
Abstract
The Forkhead box E3 (FOXE3) gene encodes a transcription factor with a forkhead/winged helix domain that is critical for development of the lens and anterior segment of the eye. Monoallelic and biallelic deleterious sequence variants in FOXE3 cause aphakia, cataracts, sclerocornea and microphthalmia in humans. We used clustered regularly interspaced short palindromic repeats/Cas9 injections to target the foxe3 transcript in zebrafish in order to create an experimental model of loss of function for this gene. Larvae that were homozygous for an indel variant, c.296_300delTGCAG, predicting p.(Val99Alafs*2), demonstrated severe eye defects, including small or absent lenses and microphthalmia. The lenses of the homozygous foxe3 indel mutants showed more intense staining with zl-1 antibody compared to control lenses, consistent with increased lens fiber cell differentiation. Whole genome transcriptome analysis (RNA-Seq) on RNA isolated from wildtype larvae and larvae with eye defects that were putative homozygotes for the foxe3 indel variant found significant dysregulation of genes expressed in the lens and eye whose orthologues are associated with cataracts in human patients, including cryba2a, cryba1l1, mipa and hsf4. Comparative analysis of this RNA-seq data with iSyTE data identified several lens-enriched genes to be down-regulated in foxe3 indel mutants. We also noted upregulation of lgsn and crygmxl2 and downregulation of fmodb and cx43.4, genes that are expressed in the zebrafish lens, but that are not yet associated with an eye phenotype in humans. These findings demonstrate that this new zebrafish foxe3 mutant model is highly relevant to the study of the gene regulatory networks conserved in vertebrate lens and eye development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Forkhead box E3 (FOXE3) gene (MIM 601094) encodes a forkhead/winged helix containing, DNA-binding transcription factor that is critical for normal development of the lens and anterior segment of the eye (Blixt et al. 2000; Brownell et al. 2000; Ormestad et al. 2002; Shi et al. 2006; Medina-Martinez et al. 2005). Biallelic, deleterious sequence variants in FOXE3 cause severe disruption to morphogenesis of the lens and anterior segment of the eye in humans and affect up to 2.5% of patients with eye defects (Williamson and FitzPatrick 2014). Defects are frequently bilateral and typically comprise aphakia, sclerocornea and microphthalmia, although anterior segment dysgenesis, congenital cataracts, aniridia and coloboma of the optic disc have also been observed (Williamson and FitzPatrick 2014; Semina et al. 2001; Gould and John 2002; Valleix et al. 2006; Iseri et al. 2009; Reis et al. 2013; Islam et al. 2015; Ullah et al. 2016; Chen et al. 2017; Saboo et al. 2017). The study of FOXE3 is thus highly relevant to the pathogenesis of human eye defects.
Foxe3 is highly conserved, with ocular expression in many vertebrates (Blixt et al. 2000, 2007; Brownell et al. 2000; Medina-Martinez and Jamrich 2007; Anand et al. 2018). In the mouse, onset of Foxe3 expression is coincident with lens induction and formation of the lens placode at embryonic day (E) 9.0 to E9.5 (Blixt et al. 2000; Brownell et al. 2000; Medina-Martinez and Jamrich 2007). Expression remains present in the lens vesicle until E12.5, when Foxe3 activity persists in the proliferative and undifferentiated cells of the anterior epithelium of the lens, but is lost from the differentiating fibers of the lens (Brownell et al. 2000; Medina-Martinez and Jamrich 2007). The Dysgenetic lens (dyl) mouse has two, spontaneous missense mutations in the DNA-binding domain of Foxe3 (p.Phe93Leu and p.Phe98Ser) that impair the ability of Foxe3 to bind to DNA and result in loss of function (Ormestad et al. 2002). Mice that are homozygous for the dyl allele have smaller lenses, cataracts, failure of the lens vesicle to close, and the anterior lens epithelium does not separate from the cornea (Brownell et al. 2000; Medina-Martinez and Jamrich 2007). In a separate murine model, a null mouse with targeted disruption of both Foxe3 alleles (Foxe3tm/tm) showed premature cessation of proliferation of the cells from the anterior lens epithelium and the mice had smaller lenses with failure of the anterior lens epithelium to separate from the cornea (Medina-Martinez et al. 2005). Lastly, the Rinshoken cataract (rct) mouse that is homozygous for a 22 basepair (bp) deletion in a cis-regulatory element upstream of Foxe3 had mild microphthalmia, congenital cataracts and severe degeneration of the lens fibers due to reduced Foxe3 expression (Wada et al. 2011).
The zebrafish lens placode increases in cell number through a delamination process and does not undergo invagination, but overall the development of the lens and expression of corresponding gene regulatory networks (GRNs) are also highly conserved in this species (Cvekl and Zhang 2017). In Danio rerio, foxe3 was detected in the ocular lens from 18 to 24 h post fertilization (hpf), corresponding to the time of lens placode induction and the start of primary lens cell differentiation. Expression remained present in the lens epithelial cells at 31 hpf (Shi et al. 2006; Kuang et al. 2016). foxe3 knockdown with antisense morpholinos (MOs) resulted in larvae with microphthalmia and reduced pupillary size compared to wildtype larvae and a minority of morphants had complete absence of the pupil (Shi et al. 2006; Swindell et al. 2008). The morphant larvae also had smaller lenses with multilayered epithelial cells and abnormal lens fiber cells (Shi et al. 2006). Interestingly, co-injection of a MO targeting p53 was able to partially rescue defects in aortic arch assembly in foxe3 knockdown zebrafish (Kuang et al. 2016).
Despite these existing animal models of decreased foxe3 function, little is understood about the GRNs conserved in vertebrate lens formation and the perturbations to such GRNs that result from foxe3 deficiency. We used Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 to create an in vivo animal model of decreased foxe3 function by targeting the protein forkhead domain in zebrafish. We then performed whole genome transcriptome analysis (RNA-Seq) to elucidate the GRNs influenced by foxe3 function in vertebrates, with the goal of better understanding the effects of deficiency of this gene on lens and eye development.
Materials and methods
All animal experiments were performed under a protocol approved by the Institute for Animal Care and Use Committee (IACUC) at UCSF (AN108657-02C). EKW strain zebrafish were maintained at 28 °C on a 14 h (h) light/10 h dark cycle.
Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 to target foxe3
We used a modification of previously described methods (Talbot and Amacher 2014). We designed a CRISPR small guide (sg)RNA targeting the forkhead domain of foxe3 (Table S1). We co-injected sgRNA and Cas9 protein into wildtype zebrafish eggs at the one cell stage and used a T7 assay and/or Sanger sequencing at 24–48 hpf to assess mutagenesis in batches of 10–20 larvae (for primers, see Table S2). F0 larvae with indel mutations were raised and fin-clipped at 6–8 weeks of age in batches of 10–20 larvae for Sanger sequencing to determine the exact indel variants present. We selected three F0 founders with small indels introducing a frameshift or eliminating a splice site in foxe3, thus predicting premature protein truncation and nonsense-mediated decay. These founder fish were outcrossed to wildtype Danio rerio and the F1 progeny were grown and genotyped to identify fish with germline transmission of the indel variants. F1 fish that were heterozygotes for indel variants in foxe3 were incrossed to obtain homozygous or compound heterozygous, F2 larvae with biallelic foxe3 mutations. From the three F0 founder fish that we raised, we selected male and female fish that were heterozygous for the c.296_300delTGCAG variant in foxe3 (Table S3) and these fish were used for subsequent incrosses and experiments. Larvae from incrosses of c.296_300delTGCAG fish were studied at 3–6 days post fertilization (dpf) for the ocular findings previously observed in patients with FOXE3 mutations and animal models of Foxe3 and foxe3 loss of function. We were not able to examine the eyes of juveniles or adults, as the fish do not survive in the zebrafish nursery system, possibly due to difficulty competing for food compared to wildtype and heterozygous larvae with normal vision. Eye size and lens morphology were measured using light microscopy. Larvae with eye defects were photographed and then genotyped by Sanger sequencing to confirm homozygosity for the foxe3 indel variants. For immunohistochemistry, larvae were fixed in 4% paraformaldehyde, washed with phosphate buffered saline (PBS) and placed in 25% (w:v) sucrose and then 35% (w:v) sucrose. Blocks were made and sectioned according to standard techniques. Sections were dried overnight and stored at -80 °C. Sections were washed three times in PBS with Tween 20 (PBST) and incubated in blocking buffer containing 4% normal goat serum (v:v), 2% normal sheep serum and 0.2% Tween in PBS for 4 h at 4 °C. Slides were incubated with a 1 in 500 dilution of zl-1 primary antibody (Zebrafish International Resource Center; https://zebrafish.org/) in blocking buffer overnight at 4 °C. Sections were then washed in PBST and incubated with antimouse IgG labeled with Texas red (M32017, Invitrogen) as secondary antibody for 4 h at 4 °C. Sections were mounted with antifade mounting medium containing 4,6-Diamidino-2-phenylindole (DAPI; Vectashield, Vectorlabs) as a counterstain and imaged with a fluorescent microscope.
Injections of wildtype foxe3 mRNA to rescue the phenotype of homozygous, foxe3 indel mutant larvae
To examine the ability of injections of wildtype foxe3 mRNA to rescue the eye phenotype observed in incrosses of heterozygous, foxe3 indel fish, we amplified the single isoform, full-length foxe3 transcript (for primers, see Table S4). Capped and polyadenylated foxe3 mRNA was prepared as previously described (mMessage mMachine® T7 Ultra kit; ThermoFischerScientific) (Chao et al. 2010). We incrossed heterozygous foxe3 indel fish and injected mRNA at concentrations of 0, 6.25 and 12.5 ng/µl into eggs at the 1–4 cell stage. Larvae were incubated and scored for eye defects at 3 dpf as above.
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) to examine dnajb1 transcription in larvae with eye defects from incrosses of heterozygous, foxe3 indel fish
We performed experiments to examine the effects of reduced foxe3 expression on dnajb1 transcription (Khan et al. 2016). After designing gene-specific primers for dnajb1a and dnajb1b (Table S5), we obtained total RNA (Qiagen RNeasy kit) at 3 dpf from wildtype EKW larvae and larvae with eye defects selected from a heterozygous, foxe3 indel incross as described above. RNA was DNase treated and cDNA was synthesized with the SuperScript III First-Strand Synthesis System (Invitrogen). Reactions were run on an ABI Prism 7500 sequence detection system and data were analyzed according to the ΔΔCt method using gapdh (NM_001115114.1) or eef1a1l1 (NM_131263.1) as an internal control according to previous methods (Wu et al. 2015).
Whole genome transcriptome analysis (RNA-Seq) to determine differential gene expression between wildtype larvae and incrossed, foxe3 indel larvae with eye defects
To investigate the effects of reduced foxe3 function on gene transcription, total RNA was extracted from pools of 20 whole-larvae at 3 dpf using the GeneJET RNA Purification Kit; we extracted from three biological replicates of wildtype EKW (control) larvae and 3 biological replicates of larvae from an incross of heterozygous foxe3 indel fish. From the indel incross, only larvae with aberrant eyes and lenses (putative homozygotes) were used for RNA extraction. We assessed RNA quality and quantity prior to making cDNA and preparing libraries for sequencing. RNA quality was reviewed with a bioanalyzer (2100 Bioanalyzer, Agilent) and libraries were prepared with an Ovation® RNA-seq system V2 kit (NuGEN). With this method, total RNA was reverse transcribed to synthesize first-strand cDNA using a combination of random hexamers and a poly-T chimeric primer. The RNA template was then partially degraded by heating and the second strand cDNA was synthesized using DNA polymerase. The double-stranded DNA was then amplified using single primer isothermal amplification (SPIA). SPIA is a linear cDNA amplification process in which RNase H degrades RNA in DNA/RNA heteroduplex at the 5′-end of the double-stranded DNA, after which the SPIA primer binds to the cDNA and the polymerase starts replication at the 3′-end of the primer by displacement of the existing forward strand. Random hexamers were then used to amplify the second-strand cDNA linearly. Finally, libraries from the SPIA-amplified cDNA were made using the Ultralow DR library kit (NuGEN). The RNA-seq libraries were analyzed for quality and primer dimers by bioanalyzer and quantified by qPCR (KAPA) prior to sequencing. High-throughput sequencing was done using SE (single-end) 50 lane(s) on a HiSeq 4000 instrument (Illumina). The datasets were deposited in NCBI GEO (GEO#).
Analysis of foxe3 indel and wildtype larvae RNA-Seq datasets
The quality of raw single-end reads of RNA-Seq dataset was tested by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to evaluate the per-base sequence quality, quality scores, sequence length distribution and overrepresented adapter/primer sequences. Based on the obtained scores data trimming and clipping was performed using “Timmomatic” (Bolger et al. 2014) and aligned against the zebrafish reference genome (GRCz10/danRer7) using a fast splice junction mapper TopHat v2.0.9 (Trapnell et al. 2009). Alignment results were then processed using Cufflinks v2.1.1 (Trapnell et al. 2009) to assemble transcripts and measure the relative abundances. Cuffdiff (Trapnell et al. 2009) was used to calculate differentially expressed genes using the genome aligned BAM files from Tophat and merged assembly generated by Cuffmerge (Trapnell et al. 2009). Statistically significant differentially expressed genes (DEGs) with log2 (fold change) greater than ± 0.5 with p value ≤ 0.05 and the mean Fragments Per Kilobase of Exon Per Million Fragments Mapped (FPKM) ≥ 1 in all datasets were identified by comparing foxe3 indel mutant with wildtype control datasets using an in house Python script. Principal component analysis (PCA) was performed on global gene expression to determine the relatedness between individual replicates of wildtype and mutant datasets. DAVID v6.8 (Database for Annotation, Visualization and Integrated Discovery) (Huang et al. 2009) was used to investigate biologically relevant functional clustering of DEGs in foxe3 indel mutant datasets. The results for enriched gene ontology (GO) for DEGs are presented as bar plots. We also performed comparative analysis of the DEGs identified in the foxe3 indel mutant RNA-seq data with previously reported microarray data (GSE9711) from transgenic mouse lenses over-expressing Foxe3 in fiber cells under the control of Cryaa-promoter (Landgren et al. 2008). In addition, we also performed comparative analysis of foxe3 indel mutant DEGs (1.5-fold up- or down-regulated) with genes exhibiting enriched expression in mouse embryonic day (E)12.5 lens according to iSyTE (Lachke et al. 2012; Kakrana et al. 2018).
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) to verify RNA-Seq results
We used qRT-PCR to validate the altered expression between wildtype and foxe3 indel larvae from the RNA-Seq experiments. For these studies, we used the same RNA samples that were previously submitted for RNA-Seq analysis. 1 ng cDNA was amplified using Universal SybrGreen mastermix containing Rox (Roche) and gene specific primers (Tables S5 and S6) at final concentrations of 2.5 µM. Reactions were run on a StepOne cycler (Thermo Fischer Scientific) and analyzed with StepOne™ Software and ExpressionSuite software (Thermo Fisher Scientific) according to the ΔΔCt method. We used gapdh or eef1a1l1 as internal control genes as above. All experiments were performed on three biological replicates from wildtype EKW (control) larvae or larvae obtained from the incrossed foxe3 indel heterozygotes, and each reaction was run in triplicate.
Results
Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 targeting foxe3
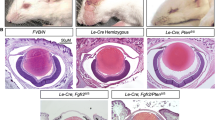
We used CRISPR/Cas9 to create three indel variants in foxe3 in independent, wildtype F0 zebrafish (Fig. S1). All of the variants predicted loss of function for the one isoform of the zebrafish gene (Table S3). We studied larvae that were homozygous for the indel variant c.296_300delTGCAG in foxe3. These larvae demonstrated an ocular phenotype, with reduced eye size and small, deformed or absent lenses (Fig. 1). The foxe3 phenotype followed an autosomal recessive inheritance pattern, with defective lens formation present in 17/57 (29.8%) and 32/134 (23.9%) larvae at 3 dpf in a total of two independent crosses of fish that were heterozygous for the foxe3 indel. Penetrance was directly verified by sequencing eight fish with aberrant lens formation and eight fish with normal-appearing eyes by light microscopy at 6 dpf. All of the larvae (8/8) with abnormal lenses were homozygous for the foxe3 frameshift indel, while 6/8 fish with normal appearing eyes were heterozygous for the indel and 2/8 were wildtype, indicating that the phenotype is highly penetrant. The eye findings were visible with light microscopy from 3 dpf and the indel foxe3 mutant larvae appeared otherwise normal externally (Fig. 1). The phenotype observed was similar to that previously described with antisense MOs targeting foxe3 that produced larvae with small lenses (Shi et al. 2006; Swindell et al. 2008). A comparison of eye diameter and eye diameter divided by body length for wildtype larvae and larvae with eye defects that were putative homozygotes for the foxe3 indel mutation showed a significant decrease in eye size, consistent with microphthalmia, in the mutant larvae for both measurements at 6 dpf (Fig. 2). We also used zl-1 antibody to detect the effects of reduced foxe3 expression on lens fiber protein and showed increased staining in the smaller lenses of the foxe3 indel mutant larvae compared to wildtype controls (Fig. 3). As zl-1 stains differentiating lens fibers (Greiling et al. 2010), this result is consistent with increased fiber cell differentiation in foxe3 indel mutant larvae compared to wildtype controls.

