
Abstract
Ectodermal dysplasia is a highly heterogeneous group of disorders that variably affect the derivatives of the ectoderm, primarily skin, hair, nails and teeth. TP63, itself mutated in ectodermal dysplasia, links many other ectodermal dysplasia disease genes through a regulatory network that maintains the balance between proliferation and differentiation of the epidermis and other ectodermal derivatives. The ectodermal knockout phenotype of five mouse genes that regulate and/or are regulated by TP63 (Irf6, Ikkα, Ripk4, Stratifin, and Kdf1) is strikingly similar and involves abnormal balance towards proliferation at the expense of differentiation, but only the first three have corresponding ectodermal phenotypes in humans. We describe a multigenerational Saudi family with an autosomal dominant form of hypohidrotic ectodermal dysplasia in which positional mapping and exome sequencing identified a novel variant in KDF1 that fully segregates with the phenotype. The recapitulation of the phenotype we observe in this family by the Kdf1−/− mouse suggests a causal role played by the KDF1 variant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Skin is the largest organ in the body and serves a multitude of physiological roles including barrier function and thermal regulation. Epidermis, the skin’s outer layer, is derived from the embryonic ectoderm through a series of steps that involve commitment, proliferation and differentiation (Blanpain and Fuchs 2009; Fuchs 1990; Niemann and Watt 2002). After achieving complete maturation, the epidermis is continuously regenerated throughout life to replenish skin cells that are sloughed at a rate of 2 million per hour (Milstone 2004). The mechanisms that underpin the development and maintenance of the epidermis are the subject of intense investigation, the ultimate goal of which is to better understand the disease mechanisms and to promote skin health.
Ectodermal dysplasia is a collective term that encompasses developmental disorders of the skin and its appendages as well as other ectoderm-derived tissues e.g., teeth, hair, nails and craniofacial structures. Although ectodermal dysplasia is broadly classified as hidrotic (when sweat glands function properly) and hypohidrotic, the phenotype is usually highly variable even within classes (Wright et al. 2014). On the other hand, recent molecular classifications provide a more meaningful categorization that allows for comparison based on molecular pathoetiology (Pagnan and Visinoni 2014).
Many genes are known to be mutated in ectodermal dysplasia ranging from the more common e.g., EDA to the very rare e.g., COG6 (Döffinger et al. 2001; Shaheen et al. 2013). Despite the marked progress made in recent years in deciphering the genetics of ectodermal dysplasia, most patients remain without a molecular diagnosis. TP63 is a particularly interesting ectodermal dysplasia gene, because it encodes a master regulator that serves as a molecular hub for virtually all known regulatory networks that control skin development and maintenance (Koster et al. 2004; Koster and Roop 2004; Truong et al. 2006). Not surprisingly, many of the genes that cause ectodermal dysplasia and other disorders of the integument encode proteins that regulate and/or are regulated by p63 (Koster 2010; Mitchell et al. 2012). Therefore, genes that are necessary for p63 regulation are attractive candidates for human skin diseases, especially when compelling animal models are available. In this study, we describe unbiased positional mapping in a large human family with a novel, autosomal dominant form of ectodermal dysplasia that led us to identify a novel, likely pathogenic variant in KDF1, encoding a recently described keratinocyte differentiation factor that acts upstream of p63 (Lee et al. 2013). The recapitulation of the human phenotype by the two available knockout mouse models supports a potentially causal link between the variant we identified and this novel form of ectodermal dysplasia.
Materials and methods
Human subjects
All patients were evaluated clinically by board-certified clinical geneticists and dermatologists. Clinical evaluation involved history taking, including family history, physical examination, electronic microscopic examination of hair shafts, histopathological examination of the skin and clinical exome sequencing. When clinical exome sequencing failed to reveal a likely causal mutation (for details about potential causes, please refer to (Shamseldin et al. 2016)), the entire family was recruited under an IRB-approved research protocol (KFSHRC RAC# 2121053) with informed consent. Blood was obtained from all affected and unaffected family members for subsequent genetic studies. Clinical photographs were obtained with a separate consent.
Positional mapping, exome sequencing and variant filtering
Axiom SNP chip genotyping was performed on all affected and unaffected members of the pedigree. AutoSNPa was used to exclude the remote possibility of pseudodominant inheritance, and DominantMapper was implemented to identify haplotypes that are exclusively shared by the affected members assuming a fully penetrant model (Carr et al. 2011). Exome capture was performed using TruSeq Exome Enrichment kit (Illumina) following the manufacturer’s protocol. Samples were prepared as an Illumina sequencing library, and in the second step, the sequencing libraries were enriched for the desired target using the Illumina Exome Enrichment protocol. The captured libraries were sequenced using Illumina HiSeq 2000 Sequencer. The reads were mapped against UCSC hg19 (http://genome.ucsc.edu/) by BWA (http://bio-bwa.sourceforge.net/). The SNPs and Indels were detected by SAMtools (http://samtools.sourceforge.net/). Variants from WES were filtered such that only novel (or very low frequency <0.1%), coding/splicing, homozygous variants that are within the candidate haplotypes of the affected individual and are predicted to be pathogenic were considered as likely causal variants. Frequency of variants was determined using publicly available variant databases (1000 Genomes, Exome Variant Server and ExAC) as well as a database of 817 in-house ethnically-matched exomes. Pathogenicity was likely if the mutation is loss-of-function (splicing/truncating) or, in the case of missense/in-frame indels, removes a highly conserved amino acid and is predicted to be pathogenic by the three in silico prediction modules PolyPhen, SIFT and CADD.
Computational structural analysis
Sequences were retrieved from the UniProt database. BLAST and SwissModel (Arnold et al. 2006) were used to search for suitable structural templates in the protein data bank (PDB). SwissModel and RaptorX (Kallberg et al. 2014) were used to produce homology models. Models were manually inspected, and mutations evaluated, using the PyMOL program (pymol.org). Disorder and secondary structure elements were predicted using RaptorX. Quark (Xu and Zhang 2012) was used for ab inito structure predictions. The presence of transmembrane helices and signal peptides was assessed using Phobius (Kall et al. 2004). Functional information was compiled from various resources, including UniProt and literature searches.
Results
Clinical report
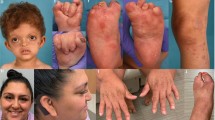
A 30-year-old female, product of a non-consanguineous marriage was referred for evaluation because of suspected ectodermal dysplasia. She complained of thin eyebrows, absent teeth (status post dental implantation at age 23 years), nail changes, recurrent abscesses in the axillae and groin and decreased sweating. She was healthy otherwise. Her family history was significant for an affected father and multiple affected siblings who shared the same phenotype except for a sister who additionally had cleft palate status post surgical repair (Fig. 1; Table 1). Furthermore, she had one affected son who, in addition to his mother’s features, had natal teeth status post extraction at 2 weeks of age. Physical examination revealed lusterless scalp hair without scarring alopecia, thinned eyebrows and lack of natural teeth. She had keratosis pilaris, marked accentuation of palmar creases, and multiple scars of old healed lesions of hidradenitis suppurativa. Fingernails were normal, while toenails showed pincer nail deformity in the big toes and mild subungual hyperkeratosis in the others (Fig. 1). Complete blood count, liver profile and bone profile were normal. Lumbar spine coccyx X-ray showed sclerotic changes in the right femoral neck, and vertebral endplates with multiple stress fractures of long bones. Bone densitometry was normal. Her abdominal and renal ultrasounds were unremarkable. Table 1 summarizes the clinical features of all affected members. A skin biopsy from her 16-year-old affected brother showed hypoplastic sweat glands with epidermal orthokeratosis and papillomatosis, and some hair follicles showed follicular plug with distorted infundibulum. These findings were consistent with hypohidrotic ectodermal dysplasia and keratosis pilaris, respectively. Clinical exome sequencing did not reveal a likely causal variant in any known ectodermal dysplasia gene and did not suggest any likely candidate gene.

a Pedigree of the study family. b Facial dysmorphism (II:8) in the form of thinning of lateral eyebrows, saddle nose, and short philtrum. c Keratosis pilaris and scars of hidradenitis suppurativa (II:8). d Prominent hair follicles with resolving hidradenitis suppurativa lesion (II:7). e Natal teeth (III:2). f Absence of teeth (I:1). g Dystrophic toenails (II:8)
Identification of a novel variant in KDF1
Lack of consanguinity, vertical transmission and involvement of both genders in the study family were highly consistent with an autosomal dominant inheritance. This was further supported by autozygosity mapping that was not consistent with pseudodominant inheritance. Assuming a fully penetrant model, we searched for all haplotypes that are exclusively shared by all seven affected members (Fig. 2). Within these candidate haplotypes, exome sequencing revealed 682 heterozygous coding/splicing variants including 77 that are absent in dbSNP (Fig. 2). Of these, there were four variants that are absent in 817 Saudi exomes and in ExAC (Table S1). Fortunately, there are available knockout mouse for three of these genes. The mouse phenotype for ASTN1 and ARF4 knockout did not involve any ectodermal findings, making the variants identified in these two genes unlikely candidates. No mouse models are available for IFIT5; so, the variant we identified therein (NM_012420:exon2:c.823G>T:p.A275S) remained a positional candidate with no further biological support from the literature. On the other hand, the phenotype of Kdf1−/− was highly reminiscent of the phenotype we report in the study family (see below). The variant therein (NM_152365:exon2:c.753C>A:p.F251L) was predicted to be pathogenic by PolyPhen (0.99), SIFT (0.01) and CADD (26). Sanger sequencing confirmed that all affected members were heterozygous for the change but none of the unaffected members. As expected from the relatively small family size, LOD score under the fully penetrant dominant model for this variant (1.8) did not reach significance.

a Chromosome ideograms with the candidate haplotypes as indicated by DominantMapper denoted by red lines. b Filtering scheme of the exome variants. c Cartoon of KDF1 and sequence chromatogram of the candidate variant. d Multisequence alignment showing strong conservation of the affected amino acid residue
We used structural bioinformatics to investigate the mechanistic basis for the F251L mutation. There is no homologous experimental structure available that would serve as a good-confidence support for modeling the 3D structure of KDF1, or larger fragments thereof. Prediction of secondary structure and disorder shows that KDF1 is a largely flexible protein with only a few isolated secondary structures, mostly helices (Figure S1). No transmembrane helices are predicted. This indicates that KDF1 functions as a protein-binding adaptor, scaffold and/or cofactor, rather than an enzyme or transmembrane transporter or receptor. F251 is located in a region that is strongly predicted to form a 25-residue-long helical segment. Ab initio 3D structure prediction suggested that this helical segment is located in a 90-residue-long domain that may adopt a small folded region (Figure S1). Within this region, F251 is predicted to be buried (solvent inaccessible), and be part of a hydrophobic patch on an amphipathic helical region. S218, located at the N-terminal end of the predicted helical fragment, has been reported to be phosphorylated. Therefore, S218 interacts with kinases, and might modulate binding functions (to membrane or ligands) of the helical fragment containing F251. F251 might, therefore, be involved in stabilising a hydrophobic core of a small domain and/or be part of a helical ligand-binding motif. In either case, its substitution by the much smaller leucine might perturb its (intra- or intermolecular) interactions, and, hence, compromise the function of KDF1.
Discussion
Skin development is highly conserved between humans and mouse, which makes the latter an attractive model for studying the pathoetiology of skin developmental disorders using forward and reverse genetics approaches. Recently, Lee and colleagues reported the identification of a mouse mutant they called shorthand (shd) in an ENU mutagenesis screen (Lee et al. 2013). Grossly, shd had taught thick skin. However, that skin had severely impaired barrier function, and histological examination revealed marked disorganization of the layers of the epidermis. Specifically, the outermost layer of the epidermis (cornified layer) is lacking and the rest of the suprabasal layers have mixed identities. The basal layer itself was intact but with markedly increased mitotic index compared to controls. The phenotype is highly reminiscent of three other mutants: Irf6, Ikka and Stratifin, and to a lesser extent Ripk4. They mapped shd to a splicing mutation in a poorly characterized gene, which they termed Kdf1 for keratinocyte differentiation factor 1. Reassuringly, a reverse genetics approach whereby Kdf1 was knocked out using a genetrap vector recapitulated the phenotype of shd. In view of the resemblance between shd and the knockout mice for Stratifin, genetic interaction between Kdf1 and Staratifin was tested, and double heterozygotes were found to, indeed, recapitulate many phenotypic aspects of single homozygotes. Of note, Kdf1 knockout mice showed enhanced expression of p63, and deficiency of p63 was found to rescue shd, which is otherwise uniformly lethal due to esophageal adhesions and cleft palate (Lee et al. 2013).
The study by Lee and colleagues clearly show a non-redundant role of KDF1 in skin development that is mediated at least in part by its regulatory influence on p63. KDF1 has no clear homologues despite its very strong conservation in mammals and, to a lesser extent, in other animals. Therefore, the genetic interaction with the much more extensively studied Stratifin offers an opportunity to place KDF1 in a molecular developmental context. Stratifin (also known as 14-3-3σ) is a tumor suppressor that was originally identified as an exclusive epithelial marker (Prasad et al. 1992). Stratifin is a target of p63 but it also exerts a feedback effect and targets p63 to proteasome degradation (Fomenkov et al. 2004; Westfall et al. 2003). Mice deficient in stratifin display a remarkably similar histological phenotype to shd, i.e., thickened skin with enhanced proliferation and impaired proliferation that is similarly rescued by p63 deficiency (Li et al. 2011).
Although milder, the phenotype we describe in this study bears remarkable resemblance to that of shd, which suggests that the candidate variant we identified in KDF1 may be causally linked to the phenotype. Unfortunately, Kdf1−/− die shortly after birth; so, it is not possible to comment on the dental and hair phenotype that are prominent in the human patients we describe here. Also, Lee and colleagues did not comment on the morphology of sweat glands in their histological examination of the skin although they did demonstrate strong expression of Kdf1 in developing hair follicles (Lee et al. 2013). However, the highly similar phenotype between kdf1−/− and stratifin−/−, and the marked hair loss (sweat gland morphology was not reported) in stratifin−/+ prompting the name repeated epilation (stratifin mutant is semidominant) (Guenet et al. 1979; Herron et al. 2005; Holbrook et al. 1982; Li et al. 2005) suggest that KDF1 deficiency is similarly likely to cause hair loss. The dominant nature of the variant we identified in humans can be reconciled with the apparently haplosufficient kdf1−/+ mice by invoking interspecies differences.
In conclusion, we describe a family with a novel, autosomal dominant form of ectodermal dysplasia that we suggest is the mild human equivalent of the shd mutant phenotype in mouse based on the finding of a novel KDF1 variant by positional mapping and exome sequencing. Future patients with KDF1 mutations, including those with copy number losses involving KDF1, will help delineate the phenotype of KDF1 deficiency in humans and its mutational mechanism.
References
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201
Blanpain C, Fuchs E (2009) Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol 10:207–217
Carr IM, Johnson CA, Markham AF, Toomes C, Bonthron DT, Sheridan EG (2011) DominantMapper: rule-based analysis of SNP data for rapid mapping of dominant diseases in related nuclear families. Hum Mutat 32:1359–1366
Döffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis-Girod S, Blanche S (2001) X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB signaling. Nat Genet 27:277–285
Fomenkov A, Zangen R, Huang Y-P, Osada M, Guo Z, Fomenkov T, Trink B, Sidransky D, Ratovitski EA (2004) RACK1 and stratifin target ΔNp63α for a proteasome degradation in head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 3:1285–1295
Fuchs E (1990) Epidermal differentiation: the bare essentials. The Journal of Cell Biology 111:2807–2814
Guenet J, Salzgeber B, Tassin M (1979) Repeated epilation: a genetic epidermal syndrome in mice. J Hered 70:90–94
Herron BJ, Liddell RA, Parker A, Grant S, Kinne J, Fisher JK, Siracusa LD (2005) A mutation in stratifin is responsible for the repeated epilation (Er) phenotype in mice. Nat Genet 37:1210–1212
Holbrook KA, Dale BA, Brown KS (1982) Abnormal epidermal keratinization in the repeated epilation mutant mouse. J Cell Biol 92:387–397
Kall L, Krogh A, Sonnhammer EL (2004) A combined transmembrane topology and signal peptide prediction method. J Mol Biol 338:1027–1036. doi:10.1016/j.jmb.2004.03.016
Kallberg M, Margaryan G, Wang S, Ma J, Xu J (2014) RaptorX server: a resource for template-based protein structure modeling. Methods Mol Biol 1137:17–27. doi:10.1007/978-1-4939-0366-5_2
Koster MI (2010) p63 in skin development and ectodermal dysplasias. J Investig Dermatol 130:2352–2358
Koster MI, Roop DR (2004) The role of p63 in development and differentiation of the epidermis: tanioku kihei memorial lecture. J Dermatol Sci 34:3–9
Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR (2004) p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev 18:126–131
Lee S, Kong Y, Weatherbee SD (2013) Forward genetics identifies Kdf1/1810019J16Rik as an essential regulator of the proliferation–differentiation decision in epidermal progenitor cells. Dev Biol 383:201–213
Li Q, Lu Q, Estepa G, Verma IM (2005) Identification of 14-3-3σ mutation causing cutaneous abnormality in repeated-epilation mutant mouse. Proc Natl Acad Sci 102:15977–15982
Li Q, Sambandam SA, Lu HJ, Thomson A, Kim S-h LuH, Xin Y, Lu Q (2011) 14-3-3σ and p63 play opposing roles in epidermal tumorigenesis. Carcinogenesis 32:1782–1788
Milstone LM (2004) Epidermal desquamation. J Dermatol Sci 36:131–140
Mitchell K, O’Sullivan J, Missero C, Blair E, Richardson R, Anderson B, Antonini D, Murray JC, Shanske AL, