carrying a homozygous missense variant in ACTL6B (NM_016188: c.893G>A;p.Arg298Gln), presented unspecified seizures associated with a severe intellectual disability, microcephaly, and autistic behavior (Karaca et al. 2015) Another patient with a homozygous stoploss mutation of ACTL6B gene (NM_016188: c.1279delT; p.*427Aspext*32) was identified in a WES analysis of 22 Rett syndrome patients without coding mutations in MECP2, CDKL5, and FOXG1 genes, but no other clinical, EEG, or neuroimaging data were described (Sajan et al. 2017). More recently, a 13-month-old girl born to consanguineous parents was reported with a homozygous non-sense mutation of ACTL6B gene (NM_016188.4:c.999T>A;p.Cys333*) (Maddirevula et al. 2018). She presented hyperekplexia and global developmental delay. MRI revealed a picture rather different from that of our patients, characterized by agenesis of corpus callosum, mild ventricular dilation, mild atrophic changes, minimally simplified gyral pattern with prominence of the sulci, and mild posterior colpocephaly. There was history of a similarly affected brother who died at 2 months of age.
Our data on these three new cases support the pathogenic role of the ACTL6B gene in a specific severe neurodevelopmental syndrome and delineate the associated clinical presentation characterized by an early onset drug-resistant DEE associated with microcephaly, brain hypomyelination, and cerebellar atrophy.
Although the limited number of patients carrying ACTL6B gene mutations does not allow to draw any genotype–phenotype correlation, differences in the severity of the encephalopathy either caused by non-sense, stoploss, or missense homozygous variants are not evident. It still remains plausible that milder ACTL6B mutations may partly preserve the nBAF activity, leading to a milder phenotype.
Interestingly, putative dominant ACTL6B variants have been occasionally seen in large studies on individuals with neurodevelopmental disorders (Krupp et al. 2017; Lelieveld et al. 2017). In our study, heterozygous individuals had no history of neurological disorders, either suggesting that the reported heterozygous ACTL6B variants were not per se causative or operate through a different (dominant) mechanism. Of note, ACTL6B lies between a differentially methylated region recently identified on chromosome 7q22.2 (Hannula-Jouppi et al. 2014) and a cluster of imprinted genes at chromosome 7q21.3, raising the question whether a preferential allelic expression of ACTL6B might explain a different clinical outcome in heterozygous individuals. However, our study does not support this hypothesis, since healthy heterozygous individuals II:3 and II:4 in family A received, respectively, a maternal and a paternal ACTL6B wild-type allele.
In conclusion, ACTL6B gene mutations probably represent a new mendelian cause for the early onset DEE and their role might be suspected when drug-resistant epileptic spasms and tonic seizures and severe motor and cognitive deficits are associated with some rather peculiar features, such as an interictal multifocal EEG pattern prevalently localized over the posterior regions, brain hypomyelination, and cerebellar atrophy. Clearly, further studies are needed to confirm the involvement of this gene in DEEs and to fully characterize the associated clinical spectrum. We also suggest that ACTL6B should be considered in genetic testing for DEEs.
References
Berg AT, Chakravorty S, Koh S, Grinspan ZM, Shellhaas RA, Saneto RP, Wirrell EC, Coryell J, Chu CJ, Mytinger JR, Gaillard WD, Valencia I, Knupp KG, Loddenkemper T, Sullivan JE, Poduri A, Millichap JJ, Keator C, Wusthoff C, Ryan N, Dobyns WB, Hegde M (2018) Why West? Comparisons of clinical, genetic and molecular features of infants with and without spasms. PLoS One 13:e0193599. https://doi.org/10.1371/journal.pone.0193599
Bögershausen N, Wollnik B (2018) Mutational landscapes and phenotypic spectrum of SWI/SNF-related intellectual disability disorders. Front Mol Neurosci 11:252. https://doi.org/10.3389/fnmol.2018.00252
