
Abstract
We review the structures and functions of ADARs and their involvements in human diseases. ADAR1 is widely expressed, particularly in the myeloid component of the blood system, and plays a prominent role in promiscuous editing of long dsRNA. Missense mutations that change ADAR1 residues and reduce RNA editing activity cause Aicardi–Goutières Syndrome, a childhood encephalitis and interferonopathy that mimics viral infection and resembles an extreme form of Systemic Lupus Erythmatosus (SLE). In Adar1 mouse mutant models aberrant interferon expression is prevented by eliminating interferon activation signaling from cytoplasmic dsRNA sensors, indicating that unedited cytoplasmic dsRNA drives the immune induction. On the other hand, upregulation of ADAR1 with widespread promiscuous RNA editing is a prominent feature of many cancers and particular site-specific RNA editing events are also affected. ADAR2 is most highly expressed in brain and is primarily required for site-specific editing of CNS transcripts; recent findings indicate that ADAR2 editing is regulated by neuronal excitation for synaptic scaling of glutamate receptors. ADAR2 is also linked to the circadian clock and to sleep. Mutations in ADAR2 could contribute to excitability syndromes such as epilepsy, to seizures, to diseases involving neuronal plasticity defects, such as autism and Fragile-X Syndrome, to neurodegenerations such as ALS, or to astrocytomas or glioblastomas in which reduced ADAR2 activity is required for oncogenic cell behavior. The range of human disease associated with ADAR1 mutations may extend further to include other inflammatory conditions while ADAR2 mutations may affect psychiatric conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thirty years ago, the first two biochemical studies describing ADAR (Adenosine deaminase acting on RNA), enzymatic activity were published (Bass and Weintraub 1987; Rebagliati and Melton 1987). It was initially expected that this double-stranded (ds) RNA unwinding activity was due to an RNA helicase. However, within a year, it was shown that ‘unwinding’ activity was due to weakened dsRNA base-pairing because adenosines were hydrolytically deaminated at position C6 to inosines (Bass and Weintraub 1988; Polson and Bass 1994). ADARs are very widespread and found in all metazoan species so far examined. Mammalian genomes have five genes encoding ADAR proteins; ADAR1 and ADAR2 are active deaminase enzymes, ADAR3 has no known editing activity and two further, closely related, testis-specific ADAD1 and ADAD2 proteins lack key catalytic residues.
As in the case of RNA splicing and other types of RNA processing, human mutations affecting ADAR RNA editing can be trans-acting when they affect the ADAR enzymatic machinery or cis-acting when they affect the editing of a particular edited transcript. This review will focus on effects of trans-acting ADAR mutations or changes in ADAR proteins in human disease. A recent publication provides the first example of a human disease variant affecting a specific RNA editing substrate. Three potassium channel Kv1.1 mutations associated with the human disorder Episodic Ataxia Type-1 are located within the RNA duplex structure required for RNA editing and decrease RNA editing both in vitro and using an in vivo assay; editing also changes amino acids in this case (Ferrick-Kiddie et al. 2017). Other sequence variants that affect individual RNA editing events in cis have been detected in genome-wide investigations of RNA editing in normal tissues (Park et al. 2017).
ADAR1
ADAR1 protein-RNA interactions
The ADAR1 gene encodes a constitutively expressed ADAR1 p110 isoform, and a larger IFN inducible ADAR1 p150 isoform (Kim et al. 1994; O’Connell et al. 1995; Patterson and Samuel 1995). The ADAR1 isoforms share the C-terminal catalytic deaminase domain, three dsRNA-binding domains (dsRBD), and one N-terminal Z DNA-binding domain (Zβ); the P150 isoform has an additional N-terminal Z DNA-binding domain (Zα) (Fig. 1).

Domain structures of human ADARs. All ADARs have similar domain organizations with a C-terminal deaminase domain (red) and different numbers of dsRNA-binding domains (dsRBDs) (blue). In addition, ADAR3 has an RG rich region at the N-terminus (brown). Two major isoforms of hADAR1 protein exist, the constitutively expressed nuclear ADAR1 p110 isoform, and the interferon inducible ADAR1 p150 cytoplasmic isoform. Both isoforms have the Zβ domain (ZBD) (yellow) and a nuclear localization sequence (NLS) (pink). The Zα DNA-binding domain and the nuclear export sequence (NES) (green) are present at the N-terminus of the longer ADAR1p150 isoform
Most of our understanding of how ADAR1 interacts with RNA comes from the extensive studies on binding by the deaminase domain and the dsRBDs of ADAR2 (see “ADAR2” below). The additional type of domain in ADAR1 is the Z DNA-binding domain first identified in this protein. One of the suggested functions of this domain is to guide ADAR1 to actively transcribed genes as this is where Z DNA is thought to occur (Herbert and Rich 1999). The role of the Z DNA-binding domains in ADAR1 function is not yet clear.
The crystal structure of the Zβ DNA-binding domain revealed that it consists of a three α helix bundle, flanked on both sites by antiparallel β sheets, the topology being α β α α β β, representing a helix-turn-helix β-sheet fold (Athanasiadis et al. 2005; Schade et al. 1999; Schwartz et al. 1999). It was found that the Zβ domain does not interact with Z DNA, even though this domain is conserved in higher vertebrate ADAR1 proteins (Schwartz et al. 1999), and present also in the p110 isoform. The ADAR1 Zα domain binds Z DNA and also has the ability to bind Z dsRNA (Brown et al. 2000), where, depending on sequence, a transition from right-handed A to left-handed Z form is also possible; dsRNA may be a key natural target of Z DNA-binding domains. The Z DNA-binding domain is complementary in shape and electrostatic properties to the sugar-phosphate backbone of the Z DNA strand. The Z DNA-binding domain is structurally related to the winged helix-turn-helix DNA-binding domains that recognize specific sequences in the major groove of B DNA; however, the recognition helix is not used in the same way for Z DNA-binding.
Human ADAR1 localizes to the cytoplasm, nucleus and nucleolus, with high concentrations also in nuclear speckles (Desterro et al. 2003). The nuclear localization sequence (NLS) is located in the third dsRBD (Strehblow et al. 2002) whereas the nuclear export signal (NES) is at the N-terminus (Poulsen et al. 2001). Thus, the ADAR1p150 isoform shuttles and is predominantly cytoplasmic whereas the constitutive p110 isoform is mainly nuclear. The Zα domain in ADAR1p150 causes it to localize to cytoplasmic stress granule (Ng et al. 2013). Localization of ADAR1 p150 to stress granules does not require enzymatic activity as active and inactive mutants both localize. The Zα domain has homology with Zα domains from E3L and ZBP1, which also localize to stress granules.
The up-regulation of ADAR1 transcripts during inflammation causes the production of different isoforms with different localizations within the cell (Yang et al. 2003b). These include another isoform, p80, starting from M519 and lacking Zβ and dsRBD l, generated by alternative splicing of exon 2. Antibodies against the C-terminus detected p150, p110 and p80 in mouse splenocytes and HeLa cells. Two additional variants of p80 and p110 that result from alternative splicing at the end of dsRBD 3 increased during treatment with ConA or IL-2 (Yang et al. 2003b). These variants localized differently from p80 and p110 in cytoplasm and nucleus in transfected HeLa and NIH3T3 cells expressing EGFP-tagged proteins, suggesting that these different isoforms are targeted to different locations during inflammation.
Aicardi–Goutières Syndrome and mouse Adar1 mutant models
A to I RNA editing is the most abundant RNA modification in the human transcriptome (Athanasiadis et al. 2004; Qiu et al. 2016); it is tissue specific (Picardi et al. 2015) and changes dynamically during early embryogenesis (Qiu et al. 2016). ADAR1 site-specifically edits some of the same sites as ADAR2 in CNS transcripts; ADAR1 appears to be mainly subsidiary to ADAR2 for site-specific editing within the CNS where both ADARs are present though not in other tissues outside the CNS, where ADAR1 predominates. However, across the human transcriptome A to I editing is observed at far more sites in non-coding sequences than in coding sequences (Athanasiadis et al. 2004; Bahn et al. 2015; Peng et al. 2012; Qiu et al. 2016; Wang et al. 2013). It appears to be mainly ADAR1 that is involved in this promiscuous RNA editing; A to I editing in transcripts encoding repetitive Alu elements is the most abundant. Editing in hairpin-forming inverted Alu repeats in 3′ UTRs and intronic sequences in pre-mRNAs as well as in noncoding RNAs contributes over 99% of ADAR editing sites (Bahn et al. 2015; Bazak et al. 2014).