Homozygous foxe3 indel larvae show eye abnormalities with aberrant lens formation at 6 days post fertilization. a Photograph of wildtype larva at 6 days post fertilization (dpf), showing normal eye formation (115×). b Photograph of wildtype larva at 6 dpf, showing normal exterior (25×). c Photograph of foxe3 indel mutant larva at 6 dpf, showing eye defect with small pupil (115×). d Photograph of foxe3 indel mutant larva at 6 dpf, showing apparently normal exterior (25×)

Measurements of eye diameter and eye diameter/body length show reduced eye size in homozygous foxe3 indel larvae at 6 days post fertilization. a Measurements of eye diameter at 6 dpf in 9 wildtype larvae (blue bar) and 8 larvae with eye defects from an incross of heterozygous, foxe3 indel fish (green bar). Plotted values represent the average of all measurements. Error bars are one standard error of the mean. A two-tailed, heteroscedastic T test showed that eye size is significantly smaller in the foxe3 indel mutant fish with eye defects (p = 0.017), indicated by asterisk. b Measurements of eye diameter divided by body length at 6 dpf in 8 wildtype larvae (blue bar) and 8 larvae with eye defects from an incross of heterozygous, foxe3 indel fish (green bar). Plotted values are averages of all measurements. Error bars are one standard error of the mean. A two-tailed, heteroscedastic T test showed eye size divided by body length is significantly smaller in the foxe3 indel mutant fish with eye defects (p = 0.0037), indicated by asterisk. (Color figure online)

Homozygous, foxe3 indel mutant larvae show smaller ocular lenses and increased staining with anti-lens fiber cell marker antibody zl-1 compared to wildtype controls at 6 days post fertilization. Immunohistochemistry performed with zl-1 antibody and visualized with Texas Red at 6 days post fertilization (dpf) demonstrates smaller lenses with more intense staining of fiber cells in foxe3 mutant larvae compared to wildtype controls. a Right eye stained with DAPI and zl-1 (Texas red) from EKW control larva. b Right eye stained with DAPI and zl-1 (Texas red) from homozygous foxe3 indel larva, showing smaller lens with more intense zl-1 staining. c Left eye stained with DAPI and zl-1 (Texas red) from EKW control larva. d Left eye stained with DAPI and zl-1 (Texas red) from homozygous foxe3 indel larva, showing smaller lens with more intense zl-1 staining. e Right eye stained with zl-1 (Texas red) from EKW control larva. f Right eye stained with zl-1 (Texas red) from homozygous foxe3 indel larva, showing smaller lens with more intense zl-1 staining. g Left eye stained with zl-1 (Texas red) from EKW control larva. h Left eye stained with zl-1 (Texas red) from homozygous foxe3 indel larva, showing smaller lens with more intense zl-1 staining. (Color figure online)
Injections of wildtype foxe3 mRNA to rescue the phenotype of foxe3 indel mutant larvae
To demonstrate that the CRISPR/Cas9 injections targeted the foxe3 gene, we attempted phenotypic rescue by injecting full length, wildtype Danio rerio foxe3 mRNA into the eggs obtained from incrosses of heterozygous, foxe3 indel fish. Without injection of mRNA, the heterozygous incrosses could typically be expected to result in approximately 25% larvae with eye defects when scored by light microscopy at 3 dpf due to the autosomal recessive inheritance of the phenotype and the high penetrance of the ocular findings as described above. However, with RNA injections of 12.5 ng/µl, the proportion of larvae manifesting eye defects decreased to 18%, indicating partial rescue, although this difference was not statistically significant (Table S7). We did not attempt injections of higher concentrations of RNA due to toxicity observed at this dose (data not shown). The partial rescue and the similarity in phenotype between larvae injected with antisense MOs targeting foxe3 and our homozygous foxe3 indel mutant fish implies that the ocular phenotype that we observed was due to disruption of foxe3.
qRT-PCR to examine expression of dnajb1a and dnajb1b in homozygous, foxe3 indel mutant larvae
We first performed qRT-PCR to examine the expression of dnajb1a and dnajb1b in the foxe3 indel mutant larvae with eye defects due to previously published results that showed downregulation of DNAJB1 expression in human cells with wildtype FOXE3 compared to cells with truncated FOXE3 after whole transcriptome analysis, implying that FOXE3 downregulates DNAJB1 (Khan et al. 2016). The qRT-PCR result for dnajb1b showed no statistically significant difference in expression, but expression of dnajb1a was upregulated in the foxe3 indel mutant fish with eye defects compared to wildtype fish (Fig. 4; p = 0.03). This result is consistent with the finding that antisense morpholino injections targeting dnajb1a result in larvae with microphthalmia and cataracts (Khan et al. 2016), suggesting that this gene is an important downstream effector of foxe3 in the zebrafish lens. The qRT-PCR results also confirmed reduced foxe3 expression in the foxe3 indel mutants with eye defects, compared to wildtype larvae, although there appeared to be residual foxe3 transcription. We were unable to obtain a suitable antibody to perform Western blotting to confirm loss of function of foxe3 in the larvae with eye defects.

Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) shows upregulation of dnajb1a in foxe3 indel mutant larvae. RNA was extracted at 3 days post fertilization (dpf) from pools of control larvae and larvae with lens defects (putative homozygotes) obtained from an incross of fish that were heterozygous for an indel variant in foxe3. After cDNA synthesis, two pairs of gene-specific primers were used to amplify dnajb1a and dnjab1b and one pair of gene-specific primers was used to amplify foxe3. qRT-PCR was performed and analyzed using the ΔΔCt method with gapdh as an internal control gene. The results show log2 expression fold change plotted for dnajb1a, dnajb1b, and foxe3 and, in addition to confirming downregulation of foxe3, demonstrate upregulation of dnajb1a (p = 0.03), but not dnajb1b (p = 0.1), in larvae with eye defects from the foxe3 indel mutant incross. All values are an average of three biological replicates with three replicates for each experiment and the error bars indicate the standard error of the mean
Whole genome transcriptome analysis (RNA-Seq) to determine differential gene expression between wildtype larvae and incrossed, foxe3 indel larvae with eye defects
To better understand the function of foxe3 expression in the lens, we performed whole genome RNA-Seq using whole larvae from wildtype controls and larvae with eye defects from a foxe3 indel incross. In total, 255,880,576 single end reads were generated from three replicates each for control and mutant. Of these, 199,380,411 mapped to the zebrafish reference genome (Table 1). PCA analysis demonstrated that the foxe3 indel mutant datasets clustered distinctly compared to wildtype control (Fig. 5a). Using a log2 (fold change) cutoff of greater than ± 0.5 at p value ≤ 0.05 and the mean FPKM ≥ 1, we could identify 2170 DEGs (Fig. 5). This included 584 upregulated and 1586 downregulated DEGs (Table S8). Next, we analyzed these DEGs with the bioinformatics tool (DAVID bioinformatics resource) and identified several gene ontology (GO) clusters that may be relevant to the defects observed in the mutant (Fig. 6a, b) (Table S10).

RNA-Seq analysis of foxe3 indel mutant zebrafish. a Principal component analysis (PCA) plot analysis of foxe3 indel mutant and wildtype RNA-Seq datasets. Principal component analysis (PCA) of foxe3 indel mutant and wildtype RNA-Seq datasets showed mutant (1, 2 and 3) and wildtype (4, 5 and 6) datasets as distinct clusters. b Differentially expressed genes in foxe3 indel mutant compared to control. A plot of fold change (FC) for differentially expressed genes in foxe3 indel mutant against mean expression levels in fragments per kilobase of transcript per million mapped reads (FPKM), highlighting downregulated (green) and upregulated (red) genes at 3 dpf. (Color figure online)

Gene ontology (GO) enrichment analysis of differentially expressed genes in foxe3 indel mutant zebrafish. a GO analysis of statistically significant differentially expressed genes (DEGs) in foxe3 indel mutant zebrafish was performed to identify enriched categories in up-regulated (n = 584) and, b down-regulated (n = 1586) candidate genes. Cluster enrichment p value < 0.05
We next compared the foxe3 indel mutant DEGs with orthologous genes enriched in mouse E12.5 lens according to the bioinformatics database resource iSyTE2.0 (integrated Systems Tool for Eye gene discovery) (Kakrana et al. 2018), which has led to the identification of high-priority candidate genes in lens development (Siddam et al. 2018). We found that 16.6% (n = 181 genes) that are 1.5-fold up- or down-regulated in the foxe3 indel mutant fish have mouse orthologs that exhibit enriched expression at ≥1.5-fold (n = 1088) in the lens at E12.5 in iSyTE (Table S11). Interestingly, of these 181 DEGs, over 70% genes (n = 128) are down-regulated in foxe3 mutant zebrafish and are enriched in the E12.5 mouse lens. Among the foxe3 down-regulated genes that are also iSyTE lens-enriched are interesting candidates such as grifin (galectin family protein in lens; Ogden et al. 1998), mipb (associated with cataract; Froger et al. 2010), crygmx (lens gamma crystallin; Greiling et al. 2009), cryba2b (associated with cataract; Reis et al. 2013), tmod1 (associated with lens fiber skeleton organization; Nowak et al. 2009), slc16a12a (associated with cataract; Kloeckener-Gruissem et al. 2008), six5 (associated with cataract; Sarkar et al. 2000), aldh1a3 (associated with microphthalmia/anophthalmia; Yahyavi et al. 2013) as well as new uncharacterized candidates such as rnf10 and tcf7, among many others.
Further, we also performed a comparative analysis of the DEGs identified in the foxe3 indel mutant fish with microarray-identified DEGs in the lens of a transgenic mouse model wherein Foxe3 is over-expressed in the fiber cells via the Cryaa-promoter (Landgren et al. 2008 PMID: 18539941). Because the experimental context in the present study is foxe3 loss-of-function (foxe3 indel mutant zebrafish) while that of the previous study is Foxe3 gain-of-function (Foxe3 over-expression in mouse lens), we expect that genes that are down-regulated in our study are likely to be identified as up-regulated in the Landgren et al. (2008) study. Indeed, we find that 13.8% genes (n = 154) in the foxe3 indel mutant fish correlate with the 1113 mouse orthologs in this manner between the two studies (Table S12). Thus, although these comparisons are between whole embryonic tissue of foxe3 indel mutant fish RNA-seq DEGs and wild-type (iSyTE) or Foxe3-over-expressed mouse lens tissue, they identify many new Foxe3 downstream targets whose positive regulation by this key transcription factor is conserved between fish and mouse.
foxe3 regulates expression of genes involved in lens development
The results of the whole transcriptome analysis (RNA-Seq) on RNA taken from pooled, putative homozygous foxe3 mutant larvae and wildtype larvae demonstrated significant misregulation of several genes that are expressed in the lens and that have human orthologues associated with eye defects, including more than 20 genes that are associated with cataracts (Table 2 and Tables S9 and S13; Fig. 7). We found significant upregulation of crystallin genes (crybb1, cryba4, cryba2a, cryba1l1, crygn1, crygn2, crygmxl2), glutamine synthetase domain containing lens protein encoding gene (lgsn), lens fiber membrane intrinsic protein (lim2.2), transcription factor genes (heat shock factor 4 (hsf4), early growth response 1 (egr1)), as well as genes with potential function in transcription (e.g. GO:0003700 ~ transcription factor activity and GO:0043565 ~ sequence-specific DNA binding) (Fig. 6a) (Table S10). The downregulated genes were found to be enriched in the GO categories for GO:0015254 ~ glycerol channel activity, GO:0006508 ~ proteolysis, GO:0008236 ~ serine-type peptidase activity, GO:0008289 ~ lipid binding, GO:0042632 ~ cholesterol homeostasis and GO:0051006 ~ positive regulation of lipoprotein lipase activity (Fig. 6b) (Table S10). The dysregulation of the candidate genes aqp3a, ctrb1, crygmx, cryba2b, cdh1, cp, cx43.4, mipa, fmodb and fgfbp1b was independently validated with qRT-PCR (Fig. 7). Interestingly, several of the genes up-regulated in the foxe3 indel mutant are normally found to be enriched in fiber cells in agreement with the function of foxe3 in maintenance of epithelial cell fate in the mouse.

Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) showing significant upregulation of lgsn, cryba2a and cryba1l1, with significant downregulation of mipa, fmodb and cx43.4. RNA was extracted at 3 days post fertilization (dpf) from pools of control larvae and larvae with lens defects (putative homozygotes) obtained from an incross of fish that were heterozygous for an indel variant in foxe3. After cDNA synthesis, gene-specific primers were used to amplify each gene. qRT-PCR was performed and analyzed using the ΔΔCt method and gapdh or eef1a1l1 as an internal control gene. The results showed significant upregulation of lgsn, cryba2a and cryba1l1, with significant downregulation of mipa, fmodb and cx43.4. All values are an average of 3 biological replicates with 3 replicates for each experiment and the error bars indicate the standard error of the mean. * indicates statistical significance (p < 0.05)
Discussion
We used CRISPR/Cas9 technology to create an in vivo model of reduced foxe3 function by targeting the protein forkhead domain in zebrafish. Larvae that are homozygous for the indel variant c.296_300delTGCAG, predicting p.(Val99Alafs*2) (NM_001079682.2), in foxe3 have smaller eyes and defective lens formation at 72 hpf; (Figs. 1, 2), resembling the findings observed with prior antisense MO injections targeting foxe3 in zebrafish (Shi et al. 2006; Swindell et al. 2008). This phenotype demonstrated an autosomal recessive pattern of inheritance and was fully penetrant. Immunohistochemistry with zl-1 antibody that stains differentiating lens fibers showed increased expression in the indel mutant larvae compared to wildtype controls. We performed whole genome transcriptome analysis (RNA-Seq) on RNA taken from whole, pooled foxe3 mutant larvae with eye defects and wildtype larvae and found significant dysregulation of genes that were expressed in the lens and that have human orthologues associated with cataracts, including upregulation of cryba2a, cryba1l1, crygmxl2, lgsn and hsf4 and downregulation of mipa, fmodb and cx43.4.
The human orthologue of cryba2a, CRYBA2, is expressed during early lens development in zebrafish and has been associated with autosomal dominant cataracts in humans (Reis et al. 2013). cryba1l1 is also expressed in the lens and the related gene, CRYBA1, is well known to be associated with human cataracts (Kannabiran et al. 1998; Khan et al. 2015). Additional crystallin genes also expressed in the lens that were dysregulated in the foxe3 indel larvae with eye defects included cryba4, crygmxl2, crybb1, crygn2, cryba1b that were upregulated and cryba2b that was downregulated (Table S8). As lens fiber differentiation involves an accumulation of crystallin proteins (Cvekl et al. 2015), the increased expression of crystallin genes in the foxe3 indel larvae is consistent with the hypothesis that loss of foxe3 augments differentiation of lens fibers, as per the finding that ectopic up-regulation of Foxe3 in murine lenses results in down-regulation of fiber expression genes, including those encoding crystallins (Landgren et al. 2008).
Other upregulated genes in the present study that support this hypothesis include hsf4, a transcription factor that is expressed in lens fiber cells and regulates the expression of heat-shock proteins, including lens B-crystallin (Somasundaram and Bhat 2004; Anand and Lachke 2017; Anand et al. 2018). Deleterious variants located in the DNA-binding domain of this gene cause autosomal dominant cataracts and autosomal recessive cataracts when within hydrophobic repeats (HR-A/B) or downstream of the hydrophobic repeats (for review, see Anand et al. 2018). Targeted Hsf4 deletion or mutation in mice also results in lens fiber cell defects and cataracts, with down regulation of the γ-crystallin genes in the early postnatal lens (Fujimoto et al. 2004; Shi et al. 2009; Anand et al. 2018). lgsn, encoding lengsin, is a member of the glutamine synthetase (GS) family of proteins that is moderately abundant in the vertebrate lens and exhibits a higher, localized concentration in differentiating lens fiber cells (Grassi et al. 2006; Wyatt et al. 2006). Recombinant murine lengsin is catalytically inactive, and thus lengsin is considered most likely to have a structural or chaperone-like role similar to that of the crystallin proteins in the lens (Wyatt et al. 2006). A function in maintaining lens transparency has been hypothesized (Grassi et al. 2006), but as yet there is no known relationship to human disease.
In relation to the genes that were found to be downregulated, major intrinsic protein of lens fiber a (mipa; also known as aquaporin 0, or aqpo) is expressed in the eye and lens vesicle (Greiling et al. 2010; Sorokina et al. 2011). This gene has been studied in Danio rerio and MO knockdown of this gene results in cataract formation, indicating that it is critical for normal lens development and transparency (Froger et al. 2010). Mutations in this gene are also a notable cause of autosomal dominant cataracts in humans (Chepelinsky 2009). Fibromodulin (fmodb) is a member of the small leucine-rich proteoglycan (SLRP) group of extracellular matrix that interacts with type I and type II collagen fibrils and inhibits fibrillogenesis in vitro (Chen et al. 2010; Amjadi et al. 2013; Steinhart et al. 2014). Fibromodulin null mice demonstrate thinner sclera and reduced diameter of collagen fibers in sclera that potentially alters susceptibility to the damage resulting from raised intraocular pressure (Chen et al. 2010; Amjadi et al. 2013). Double knockout mice with null alleles for fibromodulin and lumican (Lum(−/−)Fmod(−/−)) have high myopia, increased axial length, thin sclera and retinal detachment, and thus it has been hypothesized that fibromodulin may be relevant to the development of myopia in humans (Chakravarti et al. 2003), although no pathogenic variants have been demonstrated to date. Connexin 43 (cx43), connexin 46 and connexin 50 were also specifically expressed in the lens and Cx43 was present in lens epithelial cells and differentiating lens fiber cells prior to downregulation during differentiation (Behnam et al. 2016). Mutations in CX46 and CX50 were also associated with autosomal dominant and autosomal recessive congenital cataracts and age-related cataracts in humans (Pichi et al. 2016; Berthoud and Ngezahayo 2017).
Our results suggest that our foxe3 indel model is highly relevant to the study of lens development and has significant potential to discover insights into the GRNs that are perturbed by reduced foxe3 function and are conserved across vertebrates, thus allowing the identification of core foxe3 regulated gene circuits. Our results are consistent with the hypothesis that foxe3 inactivation is required for fiber differentiation, in that a deficiency of foxe3 increases the expression of genes involved in this process. Two unsolved factors remain. We used RNA from whole zebrafish and thus expression in other tissues may have influenced our RNA-Seq results, although it should be noted that many of the genes that were dysregulated in the foxe3 indel mutant larvae exhibit lens-specific expression, with established roles in lens biology (Lachke et al. 2012; Kakrana et al. 2018). In the future, experiments could be performed on RNA extracted from the head or eye only. Similarly, we were unable to obtain a suitable antibody to determine the amount of residual protein in the foxe3 indel mutant fish with eye defects; in view of our qRT-PCR results, we cannot differentiate between a partial loss of function and a total null model.
Conclusion
We have used CRISPR/Cas9 to target the forkhead domain of the zebrafish foxe3 gene, establishing a new Danio rerio model of foxe3 deficiency that manifests smaller eyes and lens defects, with lenses exhibiting increased staining with zl-1 antibody, consistent with increased lens fiber cell differentiation. Whole genome transcriptome analysis showed that this model had significant dysregulation of genes that are expressed in the lens and eye whose orthologues are associated with cataracts in human patients, including the fiber cell-expressed factors such as the crystallin genes cryba2a and cryba1l1, as well as mipa and hsf4. Significant upregulation of lgsn and crygmxl2 and downregulation of fmodb and cx43.4, genes that are expressed in the zebrafish lens, but that are not yet known to be associated with an eye phenotype in humans, was also noted. These results were consistent with the known role for foxe3 in the anterior epithelium of the lens prior to differentiation of lens fibers, demonstrating that the zebrafish mutant model is highly relevant to the study of the conserved gene regulatory networks involved in vertebrate lens and eye development.
References
Amjadi S, Mai K, McCluskey P, Wakefield D (2013) The role of lumican in ocular disease. ISRN Ophthalmol 2013:632302
Anand D, Lachke SA (2017) Systems biology of lens development: a paradigm for disease gene discovery in the eye. Exp Eye Res 156:22–33
Anand D, Agrawal SA, Slavotinek A, Lachke SA (2018) Mutation Update of Transcription Factor Genes FOXE3, HSF4, MAF and PITX3 causing ocular defects. Hum Mutat 39:471–494
Behnam M, Imagawa E, Chaleshtori AR, Ronasian F, Salehi M, Miyake N, Matsumoto N (2016) A novel homozygous mutation in HSF4 causing autosomal recessive congenital cataract. J Hum Genet 61:177–179
Berthoud VM, Ngezahayo A (2017) Focus on lens connexins. BMC Cell Biol 18(Suppl 1):6
Blixt A, Mahlapuu M, Aitola M, Pelto-Huikko M, Enerbäck S, Carlsson P (2000) A forkhead gene, FoxE3, is essential for lens epithelial proliferation and closure of the lens vesicle. Genes Dev 14:245–254
Blixt A, Landgren H, Johansson BR, Carlsson P (2007) Foxe3 is required for morphogenesis and differentiation of the anterior segment of the eye and is sensitive to Pax6 gene dosage. Dev Biol 302:218–229
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120
Brownell I, Dirksen M, Jamrich M (2000) Forkhead Foxe3 mapes to the dysgenetic lens locys and is critical in lens development and differentiation. Genesis 27:81–93
Chakravarti S, Paul J, Roberts L, Chervoneva I, Oldberg A, Birk DE (2003) Ocular and scleral alterations in gene-targeted lumican-fibromodulin double-null mice. Invest Ophthalmol Vis Sci 44:2422–2432
Chao R, Nevin L, Agarwal P, Riemer J, Bai X, Delaney A, Akana M, JimenezLopez N, Bardakjian T, Schneider A, Chassaing N, Schorderet DF, FitzPatrick D, Kwok PY, Ellgaard L, Gould DB, Zhang Y, Malicki J, Baier H, Slavotinek A (2010) A male with unilateral microphthalmia reveals a role for TMX3 in eye development. PLoS One 5:e10565
Chen S, Oldberg A, Chakravarti S, Birk DE (2010) Fibromodulin regulates collagen fibrillogenesis during peripheral corneal development. Dev Dyn 239:844–854
Chen J, Wang Q, Cabrera PE, Zhong Z, Sun W, Jiao X et al (2017) Molecular genetic analysis of Pakistani families with autosomal recessive congenital cataracts by homozygosity screening. Invest Ophthalmol Vis Sci 58:2207–2217
Chepelinsky AB (2009) Structural function of MIP/aquaporin 0 in the eye lens: genetic defects lead to congenital inherited cataracts. Handb Exp Pharmacol 190:265–297
Cvekl A, Zhang X (2017) Signaling and gene regulatory networks in mammalian lens development. Trends Genet 33:677–702
Cvekl A, McGreal R, Liu W (2015) Lens development and crystallin gene expression. Prog Mol Biol Transl Sci 134:129–167
Froger A, Clemens D, Kalman K, Németh-Cahalan KL, Schilling TF, Hall JE (2010) Two distinct aquaporin 0 s required for development and transparency of the zebrafish lens. Invest Ophthalmol Vis Sci 51:6582–6592
Fujimoto M, Izu H, Seki K, Fukuda K, Nishida T, Yamada S, Kato K, Yonemura S, Inouye S, Nakai A (2004) HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J 23:4297–4306
Gould DB, John SW (2002) Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum Mol Genet 11:1185–1193
Grassi F, Moretto N, Rivetti C, Cellai S, Betti M, Márquez AJ et al (2006) Structural and functional properties of lengsin, a pseudo-glutamine synthetase in the transparent human lens. Biochem Biophys Res Commun 350:424–429
Greiling TM, Houck SA, Clark JI (2009) The zebrafish lens proteome during development and aging. Mol Vis 15:2313–2125
Greiling TM, Aose M, Clark JI (2010) Cell fate and differentiation of the developing ocular lens. Invest Ophthalmol Vis Sci 51:1540–1546
Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Iseri SU, Osborne RJ, Farrall M, Wyatt AW, Mirza G, Nürnberg G et al (2009) Seeing clearly: the dominant and recessive nature of FOXE3 in eye developmental anomalies. Hum Mutat 30:1378–1386
Islam L, Kelberman D, Williamson L, Lewis N, Glindzicz MB, Nischal KK et al (2015) Functional analysis of FOXE3 mutations causing dominant and recessive ocular anterior segment disease. Hum Mutat 36:296–300
Kakrana A, Yang A, Anand D, Djordjevic D, Ramachandruni D, Singh A, Huang H, Ho JWK, Lachke SA (2018) iSyTE 2.0: a database for expression-based gene discovery in the eye. Nucleic Acids Res 46(D1):D875–D885
Kannabiran C, Rogan PK, Olmos L, Basti S, Rao GN, Kaiser-Kupfer M et al (1998) Autosomal dominant zonular cataract with sutural opacities is associated with a splice mutation in the betaA3/A1-crystallin gene. Mol Vis 4:21
Khan AO, Aldahmesh MA, Alkuraya FS (2015) Phenotypes of recessive pediatric cataract in a cohort of children with identified homozygous gene mutations (An American Ophthalmological Society Thesis). Trans Am Ophthalmol Soc 113:T7
Khan SY, Vasanth S, Kabir F, Gottsch JD, Khan AO, Chaerkady R et al (2016) FOXE3 contributes to Peters anomaly through transcriptional regulation of an autophagy-associate protein termed DNAJB1. Nat Commun 7:10953
Kloeckener-Gruissem B, Vandekerckhove K, Nürnberg G, Neidhardt J, Zeitz C, Nürnberg P et al (2008) Mutation of Solute Carrier SLC16A12 Associates with a Syndrome Combining Juvenile Cataract with Microcornea and Renal Glucosuria. Am J Hum Genet 82:772–779
Kuang S-Q, Medina-Martinez O, Guo D, Gong L, Regalado ES, Reynolds CL et al (2016) FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J Clin Invest 126:948–961
Lachke SA, Ho JW, Kryukov GV, O’Connell DJ, Aboukhalil A, Bulyk ML et al (2012) iSyTE: integrated Systems Tool for Eye gene discovery. Invest Ophthalmol Vis Sci 53:1617–1627
Landgren H, Blixt A, Carlsson P (2008) Persistent FoxE3 expression blocks cytoskeletal remodeling and organelle degradation during lens fiber differentiation. Invest Ophthalmol Vis Sci 49:4269–4277
Medina-Martinez O, Jamrich M (2007) Foxe view of lens development and disease. Development 134:1455–1463
Medina-Martinez O, Brownell I, Amaya-Manzanares F, Hu Q, Behringer RR, Jamrich M (2005) Severe defects in proliferation and differentiation of lens cells in Foxe3 null mice. Mol Cell Biol 25:8854–8863
Nowak RB, Fischer RS, Zoltoski RK, Kuszak JR, Fowler VM (2009) Tropomodulin1 is required for membrane skeleton organization and hexagonal geometry of fiber cells in the mouse lens. J Cell Biol 186:915–928
Ogden AT, Nunes I, Ko K, Wu S, Hines CS, Wang AF et al (1998) GRIFIN, a novel lens-specific protein related to the galectin family. J Biol Chem 273:28889–28896
Ormestad M, Blixt A, Churchill A, Martinsson T, Enerbäck S, Carlsson P (2002) Foxe3 haploinsufficiency in mice: a model for Peters’ anomaly. Invest Ophthalmol Vis Sci 43:1350–1357
Pichi F, Lembo A, Serafino M, Nucci P (2016) Genetics of congenital cataract. Dev Ophthalmol 57:1–14
Reis LM, Tyler RC, Muheisen S, Raggio V, Salviati L, Han DP et al (2013) Whole exome sequencing in dominant cataract identifies a new causative factor, CRYBA2, and a variety of novel alleles in known genes. Hum Genet 132:761–770
Saboo US, Penke D, Mahindrakar A, Uddaraju M, Sankurathri C, Gong X et al (2017) Exome sequencing reveals novel homozygous FOXE3 mutation in microphthalmos with staphylomatous malformation. Ophthalmic Genet 38:295–297
Sarkar PS, Appukuttan B, Han J, Ito Y, Ai C, Tsai W et al (2000) Heterozygous loss of Six5 in mice is sufficient to cause ocular cataracts. Nat Genet 25:110–114
Semina EV, Brownell I, Mintz-Hittner HA, Murray JC, Jamrich M (2001) Mutations in the human forkhead transcription factor FOXE3 associated with anterior segment ocular dysgenesis and cataracts. Hum Mol Genet 10:231–236
Shi X, Luo Y, Howley S, Dzialo A, Foley S, Hyde DR et al (2006) Zebrafish foxe3: roles in ocular lens morphogenesis through interaction with pitx3. Mech Dev 123:761–782
Shi X, Cui B, Wang Z, Weng L, Xu Z, Ma J et al (2009) Removal of Hsf4 leads to cataract development in mice through down-regulation of gamma S-crystallin and Bfsp expression. BMC Mol Biol 10:10
Siddam AD, Gautier-Courteille C, Perez-Campos L, Anand D, Kakrana A, Dang CA, Legagneux V, Méreau A, Viet J, Gross JM, Paillard L, Lachke SA (2018) The RNA-binding protein Celf1 post-transcriptionally regulates p27Kip1 and Dnase2b to control fiber cell nuclear degradation in lens development. PLoS Genet 14(3):e1007278
Somasundaram T, Bhat SP (2004) Developmentally dictated expression of heat shock factors: exclusive expression of HSF4 in the postnatal lens and its specific interaction with alphaB-crystallin heat shock promoter. J Biol Chem 279:44497–44503
Sorokina EA, Muheisen S, Mlodik N, Semina EV (2011) MIP/Aquaporin 0 represents a direct transcriptional target of PITX3 in the developing lens. PLoS One 6:e21122
Steinhart MR, Cone-Kimball E, Nguyen C, Nguyen TD, Pease ME, Chakravarti S et al (2014) Susceptibility to glaucoma damage related to age and connective tissue mutations in mice. Exp Eye Res 119:54–60
Swindell EC, Zilinski CA, Hashimoto R, Shah R, Lane ME, Jamrich M (2008) Regulation and function of foxe3 during early zebrafish development. Genesis 46:177–183
Talbot JC, Amacher SL (2014) A streamlined CRISPR pipeline to reliably generate zebrafish frameshifting alleles. Zebrafish 11:583–585
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-SEq. Bioinformatics 25:1105–1111
Ullah E, Nadeem Saqib MA, Sajid S, Shah N, Zubair M, Khan MA et al (2016) Genetic analysis of consanguineous families presenting with congenital ocular defects. Exp Eye Res 146:163–171
Valleix S, Niel F, Nedelec B, Algros MP, Schwartz C, Delbosc B et al (2006) Homozygous nonsense mutation in the FOXE3 gene as a cause of congenital primary aphakia in humans. Am J Hum Genet 79:358–364
Wada K, Maeda YY, Watanabe K, Oshio T, Ueda T, Takahashi G et al (2011) A deletion in a cis element of Foxe3 causes cataracts and microphthalmia in rct mice. Mamm Genome 22:693–702
Williamson KA, FitzPatrick DR (2014) The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur J Med Genet 57:369–380
Wu D, Mandal S, Choi A, Anderson A, Prochazkova M, Perry H et al (2015) DLX4 is associated with orofacial clefting and abnormal jaw development. Hum Mol Genet 24:4340–4352
Wyatt K, White HE, Wang L, Bateman OA, Slingsby C, Orlova EV, Wistow G (2006) Lengsin is a survivor of an ancient family of class I glutamine synthetases re-engineered by evolution for a role in the vertebrate lens. Structure 14:1823–1834
Yahyavi M, Abouzeid M, Gawdat G, de Preux A-S, Xiao T, Bardakjian T et al (2013) ALDH1A3 loss of function causes bilateral anophthalmia/microphthalmia and hypoplasia of the optic nerve and optic chiasm. Hum Mol Genet 22:3250–3258
Acknowledgements
Library preparation and QCs for sequencing was conducted by Yanxia Hao, Jim McGuire and Natasha Carli, PhD, at the Gladstone Genomics Core.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
None of the authors have potential conflicts of interest to disclose.
Human rights and animal participants
All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2018_1884_MOESM7_ESM.tif
Fig. S1. Chromatograms demonstrating indel variants in F1 fish after CRISPR/Cas9 injections targeting foxe3 (TIF 734 KB)
Rights and permissions
About this article

Cite this article
Krall, M., Htun, S., Anand, D. et al. A zebrafish model of foxe3 deficiency demonstrates lens and eye defects with dysregulation of key genes involved in cataract formation in humans. Hum Genet 137, 315–328 (2018). https://doi.org/10.1007/s00439-018-1884-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-018-1884-1