Carvill GL, Heavin SB, Yendle SC, McMahon JM, O’Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, Malone S, Wallace G, Stanley T, Bye AM, Bleasel A, Howell KB, Kivity S, Mackay MT, Rodriguez-Casero V, Webster R, Korczyn A, Afawi Z, Zelnick N, Lerman-Sagie T, Lev D, Moller RS, Gill D, Andrade DM, Freeman JL, Sadleir LG, Shendure J, Berkovic SF, Scheffer IE, Mefford HC (2013) Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 45:825–830. https://doi.org/10.1038/ng.2646
Chesi A, Staahl BT, Jovicic A, Couthouis J, Fasolino M, Raphael AR, Yamazaki T, Elias L, Polak M, Kelly C, Williams KL, Fifita JA, Maragakis NJ, Nicholson GA, King OD, Reed R, Crabtree GR, Blair IP, Glass JD, Gitler AD (2013) Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci 16:851–855. https://doi.org/10.1038/nn.3412
Goodwin LR, Picketts DJ (2018) The role of ISWI chromatin remodeling complexes in brain development and neurodevelopmental disorders. Mol Cell Neurosci 87:55–64. https://doi.org/10.1016/j.mcn.2017.10.008
Hannula-Jouppi K, Muurinen M, Lipsanen-Nyman M, Reinius LE, Ezer S, Greco D, Kere J (2014) Differentially methylated regions in maternal and paternal uniparental disomy for chromosome 7. Epigenetics 9:351–65. https://doi.org/10.4161/epi.27160
He N, Lin ZJ, Wang J, Wei F, Meng H, Liu XR, Chen Q, Su T, Shi YW, Yi YH, Liao WP (2018) Evaluating the pathogenic potential of genes with de novo variants in epileptic encephalopathies. Genet Med. https://doi.org/10.1038/s41436-018-0011-y
Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, Gonzaga-Jauregui C, Erdin S, Bayram Y, Campbell IM, Hunter JV, Atik MM, Van Esch H, Yuan B, Wiszniewski W, Isikay S, Yesil G, Yuregir OO, Tug Bozdogan S, Aslan H, Aydin H, Tos T, Aksoy A, De Vivo DC, Jain P, Geckinli BB, Sezer O, Gul D, Durmaz B, Cogulu O, Ozkinay F, Topcu V, Candan S, Cebi AH, Ikbal M, Yilmaz Gulec E, Gezdirici A, Koparir E, Ekici F, Coskun S, Cicek S, Karaer K, Koparir A, Duz MB, Kirat E, Fenercioglu E, Ulucan H, Seven M, Guran T, Elcioglu N, Yildirim MS, Aktas D, Alikasifoglu M, Ture M, Yakut T, Overton JD, Yuksel A, Ozen M, Muzny DM, Adams DR, Boerwinkle E, Chung WK, Gibbs RA, Lupski JR (2015) Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron 88:499–513. https://doi.org/10.1016/j.neuron.2015.09.048
Ko A, Youn SE, Kim SH, Lee JS, Kim S, Choi JR, Kim HD, Lee ST, Kang HC (2018) Targeted gene panel and genotype-phenotype correlation in children with developmental and epileptic encephalopathy. Epilepsy Res 141:48–55. https://doi.org/10.1016/j.eplepsyres.2018.02.003
Krupp DR, Barnard RA, Duffourd Y, Evans SA, Mulqueen RM, Bernier R, Riviere JB, Fombonne E, O’Roak BJ (2017) Exonic mosaic mutations contribute risk for autism spectrum disorder. Am J Hum Genet 101:369–390. https://doi.org/10.1016/j.ajhg.2017.07.016
Lelieveld SH, Wiel L, Venselaar H, Pfundt R, Vriend G, Veltman JA, Brunner HG, Vissers L, Gilissen C (2017) Spatial clustering of de novo missense mutations identifies candidate neurodevelopmental disorder-associated genes. Am J Hum Genet 101:478–484. https://doi.org/10.1016/j.ajhg.2017.08.004
Li H (2012) Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics 28:1838–1844. https://doi.org/10.1093/bioinformatics/bts280
Lopez AJ, Wood MA (2015) Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front Behav Neurosci 9:100. https://doi.org/10.3389/fnbeh.2015.00100
Maddirevula S, Alzahrani F, Al-Owain M, Al Muhaizea MA, Kayyali HR, AlHashem A, Rahbeeni Z, Al-Otaibi M, Alzaidan HI, Balobaid A, El Khashab HY, Bubshait DK, Faden M, Yamani SA, Dabbagh O, Al-Mureikhi M, Jasser AA, Alsaif HS, Alluhaydan I, Seidahmed MZ, Alabbasi BH, Almogarri I, Kurdi W, Akleh H, Qari A, Al Tala SM, Alhomaidi S, Kentab AY, Salih MA, Chedrawi A, Alameer S, Tabarki B, Shamseldin HE, Patel N, Ibrahim N, Abdulwahab F, Samira M, Goljan E, Abouelhoda M, Meyer BF, Hashem M, Shaheen R, AlShahwan S, Alfadhel M, Ben-Omran T, Al-Qattan MM, Monies D, Alkuraya FS (2018) Autozygome and high throughput confirmation of disease genes candidacy. Genet Med. https://doi.org/10.1038/s41436-018-0138-x
Magi A, Tattini L, Palombo F, Benelli M, Gialluisi A, Giusti B, Abbate R, Seri M, Gensini GF, Romeo G, Pippucci T (2014) H3M2: detection of runs of homozygosity from whole-exome sequencing data. Bioinformatics 30:2852–2859. https://doi.org/10.1093/bioinformatics/btu401
Marom R, Jain M, Burrage LC, Song IW, Graham BH, Brown CW, Stevens SJC, Stegmann APA, Gunter AT, Kaplan JD, Gavrilova RH, Shinawi M, Rosenfeld JA, Bae Y, Tran AA, Chen Y, Lu JT, Gibbs RA, Eng C, Yang Y, Rousseau J, de Vries BBA, Campeau PM, Lee B (2017) Heterozygous variants in ACTL6A, encoding a component of the BAF complex, are associated with intellectual disability. Hum Mutat 38:1365–1371. https://doi.org/10.1002/humu.23282
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. https://doi.org/10.1101/gr.107524.110
Myers KA, Johnstone DL, Dyment DA (2019) Epilepsy genetics: current knowledge, applications, and future directions. Clin Genet 95:95–111. https://doi.org/10.1111/cge.13414
Olave I, Wang W, Xue Y, Kuo A, Crabtree GR (2002) Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes Dev 16:2509–2517. https://doi.org/10.1101/gad.992102
Sajan SA, Jhangiani SN, Muzny DM, Gibbs RA, Lupski JR, Glaze DG, Kaufmann WE, Skinner SA, Annese F, Friez MJ, Lane J, Percy AK, Neul JL (2017) Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet Med 19:13–19. https://doi.org/10.1038/gim.2016.42
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshe SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH, Zuberi SM (2017) ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 58:512–521. https://doi.org/10.1111/epi.13709
Sokpor G, Xie Y, Rosenbusch J, Tuoc T (2017) Chromatin remodeling BAF (SWI/SNF) complexes in neural development and disorders. Front Mol Neurosci 10:243. https://doi.org/10.3389/fnmol.2017.00243
Staahl BT, Crabtree GR (2013) Creating a neural specific chromatin landscape by npBAF and nBAF complexes. Curr Opin Neurobiol 23:903–913. https://doi.org/10.1016/j.conb.2013.09.003
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA (2013) From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinform 43(11 10):1–33. https://doi.org/10.1002/0471250953.bi1110s43
Vogel-Ciernia A, Matheos DP, Barrett RM, Kramar EA, Azzawi S, Chen Y, Magnan CN, Zeller M, Sylvain A, Haettig J, Jia Y, Tran A, Dang R, Post RJ, Chabrier M, Babayan AH, Wu JI, Crabtree GR, Baldi P, Baram TZ, Lynch G, Wood MA (2013) The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat Neurosci 16:552–561. https://doi.org/10.1038/nn.3359
Vogel-Ciernia A, Kramar EA, Matheos DP, Havekes R, Hemstedt TJ, Magnan CN, Sakata K, Tran A, Azzawi S, Lopez A, Dang R, Wang W, Trieu B, Tong J, Barrett RM, Post RJ, Baldi P, Abel T, Lynch G, Wood MA (2017) Mutation of neuron-specific chromatin remodeling subunit BAF53b: rescue of plasticity and memory by manipulating actin remodeling. Learn Mem 24:199–209. https://doi.org/10.1101/lm.044602.116
Wu JI, Lessard J, Olave IA, Qiu Z, Ghosh A, Graef IA, Crabtree GR (2007) Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56:94–108. https://doi.org/10.1016/j.neuron.2007.08.021
Yang H, Wang K (2015) Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc 10:1556–1566. https://doi.org/10.1038/nprot.2015.105
Acknowledgements
We thank the probands and their families for the participation in this study. For technical support, we acknowledge the participation of Drs. Silvestra Amata, Angela Spalletta, Maurizio Sturnio, and Pietro Schinocca from the Laboratory of Medical Genetics, Oasi Research Institute—IRCCS, Troina (Italy). We also thank Dr. Anthony Charles for the English language editing. This work was funded by Grant RF-2011-02350693 from the Italian Ministry of Health to Marco Fichera.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Financial disclosures
The authors report no disclosures relevant to the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fichera, M., Failla, P., Saccuzzo, L. et al. Mutations in ACTL6B, coding for a subunit of the neuron-specific chromatin remodeling complex nBAF, cause early onset severe developmental and epileptic encephalopathy with brain hypomyelination and cerebellar atrophy. Hum Genet 138, 187–198 (2019). https://doi.org/10.1007/s00439-019-01972-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-019-01972-3