Homozygous or compound heterozygous mutations in ADAR1 cause Aicardi–Goutières Syndrome (AGS) (Livingston et al. 2014; Rice et al. 2012). AGS is a rare and often fatal childhood encephalopathy with increased levels of interferon (IFN), and symptoms that mimic congenital virus infections (Crow and Manel 2015). ADAR1 mutations, including some of the AGS-associated variants, alternatively cause childhood dystonia due to striatal neurodegeneration (Livingston et al. 2014), or spastic paraplegia with corticospinal motor system degeneration (Livingston et al. 2014), in other patients. AGS is like an extreme form of Systemic Lupus Erythematosus (SLE), a much more common disease that involves periodic increases in interferon expression (Crow and Manel 2015). Mutations in innate immune nucleic acid sensors contribute to SLE and to other inflammatory diseases and further investigation may show ADAR1 involvement in other inflammatory diseases.
The AGS mutations are missense mutations mainly on the surface of the deaminase domain with one other in the N-terminal Zα domain of ADAR1 p150. The AGS mutations reduce editing activity to different extents (Mannion et al. 2014); with the exception of ADAR G1007R none of the individual mutations that occur in patients entirely eliminates ADAR1 editing activity. Many patients are from consanguineous families and are homozygous or transheterozygous for mutant combinations that together also do not eliminate all editing activity; ADAR1 G1007R is the only heterozygous mutant that causes AGS, probably because it binds dsRNA well (Heale et al. 2009; Rice et al. 2012) and may interfere with the normal protein to cause disease dominantly. Some remaining editing activity is presumably required for live birth. Based on the structure of the ADAR2 deaminase domain-Brf2 RNA complex it is clear that several of the ADAR1 deaminase domain AGS mutations change residues that make RNA contacts [for structure images see Fisher and Beal (2017)]. Mutations on other parts of the deaminase domain may affect RNA-binding by indirect interactions across the deaminase domain surface (Fisher and Beal 2017) or they may impair interactions with RLR proteins, or with other constitutive RNA helicases that may also act as dsRNA sensors before RIG-I and MDA5, which are induced by IFN (Zhang et al. 2011).
In mice the Adar1 gene (this gene is named Adar in the Mouse Genome Informatics (MGI) database, http://www.informatics.jax.org/) consist of 15 exons, Northern blot analysis revealed transcripts of 7 and 5 kb (Mittaz et al. 1997; Slavov et al. 2000), and nuclear ADAR1p110 and cytoplasmic ADAR1p150 proteins are produced. There are high expression levels in adult mice in the brain, spleen, follicular B cells and macrophages and lower levels in the pancreas, eyes and testis. After treatment with lipopolysaccharide (LPS), to induce interferon (IFN) expression, induction of both ADAR1 isoforms was observed after 4 h (Yang et al. 2003a). Mouse Adar1 null mutants are embryonic lethal by day E12.5, with aberrant IFN expression and loss of hematopoietic cells (Hartner et al. 2004, 2009; Wang et al. 2004). Apoptosis in different embryonic organs including vertebra and heart was observed in Adar1 mutant mouse embryos (Wang et al. 2004). We rescued Adar1 null mutant embryonic lethality in Adar1 Δ2−13 ;Mavs mice in which signaling from three interacting, cytoplasmic, antiviral RIG-I-like dsRNA receptors (RLRs) (Mannion et al. 2014) is prevented by loss of their shared mitochondrial antiviral signaling (MAVS) adapter protein. Adar1 Δ2−13 ;Mavs mice still die at P0.5 and the cause of death is unknown.
In another study using an Adar1 7−9 mutant allele (Pestal et al. 2015) some Adar1 7−9 ;Mavs double mutant pups survive beyond the day of birth but still die within several weeks. The Adar1 7−9 mutant allele may be able to produce a truncated ADAR1 protein with only dsRBDs and this may partly ameliorate the mutant phenotype (Walkley et al. 2012). Further evidence that ADAR1 protein or truncated ADAR1 proteins also make editing-independent contributions to suppression of aberrant innate immune responses, probably because they still bind particular RNAs, come from a recent study on an Adar1 E861A catalytically inactive mutant (Liddicoat et al. 2015). The catalytically inactive mutant is also embryonic lethal but dies about 2 days later than the Adar1 deletion mutants. Also, deletion of the IfiH1 gene encoding Mda5, the RIG I-like receptor (RLR) activated by long dsRNA, fully rescued the Adar1 E861A catalytically inactive mutant (Liddicoat et al. 2015). This IfiH1 mutant did not rescue the Adar1 Δ2−13 null mutant in our study (L.K, unpublished), nor in the other study using the Adar1 7−9 mutant allele (Pestal et al. 2015). Adar1 E861A ;Ddx58 −/− double mutants lacking RIG I, which is activated by short dsRNA, did not show rescue of Adar1 E861A mutant embryonic lethality (Liddicoat et al. 2015). Therefore, activation of MDA5 is the main reason for high expression of ISGs and embryonic lethality in Adar1 E861A. Transfection into Adar1 Δ2−13 ;Trp53 MEFs also indicate that the suppressor of this pathway is the ADAR1 p150 isoform; p110 may have other roles in development—some substrates are edited primarily by the nuclear p110 isoform, e.g., 5-HTR2C mRNA (Pestal et al. 2015).
The information from the mouse studies gives the understanding of ADAR1-mutant AGS illustrated in Fig. 2. Constitutively expressed nuclear ADAR1p110 converts A to I in dsRNA produced during cellular transcription. Edited dsRNAs are unable to activate the main cytoplasmic RIG I-like receptors (RLRs), MDA5 and RIG-I. Viral dsRNA, or cellular dsRNA that is unedited due to absence or inactivation of ADAR1, is detected in the cytoplasm by MDA-5 or RIG-I. Upon activation, these receptors interact with the MAVS adaptor protein located on the mitochondrial membrane and activate IRF3, IRF7 and NFκB. IRF3 and IRF7 phosphorylation lead to their translocation into the nucleus and induction of type I IFN. Activation of NFκB leads to transcription of proinflammatory cytokines. The interferon response includes a resolution phase (Schoggins et al. 2011); one of the proteins induced later by increased type I IFN is ADAR1 p150. This protein, mainly localized in the cytoplasm, edits all dsRNA regardless of its origin. Such edited dsRNA does not activate MDA-5 and RIG-I, and this leads to a termination of further IFN signaling. A more recent publication extends this model by showing that very early in the response to virus infection ADAR1p110 is rapidly degraded in a ubiquitin-dependent process (Li et al. 2016), presumably to amplify dsRNA detection by the antiviral sensors; both ADARp110 and ADARp150 are increased again later in the interferon-coordinated process.

Intracellular detection of dsRNA and role of ADAR1. a Constitutively expressed isoform ADAR1p110 converts A to I in dsRNA produced during transcription. Edited dsRNAs are unable to activate the main cytoplasmic RIG I-like receptors (RLRs), MDA5 and RIG-I. b Viral dsRNA or cellular dsRNA that is unedited due to absence or inactivation of ADAR1 is detected in the cytoplasm by MDA-5 or RIG-I. Upon activation, these receptors interact with the MAVS adaptor protein located on the mitochondrial membrane and activate NFκB. Activation of NFκB lead to transcription of proinflammatory cytokines. IRF3 and IRF7 phosphorylation lead to their translocation into the nucleus and induction of type I IFN. c One of the proteins induced by type I IFN is ADAR1 p150. This protein, mainly localized in the cytoplasm, edits all dsRNA. Such edited dsRNA is not detected by MDA-5 and RIG-I, and this leads to a termination of further IFN signaling
Heterozygous loss of function mutations in ADAR1 also cause Dyschromatosus Symmetrica Hereditaria (DSH1), an otherwise apparently harmless skin condition with pigmented macules due to aberrant melanocyte distributions; some DSH1 patients have been shown to also have a low aberrant expression of interferon (Rice et al. 2012). Increased basal interferon expression leading to improved resistance to infectious disease might account for DSH1 being a not-uncommon condition in certain regions of China. The innate immune system needs to respond to different infections rapidly but proportionately and the inflammation process normally resolves quickly thereafter. At the level of human population evolution this need for balance may lead to evolutionary trade-off selection (Medzhitov 2010; Okin and Medzhitov 2012; Ramos et al. 2015). For instance, SLE is particularly prevalent in African-American women; proinflammatory human genetic variants that protect against long-established major lethal pathogens such as malaria, especially in children, may be maintained in some populations even though these variants may incur inflammatory costs, especially later in life. Genetic variants affecting innate immune sensing of nucleic acids affect detection not only of viruses but also of other pathogens such as malaria and TB. Variants of ADAR1 may set different inflammatory response thresholds in different individuals that affect resistance to infectious diseases.
ADAR2
ADAR 2 protein–RNA interactions
Until recently ADAR2 proteins received more experimental attention than ADAR1 proteins. ADAR2 has been preferred for studies on site-specific ADAR–RNA interactions in vitro; it has a simpler domain structure than ADAR1 and it can be expressed to higher levels in, and purified from, heterologous cells. Therefore, studies on ADAR2 have contributed more to understanding how ADAR proteins recognize editing sites and how the editing reaction is catalysed. The RNA editing substrate at the Gria2 R/G site has been minimized to a 72 base RNA hairpin that has been the mainstay of in vitro studies to understand site-specific RNA editing by ADARs (Ohman et al. 2000; Schoft et al. 2007).
The ADAR 2 protein consists of the C-terminal deaminase domain and two dsRNA-binding domains (dsRBDs) (Fig. 1). All three domains contribute to dsRNA binding. The dsRBD domains are 65–75 amino acids long (St Johnston et al. 1992) and found in eukaryotic, prokaryotic and viral proteins (Masliah et al. 2013). The dsRBD structure is a mixed α/β fold with conserved αβββα topology (Bycroft et al. 1995). The structures of the individual human ADAR2 dsRBDs I and II alone and in complexes with portions of the mouse Gria2 R/G RNA hairpin were determined by NMR studies (Stefl et al. 2010). The mode of recognition is the interaction of helix α1 and the β1–β2 loop with successive minor grooves in a partially sequence-specific manner. Conserved positively charged residues at the N-terminus of helix α2 also interact across the major groove with the phosphodiester backbone. The RNA recognition is achieved mainly through direct interactions with 2′-hydroxyl groups of RNA by dsRBDs and also through a small number of interactions with bases (Gan et al. 2006; Matthews et al. 2016; Ramos et al. 2000; Ryter and Schultz 1998; Stefl et al. 2010).
The structure of the 45 kD deaminase domain of hADAR2 has been solved by X-ray crystallography (Macbeth et al. 2005); it forms a globular structure and is composed of two α-helices, 4 β-strands and numerous loops. The active site contains a zinc (Zn2+) ion coordinated by two cysteines and a histidine; mutation of these residues or the highly conserved active site glutamate (E396), involved in proton transfer during adenosine deamination, abolish catalytic activity. Matthews, Beal and coworkers recently used X-ray crystallography to describe the interaction of human ADAR2 deaminase domain with different dsRNAs and elucidated the unique base-flipping mechanism of the deaminase domain of ADAR2. A flipping loop approaches the target adenosine residue from the minor groove side, altering the dsRNA helical conformation by widening the major groove and shifting the bases immediately 5′ to the editing site toward the helical axis (Matthews et al. 2016). The adenosine is flipped out through the minor groove and enters the catalytic site while the glutamate of the flipping loop provides compensatory interactions with the unpaired base on the unedited strand. The ADAR proteins edit in a sequence-specific manner. Both ADAR1 and ADAR2 prefer target adenosines with 5′neighbor A or U and 3′neighbor G, i.e., a U/AAG site, where the edited base is underlined (Eggington et al. 2011; Lehmann and Bass 2000). The importance of the 5′ neighbor was proved by replacing 5′ A-U by 5′ C-G or 5′ G-C, both of which result in 80% decreases in editing of the yeast-derived Bdf2 dsRNA by ADAR2. The decrease of editing activity is a result of a steric clash between the 2-amino group of the 5′ guanosine and the glycine 489 residue in the flipping loop. The structure of a Gria2 R/G site complex with full length ADAR2 or ADAR2 dsRBD II plus deaminase domain will be necessary to define a canonical interaction pattern. This RNA is not in the complexes solved so far; also details of the RNA interactions with full length ADAR2 and with different RNA substrates may differ from those observed with deaminase domains alone. The Beal group has also shown that ADARs are able to edit the DNA strand in a DNA–RNA hybrid in vitro, as the hybrid retains the A-form helix of dsRNA (Zheng et al. 2017). The efficiency is about 15-fold lower than with dsRNA and ancillary factors may be required to edit DNA/RNA hybrids in vivo.
ADAR2 is primarily nuclear and nucleolar, similar to ADAR1p110 (Desterro et al. 2003; Sansam et al. 2003). When plasmids encoding editable transcripts are transfected into cells both ADAR1p110 and ADAR2 re-localize from the nucleolus to the nucleoplasm where they can edit the target transcripts (Desterro et al. 2003). Recently the NLS for ADAR2 was located (Behm et al. 2017); two NLS elements are close together in the amino terminus adjacent to the first dsRBD. Importin-α4 interacts with ADAR2 and is required for the nuclear localization.
ADAR2 is posttranslationally modified; it can be phosphorylated at the N-terminus by an unknown kinase and then becomes a substrate for Pin1 (peptidylprolyl cis/trans isomerase, NIMA-interacting 1) (Marcucci et al. 2011); the isomerization of proline of ADAR2 is necessary for nuclear retention of ADAR2. However even in the absence of Pin1, a large cytoplasmic accumulation of ADAR2 is not observed, due to the E3 ligase WWP2 in the cytoplasm that ubiquinates ADAR2, resulting in its degradation by the proteasome. Therefore, in the absence of both Pin1 and WWP2, ADAR2 can accumulate in the cytoplasm. Recently, it has been reported that nuclear localization is less complete and lower levels of ADAR2 are seen in the nucleus in immature neurons (Behm et al. 2017), in agreement with the observation of an increase in nuclear editing of transcripts in neurons (Veno et al. 2012). An increase in the editing level of several sites was found in vivo during mouse brain development (Wahlstedt et al. 2009) and in vitro during rat neuron maturation (Orlandi et al. 2011), but a parallel increase in the expression level of ADAR was not always found (Paupard et al. 2000; Wahlstedt et al. 2009).
Roles of ADAR2 site-specific RNA editing in CNS transcripts
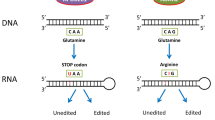
ADAR2 site-specifically edits codons of CNS transcripts, leading to substitution of a different amino acid because inosine decodes as guanosine during translation (Basilio et al. 1962). ADAR2 edits specific adenosine residues within RNA hairpins that typically are formed between a portion of an exon flanking the edited adenosine and sequences in a nearby intron (Higuchi et al. 1993). ADAR2 and RNA editing events that change specific codons in edited transcripts have important roles in the central nervous system (CNS). The glutamine to arginine (Q/R), editing sites in the mouse Gria2 and human GRIA2 transcripts encoding the ionotropic glutamate receptor A2 subunit (GluA2), are edited with over 99% efficiency (Sommer et al. 1991). GRIA2 Q/R site editing changes a key residue in the ion channel pore of this dominant α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor subunit, making AMPA receptors containing the GluA2 R subunit impermeable to calcium. Q/R sites are edited also in transcripts encoding kainate receptor subunits and have a similar effect on calcium permeability of the kainate class of glutamate receptors (Evans et al. 2017). The nomenclature for glutamate receptor subunits has changed over time (Collingridge et al. 2009); the mouse gene and the encoded protein were previously named GluR B and Glu R B, respectively.
Mice homozygous for a targeted Adar2 (named Adarb1 in the MGI database) deletion of exon 6, which encodes the C-terminal deaminase domain, developed progressive seizures as pups and died around weaning (by 21 days) (Higuchi et al. 2000). Mutations in Gria2 transcripts that prevent formation of the dsRNA structure required for editing similarly led to death of mice, with increased calcium permeability in and death of hippocampal neurons (Higuchi et al. 1993). The death of Adar2 mutant pups is prevented by combining the Adar2 mutation with a Gria2 mutant in which the arginine residue at the Q/R editing site is introduced in the genomic sequence (Higuchi et al. 2000), demonstrating that the Q/R site is the key physiological target of ADAR2 editing. Another specific editing event in GluA2 at the R/G site, has less dramatic effects on AMPA receptor function (Lomeli et al. 1994).
Glutamate excitotoxic neuron death is an important factor in certain neurodegenerations, such as the spreading spinal neurodegeneration in ALS (King et al. 2016). ADAR2 expression is proposed to decrease in ALS motor neurons and downregulation of editing at the GRIA2 Q/R site is observed in motor neurons of patients with ALS (Aizawa et al. 2016; Hideyama et al. 2012). The Adar2 mutant mice die too quickly to show more extensive neurodegeneration; however, targeted deletion of Adar2 in motor neurons in young mice leads to slow motor-neuron degeneration (Hideyama et al. 2010). When vertebrate neuronal cells are treated with excess glutamate to induce neurotoxicity, ADAR2 protein is cleaved by a calcium-activated protease, indicating that glutamate excitotoxicity is mediated in part by suppression of GluA2 Q/R site editing (Mahajan et al. 2011). Intriguingly, not all excitatory neurotransmitters but only glutamate is capable of killing vertebrate neurons by excitotoxicity. Glutamate is not excitotoxic to invertebrate neurons, which do not have editing of glutamate receptor transcripts.
Why vertebrate neurons have evolved to incur the risk of excitotoxic neuron death by glutamate if ADAR2 RNA editing fails was an unanswered question (Buckingham et al. 2008), that may now have an answer. Recent findings indicate that the purpose may be to allow regulated ADAR2 RNA editing of glutamate receptor transcripts in response to neuronal excitation. RNA editing in GluA2 changes residues at subunit interfaces and affects assembly of receptor tetramers (Greger and Esteban 2007). Glutamate receptors with Q/R site-unedited subunits more efficiently undergo intracellular membrane transport to the cell surface near synapses (Greger et al. 2002, 2017). Q/R site-edited subunits accumulate in ER and may be destroyed before reaching the cell surface, i.e., ADAR2 editing evolved to restrain glutamate signaling by synaptic scaling in response to general neuronal excitation (Penn et al. 2013). This occurs in both AMPA and kainite receptors and is important for neuronal synaptic plasticity responses (Evans et al. 2017); it remains to be determined whether editing effects on membrane trafficking apply to other classes of receptors or other edited proteins also. Loss of edited isoforms of many proteins (Graveley et al. 2011) is nonlethal in Drosophila and editing may preferentially alter surface residues of independently folding domains in many edited proteins, but this remains to be established by mapping editing events on protein structures (Solomon et al. 2016). ADAR2 is also expressed in pancreatic beta cells, where it is required for efficient secretion of insulin in response to high blood sugar (Gan et al. 2006). No trans-acting human disease mutations in ADAR2 have been described; loss of function mutations in human ADAR2 might contribute to seizures, epilepsy or neurodegenerations (Higuchi et al. 2000), based on the calcium permeability effects. ADAR2 mutations might also contribute to human diseases such as autism (Filippini et al. 2017) that may involve defects in synaptic scaling or to psychiatric illnesses because of effects on glutamate receptor transport. Lower expression of ADAR2 and lower levels of R/G site editing were also observed in patients with mood disorders (Kubota-Sakashita et al. 2014).
A recent publication shows a strong effect of ADAR2 RNA editing on circadian cycling of transcripts in liver (Terajima et al. 2017), indicating another entirely new way in which ADAR 2 variants may contribute to human disease. The authors suggest that previous efforts to detect consistent circadian effects on editing in whole Drosophila brain, even though the period mutant significantly affects editing of some sites (Hughes et al. 2012), might have been due to different clock behaviors in different brain regions. Another hint about roles of vertebrate ADARs comes from a Drosophila study showing that mutating the homolog of ADAR2 dramatically affects sleep (Robinson et al. 2016). It is possible that circadian effects on editing have complicated studies on vertebrate CNS editing. For instance, transcripts encoding a G protein-coupled serotonergic receptor, the 5-hydroxytryptamine Receptor 2C (HTR2C), are edited by both ADAR1 and ADAR2 (Burns et al. 1997). The extensively edited isoforms (5-HT2C-VGVR, 5-HT2C-VSVR) show decreased serotonin responses because of less effective coupling with the phospholipase C signaling cascade (Fitzgerald et al. 1999; Niswender et al. 1999). Many studies showed higher editing of the A site in brains of patients who committed suicide (Dracheva et al. 2008; Iwamoto and Kato 2003; Lyddon et al. 2013; Niswender et al. 2001), although other studies failed to replicate the findings. Difficulties may reflect limited numbers of brain samples or imperfect matching of samples. It also remains to be determined whether RNA editing events reported to be affected in mood disorders show circadian cycling in CNS or in other simpler tissues. It is probably advisable to record the time of day to which RNA editing data applies and to beware of circadian mismatches between samples from now on.
Other proteins that interact with the same RNAs as ADAR2 may affect the ability of ADAR2 to edit these transcripts. The ADAR3 protein is very similar to ADAR2 (the Adar3 gene is named Adarb2 in the MGI database), but no enzymatic activity has been reported for ADAR3, which is mainly expressed in the amygdala and hypothalamus region in the brain (Chen et al. 2000). ADAR3 is a dsRNA binding protein, thought to modulate the activities of ADAR1 and ADAR2, through binding and sequestering shared target transcripts (Donnelly et al. 2013; Oakes et al. 2017); other proteins inhibiting ADAR2 editing have also been identified (Filippini et al. 2017). Overexpression of Adar2 gave rise to obese mice due to overeating caused by a hypothalamic effect (Singh et al. 2007). Human variants of either ADAR2 or the hypothalamus-enriched ADAR3 could contribute to mood disorders or eating disorders.
ADARs in cancer
Elevated ADAR1 and promiscuous RNA editing in cancers
Until recently, cancer research focused mainly on the identification and dissection of the roles played by DNA mutations as important drivers for tumorigenesis and tumor progression. Recently, however, a growing body of evidence shows that both C-to-U and A-to-I RNA editing are also important contributors to cancer. The importance of integrating RNA-Seq data and genomic deep-sequencing has been revealed by recent studies (Han et al. 2015; Paz-Yaacov et al. 2015).
ADAR1 is upregulated and gives elevated, widespread RNA editing in many cancers; different tumors have distinct and characteristic RNA editing patterns compared to their normal tissue counterparts (Fumagalli et al. 2015; Han et al. 2015; Paz-Yaacov et al. 2015). Paired exome and transcriptome sequencing of normal and cancerous breast tissues (triple-negative, HER2-positive, luminal A and luminal B tumors) was carried out by Fumagalli and co-workers. The authors in this study called RNA versus DNA single nucleotide A to G differences (RDDs), with most of them located in regions encoding Alu elements. Analyzing these editing sites they reported a significant increase of editing frequency in tumor cells. ADAR1 overexpression was observed in cancer samples and was due to both DNA copy number amplification and inflammatory pathway activation in cancer cells. Although no correlation between ADAR1 and HER2 status or tumor size was found, ADAR1 silencing in breast cancer cells led to less cell proliferation and increased apoptosis. Of note, ADAR1 expression does not correlate with any specific breast cancer subtype (Fumagalli et al. 2015). ADAR1 is one of the top 5% of upregulated genes in relapsed lobular breast cancer (Shah et al. 2009). The authors demonstrated that lobular metastatic cancer accumulated not only genomic mutations but also a high frequency of non-synonymous hypothetical editing events, resulting in variant protein sequences (Shah et al. 2009).
Consistent with the above study another group found that elevated RNA editing activity is a major contributor to transcriptomic diversity in several tumors (Paz-Yaacov et al. 2015). This study also focused on Alu hyper editing sites, demonstrated that editing is significantly elevated in many different cancer types with only few tumors showing lower editing frequency compared to normal controls (Paz-Yaacov et al. 2015); increased editing also reflects ADAR1 overexpression in these tumors.
The Cancer Genome Atlas (TCGA) has enabled researchers to address important questions about RNA editing on a large scale. TCGA RNA-seq data was analyzed and RNA editing signals in 17 different cancer types detected and related to normal tissues (Han et al. 2015). While this study focused on RNA-Seq data, both Alu and non-Alu RNA editing events were taken into consideration. Most of the informative RNA editing sites were in 3′ UTRs and intronic regions, as observed previously in mouse tissues (Gu et al. 2012). The study revealed specific altered RNA editing patterns in tumor samples relative to normal tissues, which correlate best with global ADAR1 expression levels. The authors went further by focusing on a few nonsynonymous RNA editing sites that may be “master” driver events in different tumors. By correlating RNA editing, tumor subtype, stage and survival, the authors detected a number of recoding RNA editing sites with potential clinical relevance. These editing sites show marked editing differences between distinct patient groups within a cancer type, and they may represent promising biomarker candidates for further assessment (Han et al. 2015).
ADAR1 RNA site-specific editing in cancers
While recent studies provide a global picture of editing alteration in cancer, parallel investigations on how specific editing events may affect cancer are also necessary. One of the first cancers to be associated with altered A-to-I RNA editing was acute myeloid leukemia (AML) (Beghini et al. 2000), an acute, fast growing form of leukemia affecting the myeloid line of blood cells. In AML, blast cells do not develop and patients are unable to defend against infections. The PTPN6 transcript is hyper-edited in low-differentiated CD34(+)/CD117(+) blasts isolated from AML patients (Beghini et al. 2000). PTPN6 (tyrosine-protein phosphatase non-receptor type 6) is an important protein for hematopoietic development recognises and dephosphorylates tyrosine residues of several target proteins such as growth-promoting receptors, including c-Kit tyrosine kinase. In CD34(+)/CD117(+) blasts an aberrant PTPN6 transcript encoding a protein with an altered SH2 domain was observed; the PTPN6 SH2 (Src Homology 2)-domain mediates target protein recognition. The aberrant PTPN6 transcript has multiple editing events, among them an editing event at a splicing branch-point that leads to a non-functional transcript retaining intron 3. It is important to underline that PTPN6 exerts a tumor-suppressor role by down-regulating a broad spectrum of growth-promoting receptors and cytokine receptors. Expression of the non-functional PTPN6 from aberrantly edited transcripts may be a key step for hematopoietic transformation. It remains unclear which ADAR is involved.
CML (chronic myeloid leukemia), another leukemia affecting myeloid cells is slow growing and associated with the abnormal Philadelphia chromosome (Ph chromosome) carrying a Bcr-Abl fusion gene. CML has three stages: the chronic phase, the accelerated phase and blast crisis. A role for A-to-I editing in CML has been established recently (Jiang et al. 2013); whole-transcriptome sequencing of normal, chronic phase and blast crisis cells from CML patients revealed increased IFN-γ pathway gene expression, in concert with Bcr-Abl amplification. Interestingly, along with disease progression from chronic phase to blast crisis, the authors observed enhanced expression of the IFN-induced ADAR1 p150 isoform. Increased ADAR1 p150 was associated with a substantial enrichment of A-to-G changes at target sites and with RNA editing-dependent transcriptional gene modulation. For example, increased ADAR1 p150 was associated with upregulation of myeloid transcription factor PU.1 and downregulation of the erythroid transcription factor GATA1, thereby contributing to myeloid lineage skewing. Moreover, shRNA knockdown of ADAR1 in leukemia cells induced a significant reduction in leukemia stem cells in serial transplantation studies. The authors hypothesize that Bcr-Abl–mediated up-regulation of inflammatory pathway receptors could sensitize hematopoietic progenitors to inflammatory stimuli that drive ADAR1 overexpression (Jiang et al. 2013). These data suggest that ADAR1 plays a pivotal role in malignant progenitor self-renewal and is an important driver of malignant progenitor reprogramming into self-renewing leukemia stem cells. In addition ADAR1 has been shown to be important for maintenance of hematopoietic stem cells in mice (Hartner et al. 2009).
More recently, another study also underlines the urgent need for data collection on both DNA damage and post-transcriptional events such as RNA editing in cancers (Steinman et al. 2013). By transplanting Bcr-Abl marrow cells in which ADAR1 could subsequently be conditionally knocked-out, it was possible to monitor the effect of ADAR1 deletion after leukemia had been established. It was observed that ADAR1 is indeed required for leukemia cell survival in vivo and that knocking down ADAR1 results in rapid leukemic cell loss, suggesting ADAR1 as a molecular target for CML-directed therapeutics (Steinman et al. 2013). However, contrary to the earlier study, these authors reported that ADAR1 expression is independent of Bcr-Abl. Recently, it has been shown that ADAR1 drives leukemia (CML) stem cell self-renewal by impairing pri-let-7 biogenesis (Zipeto et al. 2016). Despite a long list of studies pointing at ADAR1 as a master gene in several leukemias recently another RNA-seq study has shown that both ADAR enzymes also have roles in myeloid leukemia cell differentiation (Rossetti et al. 2017).
Reduced levels of A-to-I RNA editing have been observed in metastatic melanoma. ADAR1 downregulation is mediated by cAMP response element-binding protein (CREB) in metastatic melanoma (Shoshan et al. 2015). In this cellular context, reduced levels of ADAR1 promoted tumor growth and cell proliferation, accompanied by a metastatic phenotype. Changes in melanoma gene expression profiles and miRNA expression levels were observed and can be attributed to reduced ADAR1 activity in a cell type- and context-specific manner. A decrease in editing by ADAR1 within miR-455-5p promotes melanoma growth and metastasis (Shoshan et al. 2015). RNA editing-independent changes in miRNA expression may also occur as a result of direct binding of ADAR1 to pri-miRNA or regulation of the miRNA processing machinery by ADAR1 (Nemlich et al. 2013).
In hepatocellular carcinoma, editing of the transcript encoding the antizyme inhibitor-1 (AZIN1) to give a high-frequency S/G amino acid substitution, has been reported as causative during the multistep progression from healthy hepatic progenitors (Chen et al. 2013). Notably, increased editing in AZIN1 was associated with upregulation of both ADAR1 p150 and p110 levels. Increased editing of AZIN1 transcripts promoted tumor initiation by increasing both invasiveness and tumorigenic potential. These enhanced tumorigenic properties occur as a result of amino acid substitutions that increase binding of AZIN1 to antizyme1, thereby neutralizing antizyme-mediated degradation of downstream oncoproteins such as OCD and cyclin D1. Screening for A-to-I editing in a broad spectrum of cancers identified increased AZIN1 recoding also in esophageal squamous cell carcinoma samples (ESCC) and identified ADAR1 as an oncogene and negative predictor of overall survival during ESCC progression (Qin et al. 2014).
Alteration of another A-to-I editing recoding event has been identified within a RAS homolog family member Q (RHOQ) in colorectal cancer (CRC) (Han et al. 2014). Notably, altered editing at this site was reported to be common in many tumors, including gastric and lung cancer, as well as in HCC. A-to-I editing in RHOQ transcripts resulted in an amino acid substitution from asparagine to serine that enhanced protein activity and mediated dynamic actin cytoskeletal reorganization, thereby increasing the invasive potential of the cells. Similar to this observation, edited RHOQ was associated with increased recurrence rates that were even higher when combined with mutant KRAS (G12D). Although editing of RHOQ was functionally relevant for invasiveness and recurrence in CRC, the amino acid substitution did not appear to alter either protein stability or structural folding, and further investigation is required (Han et al. 2014).
ADAR2 in cancers
A-to-I editing at the GRIA2 Q/R site has been reported to be altered in glioblastoma (also known as grade IV astrocytoma), a deadly brain cancer (Maas et al. 2001). Progressive decrease of ADAR2-mediated editing correlates with the grade of malignancy in paediatric astrocytomas (from low to high grade of malignancy) (Cenci et al. 2008). Forced expression of ADAR2 in glioblastoma cells indeed inhibited both cell proliferation and migration (Cenci et al. 2008; Galeano et al. 2013). Hypo-editing in glioblastoma, was also found in microRNAs (Alon et al. 2011). Decreased ADAR2-mediated editing within the miR-376 cluster increased glioblastoma invasiveness in vivo (Choudhury et al. 2012). Additionally, loss of ADAR2 editing in several key microRNA precursors (pri-miR221/222 and pri-miR-21) was also reported to lead to glioblastoma cell proliferation and increased migration (Tomaselli et al. 2015). The findings that ADAR2 suppresses growth and metastatic features of brain cancer cells suggest that human ADAR2 genetic variants with reduced ADAR2 expression might not only have increased excitability, seizure susceptibility and increased risk of neurodegeneration but also they may lead to greater risk for developing brain tumours. In oesophageal squamous cell carcinoma (ESCC), it was also reported that ADAR2 acts as a tumor suppressor gene as it inhibits tumor growth in vitro and in vivo by editing the IGFBP7 transcript (Chen et al. 2017). In summary, while ADAR1 acts as an oncogene, ADAR2 acts as a tumor suppressor gene in brain tumors (such as astrocytomas) and ESCC.
Conclusions
For ADAR1 we now have a better sense of the main biological role, associated with discrimination between self and non-self RNA as well as with some site-specific RNA editing events. ADAR1 mutations causing AGS are by far the most interesting human genetic finding in the ADAR RNA editing field. It is not yet clear if AGS mutations mainly affect ADAR1 RNA-binding and editing or if interactions with other proteins are affected by AGS mutations. Interactions of ADAR1 with DICER proteins, RLRs and other RNA helicases require further study. It is often difficult to predict which genetic defects will associate with particular proteins. In the case of ADAR2, this is partly because we do not know whether there is an overall biological principle that has governed the evolution of the site-specific editing performed by ADAR2. We do know, for instance, that ADAR2 itself is regulated by levels of neurotransmitters, by excitatory states of neurons/glia and by circadian clocks—we should expect to be further surprised.
Upregulation of ADAR1 in cancers may be linked to inflammation in tumors and the inflammatory status of these tumors may allow them to be targeted for immune checkpoint therapies combined with ADAR inhibitors. It remains to be established in vivo that ADARs mutates DNA, either in somatic hypermutation, class switching, aberrantly in cancers or in genomes over evolutionary time. So, despite the ADAR activity having been first identified over 30 years ago, many question still remain unanswered.
References
Aizawa H, Hideyama T, Yamashita T, Kimura T, Suzuki N, Aoki M, Kwak S (2016) Deficient RNA-editing enzyme ADAR2 in an amyotrophic lateral sclerosis patient with a FUS(P525L) mutation. J Clin Neurosci 32:128–129. doi:10.1016/j.jocn.2015.12.039
Alon S, Vigneault F, Eminaga S, Christodoulou DC, Seidman JG, Church GM, Eisenberg E (2011) Barcoding bias in high-throughput multiplex sequencing of miRNA. Genome Res 21:1506–1511
Athanasiadis A, Rich A, Maas S (2004) Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol 2:e391
Athanasiadis A, Placido D, Maas S, Brown BA 2nd, Lowenhaupt K, Rich A (2005) The crystal structure of the Zbeta domain of the RNA-editing enzyme ADAR1 reveals distinct conserved surfaces among Z-domains. J Mol Biol 351:496–507
Bahn JH, Ahn J, Lin X, Zhang Q, Lee JH, Civelek M, Xiao X (2015) Genomic analysis of ADAR1 binding and its involvement in multiple RNA processing pathways. Nat Commun 6:6355. doi:10.1038/ncomms7355
Basilio C, Wahba AJ, Lengyel P, Speyer JF, Ochoa S (1962) Synthetic polynucleotides and the amino acid code. Proc Natl Acad Sci USA 48:613–616
Bass BL, Weintraub H (1987) A developmental regulated activity that unwinds RNA duplexes. Cell 48:607–613
Bass BL, Weintraub H (1988) An unwinding activity that covalently modifies its double-strand RNA substrate. Cell 55:1089–1098
Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E, Levanon EY (2014) A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24:365–376. doi:10.1101/gr.164749.113
Beghini A, Ripamonti CB, Peterlongo P, Roversi G, Cairoli R, Morra E, Larizza L (2000) RNA hyperediting and alternative splicing of hematopoietic cell phosphatase (PTPN6) gene in acute myeloid leukemia. Hum Mol Genet 9:2297–2304
Behm M, Wahlstedt H, Widmark A, Eriksson M, Ohman M (2017) Accumulation of nuclear ADAR2 regulates A-to-I RNA editing during neuronal development. J Cell Sci. doi:10.1242/jcs.200055
Brown BA 2nd, Lowenhaupt K, Wilbert CM, Hanlon EB, Rich A (2000) The zalpha domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc Natl Acad Sci USA 97:13532–13536. doi:10.1073/pnas.240464097
Buckingham SD, Kwak S, Jones AK, Blackshaw SE, Sattelle DB (2008) Edited GluR2, a gatekeeper for motor neurone survival? BioEssays 30:1185–1192
Burns CM, Chu H, Rueter SM, Hutchinson LK, Canton H, Sanders-Bush E, Emeson RB (1997) Regulation of serotonin-2C receptor G-protein coupling by RNA editing. Nature 387:303–308
Bycroft M, Grunert S, Murzin AG, Proctor M, St Johnston D (1995) NMR solution structure of a dsRNA binding domain from Drosophila staufen protein reveals homology to the N-terminal domain of ribosomal protein S5. EMBO J 14:3563–3571
Cenci C, Barzotti R, Galeano F, Corbelli S, Rota R, Massimi L, Di Rocco C, O’Connell MA, Gallo A (2008) Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J Biol Chem 283:7251–7260
Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K (2000) A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6:755–767
Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y, Liu M, Yuan YF, Fu L, Kong KL, Qi L, Li Y, Zhang N, Tong AH, Kwong DL, Man K, Lo CM, Lok S, Tenen DG, Guan XY (2013) Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat Med 19:209–216. doi:10.1038/nm.3043
Chen YB, Liao XY, Zhang JB, Wang F, Qin HD, Zhang L, Shugart YY, Zeng YX, Jia WH (2017) ADAR2 functions as a tumor suppressor via editing IGFBP7 in esophageal squamous cell carcinoma. Int J Oncol 50:622–630. doi:10.3892/ijo.2016.3823
Choudhury Y, Tay FC, Lam DH, Sandanaraj E, Tang C, Ang BT, Wang S (2012) Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J Clin Invest 122:4059–4076. doi:10.1172/JCI62925
Collingridge GL, Olsen RW, Peters J, Spedding M (2009) A nomenclature for ligand-gated ion channels. Neuropharmacology 56:2–5
Crow YJ, Manel N (2015) Aicardi–Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15:429–440
Desterro JMP, Keegan LP, Lafarga M, Berciano MT, O’Connell M, Carmo-Fonseca M (2003) Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci 116:1805–1818. doi:10.1242/jcs.00371
Donnelly CJ, Zhang P-W, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80:415–428
Dracheva S, Patel N, Woo DA, Marcus SM, Siever LJ, Haroutunian V (2008) Increased serotonin 2C receptor mRNA editing: a possible risk factor for suicide. Mol Psychiatry 13:1001–1010. doi:10.1038/sj.mp.4002081
Eggington JM, Greene T, Bass BL (2011) Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2:319. doi:10.1038/ncomms1324
Evans AJ, Gurung S, Wilkinson KA, Stephens DJ, Henley JM (2017) Assembly, secretory pathway trafficking, and surface delivery of kainate receptors is regulated by neuronal activity. Cell Rep 19:2613–2626
Ferrick-Kiddie EA, Rosenthal JJ, Ayers GD, Emeson RB (2017) Mutations underlying Episodic Ataxia type-1 antagonize Kv1. 1 RNA editing. Sci Rep 7:41095. doi:10.1038/srep41095
Filippini A, Bonini D, Lacoux C, Pacini L, Zingariello M, Sancillo L, Bosisio D, Salvi V, Mingardi J, La Via L (2017) Absence of the Fragile X Mental Retardation Protein results in defects of RNA editing of neuronal mRNAs in mouse. RNA Biol 22:1–12. doi:10.1080/15476286.2017.1338232
Fisher AJ, Beal PA (2017) Effects of Aicardi–Goutieres syndrome mutations predicted from ADAR-RNA structures. RNA Biol 14:164–170. doi:10.1080/15476286.2016.1267097
Fitzgerald LW, Iyer G, Conklin DS, Krause CM, Marshall A, Patterson JP, Tran DP, Jonak GJ, Hartig PR (1999) Messenger RNA editing of the human serotonin 5-HT2C receptor. Neuropsychopharmacology 21:82S–90S. doi:10.1016/S0893-133X(99)00004-4
Fumagalli D, Gacquer D, Rothe F, Lefort A, Libert F, Brown D, Kheddoumi N, Shlien A, Konopka T, Salgado R, Larsimont D, Polyak K, Willard-Gallo K, Desmedt C, Piccart M, Abramowicz M, Campbell PJ, Sotiriou C, Detours V (2015) Principles governing A-to-I RNA editing in the breast cancer transcriptome. Cell Rep 13:277–289. doi:10.1016/j.celrep.2015.09.032
Galeano F, Rossetti C, Tomaselli S, Cifaldi L, Lezzerini M, Pezzullo M, Boldrini R, Massimi L, Di Rocco CM, Locatelli F, Gallo A (2013) ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene 32:998–1009. doi:10.1038/onc.2012.125
Gan Z, Zhao L, Yang L, Huang P, Zhao F, Li W, Liu Y (2006) RNA editing by ADAR2 is metabolically regulated in pancreatic islets and beta-cells. J Biol Chem 281:33386–33394
Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, Brown JB, Cherbas L, Davis CA, Dobin A, Li R, Lin W, Malone JH, Mattiuzzo NR, Miller D, Sturgill D, Tuch BB, Zaleski C, Zhang D, Blanchette M, Dudoit S, Eads B, Green RE, Hammonds A, Jiang L, Kapranov P, Langton L, Perrimon N, Sandler JE, Wan KH, Willingham A, Zhang Y, Zou Y, Andrews J, Bickel PJ, Brenner SE, Brent MR, Cherbas P, Gingeras TR, Hoskins RA, Kaufman TC, Oliver B, Celniker SE (2011) The developmental transcriptome of Drosophila melanogaster. Nature 471:473–479
Greger IH, Esteban JA (2007) AMPA receptor biogenesis and trafficking. Curr Opin Neurobiol 17:289–297
Greger IH, Khatri L, Ziff EB (2002) RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 34:759–772
Greger IH, Watson JF, Cull-Candy SG (2017) Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron 94:713–730
Gu T, Buaas FW, Simons AK, Ackert-Bicknell CL, Braun RE, Hibbs MA (2012) Canonical A-to-I and C-to-U RNA editing is enriched at 3′UTRs and microRNA target sites in multiple mouse tissues. PLoS One 7:e33720. doi:10.1371/journal.pone.0033720
Han SW, Kim HP, Shin JY, Jeong EG, Lee WC, Kim KY, Park SY, Lee DW, Won JK, Jeong SY, Park KJ, Park JG, Kang GH, Seo JS, Kim JI, Kim TY (2014) RNA editing in RHOQ promotes invasion potential in colorectal cancer. J Exp Med 211:613–621. doi:10.1084/jem.20132209
Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HM, Eterovic AK, Yuan Y, Li J, Nair N, Minelli R, Tsang YH, Cheung LW, Jeong KJ, Roszik J, Ju Z, Woodman SE, Lu Y, Scott KL, Li JB, Mills GB, Liang H (2015) The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell 28:515–528. doi:10.1016/j.ccell.2015.08.013
Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH (2004) Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 279:4894–4902
Hartner JC, Walkley CR, Lu J, Orkin SH (2009) ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10:109–115
Heale BS, Keegan LP, McGurk L, Michlewski G, Brindle J, Stanton CM, Caceres JF, O’Connell MA (2009) Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J 28:3145–3156. doi:10.1038/emboj.2009.244
Herbert A, Rich A (1999) Left-handed Z-DNA: structure and function. Genetica 106:37–47
Hideyama T, Yamashita T, Suzuki T, Tsuji S, Higuchi M, Seeburg PH, Takahashi R, Misawa H, Kwak S (2010) Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J Neurosci 30:11917–11925
Hideyama T, Yamashita T, Aizawa H, Tsuji S, Kakita A, Takahashi H, Kwak S (2012) Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol Dis 45:1121–1128. doi:10.1016/j.nbd.2011.12.033
Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH (1993) RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell 75:1361–1370
Higuchi M, Maas S, Single F, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg P (2000) Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406:78–81
Hughes ME, Grant GR, Paquin C, Qian J, Nitabach MN (2012) Deep sequencing the circadian and diurnal transcriptome of Drosophila brain. Genome Res 22:1266–1281
Iwamoto K, Kato T (2003) RNA editing of serotonin 2C receptor in human postmortem brains of major mental disorders. Neurosci Lett 346:169–172
Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, Rusert JM, Dao KH, Morris SR, Goldstein LS, Marra MA, Frazer KA, Jamieson CH (2013) ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci USA 110:1041–1046. doi:10.1073/pnas.1213021110
Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K (1994) Molecular cloning of cDNAs for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc Natl Acad Sci USA 91:11457–11461
King AE, Woodhouse A, Kirkcaldie MT, Vickers JC (2016) Excitotoxicity in ALS: overstimulation, or overreaction? Exp Neurol 275(Pt 1):162–171. doi:10.1016/j.expneurol.2015.09.019
Kubota-Sakashita M, Iwamoto K, Bundo M, Kato T (2014) A role of ADAR2 and RNA editing of glutamate receptors in mood disorders and schizophrenia. Mol Brain 7:5. doi:10.1186/1756-6606-7-5
Lehmann KA, Bass BL (2000) Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry 39:12875–12884
Li L, Qian G, Zuo Y, Yuan Y, Cheng Q, Guo T, Liu J, Liu C, Zhang L, Zheng H (2016) Ubiquitin-dependent turnover of adenosine deaminase acting on RNA 1 (ADAR1) is required for efficient antiviral activity of type I interferon. J Biol Chem 291:24974–24985
Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR (2015) RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349(6252):1115–1120. doi:10.1126/science.aac7049
Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CG, Falconer A, Forte G, Jenkinson EM, Kasher PR, Szynkiewicz M, Rice GI, Crow YJ (2014) A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet 51:76–82. doi:10.1136/jmedgenet-2013-102038
Lomeli H, Mosbacher J, Melcher T, Höger T, Geiger JR, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH (1994) Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266:1709–1713
Lyddon R, Dwork AJ, Keddache M, Siever LJ, Dracheva S (2013) Serotonin 2c receptor RNA editing in major depression and suicide. World J Biol Psychiatry 14:590–601. doi:10.3109/15622975.2011.630406
Maas S, Patt S, Schrey M, Rich A (2001) Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci USA 98:14687–14692
Macbeth MR, Schubert HL, VanDemark AF, Lingam AT, Hill CP, Bass BL (2005) Structural biology: inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 309:1534–1539. doi:10.1126/science.1113150
Mahajan SS, Thai KH, Chen K, Ziff E (2011) Exposure of neurons to excitotoxic levels of glutamate induces cleavage of the rna editing enzyme, adenosine deaminase acting on RNA 2, and loss of glur2 editing. Neuroscience. doi:10.1016/j.neuroscience.2011.05.027
Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, Jantsch MF, Dorin J, Adams IR, Scadden AD, Ohman M, Keegan LP, O’Connell MA (2014) The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9:1482–1494. doi:10.1016/j.celrep.2014.10.041
Marcucci R, Brindle J, Paro S, Casadio A, Hempel S, Morrice N, Bisso A, Keegan LP, Del Sal G, O’Connell MA (2011) Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J 30:4211–4222. doi:10.1038/emboj.2011.303
Masliah G, Barraud P, Allain FH (2013) RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci 70:1875–1895. doi:10.1007/s00018-012-1119-x
Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ, Beal PA (2016) Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol 23:426–433. doi:10.1038/nsmb.3203
Medzhitov R (2010) Inflammation 2010: new adventures of an old flame. Cell 140:771–776
Mittaz L, Antonarakis SE, Higuchi M, Scott HS (1997) Localization of a novel human RNA-editing deaminase (hRED2 or ADARB2) to chromosome 10p15. Hum Genet 100:398–400
Nemlich Y, Greenberg E, Ortenberg R, Besser MJ, Barshack I, Jacob-Hirsch J, Jacoby E, Eyal E, Rivkin L, Prieto VG, Chakravarti N, Duncan LM, Kallenberg DM, Galun E, Bennett DC, Amariglio N, Bar-Eli M, Schachter J, Rechavi G, Markel G (2013) MicroRNA-mediated loss of ADAR1 in metastatic melanoma promotes tumor growth. J Clin Investig 123:2703–2718. doi:10.1172/JCI62980
Ng SK, Weissbach R, Ronson GE, Scadden AD (2013) Proteins that contain a functional Z-DNA-binding domain localize to cytoplasmic stress granules. Nucleic Acids Res 41:9786–9799. doi:10.1093/nar/gkt750
Niswender CM, Copeland SC, Herrick-Davis K, Emeson RB, Sanders-Bush E (1999) RNA editing of the human serotonin 5-hydroxytryptamine 2C receptor silences constitutive activity. J Biol Chem 274:9472–9478
Niswender CM, Herrick-Davis K, Dilley GE, Meltzer HY, Overholser JC, Stockmeier CA, Emeson RB, Sanders-Bush E (2001) RNA editing of the human serotonin 5-HT2C receptor. Alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology 24:478–491
Oakes E, Anderson A, Cohen-Gadol A, Hundley HA (2017) Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J Biol Chem 292:4326–4335
O’Connell MA, Krause S, Higuchi M, Hsuan JJ, Totty NF, Jenny A, Keller W (1995) Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase. Mol Cell Biol 15:1389–1397
Ohman M, Kallman AM, Bass BL (2000) In vitro analysis of the binding of ADAR2 to the pre-mRNA encoding the GluR-B R/G site. RNA 6:687–697
Okin D, Medzhitov R (2012) Evolution of inflammatory diseases. Curr Biol 22:R733–R740
Orlandi C, La Via L, Bonini D, Mora C, Russo I, Barbon A, Barlati S (2011) AMPA receptor regulation at the mRNA and protein level in rat primary cortical cultures. PLoS One 6:e25350
Park E, Guo J, Shen S, Demirdjian L, Wu YN, Lin L, Xing Y (2017) Population and allelic variation of A-to-I RNA editing in human transcriptomes. Genome Biol 18:143
Patterson JB, Samuel CE (1995) Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol 15:5376–5388
Paupard MC, O’Connell MA, Gerber AP, Zukin RS (2000) Patterns of developmental expression of the RNA editing enzyme rADAR2. Neuroscience 95:869–879
Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E, Levanon EY (2015) Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep 13:267–276. doi:10.1016/j.celrep.2015.08.080
Peng Z, Cheng Y, Tan BC, Kang L, Tian Z, Zhu Y, Zhang W, Liang Y, Hu X, Tan X, Guo J, Dong Z, Liang Y, Bao L, Wang J (2012) Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol 30:253–260. doi:10.1038/nbt.2122
Penn AC, Balik A, Greger IH (2013) Reciprocal regulation of A-to-I RNA editing and the vertebrate nervous system. Front Neurosci 7:61. doi:10.3389/fnins.2013.00061
Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB (2015) Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43:933–944. doi:10.1016/j.immuni.2015.11.001
Picardi E, Manzari C, Mastropasqua F, Aiello I, D’Erchia AM, Pesole G (2015) Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep 5:14941. doi:10.1038/srep14941
Polson AG, Bass BL (1994) Preferential selection of adenosines for modification by double-stranded RNA adenosine deaminase. EMBO J 13:5701–5711
Poulsen H, Nilsson J, Damgaard CK, Egebjerg J, Kjems J (2001) CRM1 mediates the export of ADAR1 through a nuclear export signal within the Z-DNA binding domain. Mol Cell Biol 21:7862–7871
Qin YR, Qiao JJ, Chan THM, Zhu YH, Li FF, Liu H, Fei J, Li Y, Guan XY, Chen L (2014) Adenosine-to-inosine RNA editing mediated by adars in esophageal squamous cell carcinoma. Can Res 74:840–851. doi:10.1158/0008-5472.CAN-13-2545
Qiu S, Li W, Xiong H, Liu D, Bai Y, Wu K, Zhang X, Yang H, Ma K, Hou Y, Li B (2016) Single-cell RNA sequencing reveals dynamic changes in A-to-I RNA editome during early human embryogenesis. BMC Genom 17:766. doi:10.1186/s12864-016-3115-2
Ramos A, Grunert S, Adams J, Micklem DR, Proctor MR, Freund S, Bycroft M, St Johnston D, Varani G (2000) RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J 19:997–1009
Ramos PS, Shedlock AM, Langefeld CD (2015) Genetics of autoimmune diseases: insights from population genetics. J Hum Genet 60:657
Rebagliati MR, Melton DA (1987) Antisense RNA injections in fertilized frog eggs reveal an RNA duplex unwinding activity. Cell 48:599–605
Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W Jr, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, Crow YJ (2012) Mutations in ADAR1 cause Aicardi–Goutieres syndrome associated with a type I interferon signature. Nat Genet 44:1243–1248. doi:10.1038/ng.2414
Robinson J, Paluch J, Dickman D, Joiner W (2016) ADAR-mediated RNA editing suppresses sleep by acting as a brake on glutamatergic synaptic plasticity. Nat Commun 7:10512. doi:10.1038/ncomms10512
Rossetti C, Picardi E, Ye M, Camilli G, D’Erchia AM, Cucina L, Locatelli F, Fianchi L, Teofili L, Pesole G, Gallo A, Sorrentino R (2017) RNA editing signature during myeloid leukemia cell differentiation. Leukemia. doi:10.1038/leu.2017.134
Ryter JM, Schultz SC (1998) Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J 17:7505–7513
Sansam CL, Wells KS, Emeson RB (2003) Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci USA 100(24):14018–14023. doi:10.1073/pnas.2336131100
Schade M, Turner CJ, Kuhne R, Schmieder P, Lowenhaupt K, Herbert A, Rich A, Oschkinat H (1999) The solution structure of the Zalpha domain of the human RNA editing enzyme ADAR1 reveals a prepositioned binding surface for Z-DNA. Proc Natl Acad Sci USA 96:12465–12470
Schoft VK, Schopoff S, Jantsch MF (2007) Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res 35:3723–3732
Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM (2011) A diverse array of gene products are effectors of the type I interferon antiviral response. Nature 472:481
Schwartz T, Rould MA, Lowenhaupt K, Herbert A, Rich A (1999) Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science 284:1841–1845
Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A, Delaney A, Gelmon K, Guliany R, Senz J, Steidl C, Holt RA, Jones S, Sun M, Leung G, Moore R, Severson T, Taylor GA, Teschendorff AE, Tse K, Turashvili G, Varhol R, Warren RL, Watson P, Zhao Y, Caldas C, Huntsman D, Hirst M, Marra MA, Aparicio S (2009) Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461:809–813. doi:10.1038/nature08489
Shoshan E, Mobley AK, Braeuer RR, Kamiya T, Huang L, Vasquez ME, Salameh A, Lee HJ, Kim SJ, Ivan C, Velazquez-Torres G, Nip KM, Zhu K, Brooks D, Jones SJ, Birol I, Mosqueda M, Wen YY, Eterovic AK, Sood AK, Hwu P, Gershenwald JE, Robertson AG, Calin GA, Markel G, Fidler IJ, Bar-Eli M (2015) Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat Cell Biol 17:311–321. doi:10.1038/ncb3110
Singh M, Kesterson RA, Jacobs MM, Joers JM, Gore JC, Emeson RB (2007) Hyperphagia-mediated obesity in transgenic mice misexpressing the RNA-editing enzyme ADAR2. J Biol Chem 282:22448–22459
Slavov D, Crnogorac-Jurcevic T, Clark M, Gardiner K (2000) Comparative analysis of the DRADA A-to-I RNA editing gene from mammals, pufferfish and zebrafish. Gene 250:53–60
Solomon O, Eyal E, Amariglio N, Unger R, Rechavi G (2016) e23D: database and visualization of A-to-I RNA editing sites mapped to 3D protein structures. Bioinformatics 32:2213–2215
Sommer B, Köhler M, Sprengel R, Seeburg PH (1991) RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67:11–19
St Johnston D, Brown NH, Gall JG, Jantsch M (1992) A conserved double-stranded RNA-binding domain. Proc Natl Acad Sci USA 89:10979–10983
Stefl R, Oberstrass FC, Hood JL, Jourdan M, Zimmermann M, Skrisovska L, Maris C, Peng L, Hofr C, Emeson RB, Allain FH (2010) The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell 143:225–237
Steinman RA, Yang Q, Gasparetto M, Robinson LJ, Liu X, Lenzner DE, Hou J, Smith C, Wang Q (2013) Deletion of the RNA-editing enzyme ADAR1 causes regression of established chronic myelogenous leukemia in mice. Int J Cancer 132:1741–1750. doi:10.1002/ijc.27851
Strehblow A, Hallegger M, Jantsch MF (2002) Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol Biol Cell 13:3822–3835
Terajima H, Yoshitane H, Ozaki H, Suzuki Y, Shimba S, Kuroda S, Iwasaki W, Fukada Y (2017) ADARB1 catalyzes circadian A-to-I editing and regulates RNA rhythm. Nat Genet 49:146–151
Tomaselli S, Galeano F, Alon S, Raho S, Galardi S, Polito VA, Presutti C, Vincenti S, Eisenberg E, Locatelli F, Gallo A (2015) Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol 16:5. doi:10.1186/s13059-014-0575-z
Veno MT, Bramsen JB, Bendixen C, Panitz F, Holm IE, Ohman M, Kjems J (2012) Spatio-temporal regulation of ADAR editing during development in porcine neural tissues. RNA Biol 9:1054–1065. doi:10.4161/rna.21082
Wahlstedt H, Daniel C, Ensterö M, Öhman M (2009) Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res 19:978–986
Walkley CR, Liddicoat B, Hartner JC (2012) Role of ADARs in mouse development. Curr Top Microbiol Immunol 353:197–220. doi:10.1007/82_2011_150
Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K (2004) Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 279:4952–4961. doi:10.1074/jbc.M310162200
Wang IX, So E, Devlin JL, Zhao Y, Wu M, Cheung VG (2013) ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep 5:849–860. doi:10.1016/j.celrep.2013.10.002
Yang JH, Luo X, Nie Y, Su Y, Zhao Q, Kabir K, Zhang D, Rabinovici R (2003a) Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology 109:15–23
Yang JH, Nie Y, Zhao Q, Su Y, Pypaert M, Su H, Rabinovici R (2003b) Intracellular localization of differentially regulated RNA-specific adenosine deaminase isoforms in inflammation. J Biol Chem 278:45833–45842
Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu YJ (2011) DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34:866–878. doi:10.1016/j.immuni.2011.03.027
Zheng Y, Lorenzo C, Beal PA (2017) DNA editing in DNA/RNA hybrids by adenosine deaminases that act on RNA. Nucleic Acids Res 45:3369–3377. doi:10.1093/nar/gkx050
Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun HJ, Pineda G, Morris SR, Mason CN, Geron I, Barrett C, Goff DJ, Wall R, Pellecchia M, Minden M, Frazer KA, Marra MA, Crews LA, Jiang Q, Jamieson CH (2016) ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell 19:177–191. doi:10.1016/j.stem.2016.05.004
Acknowledgements
M.A. O’C has received funding from the European Union’s Seventh Framework Programme for research, technological development and demonstration under Grant agreement no 621368. A.G has received funding from the AIRC IG Grant no. 17615. We would like to thank two reviewers for detailed comments and additional references.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gallo, A., Vukic, D., Michalík, D. et al. ADAR RNA editing in human disease; more to it than meets the I. Hum Genet 136, 1265–1278 (2017). https://doi.org/10.1007/s00439-017-1837-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-017-1837-0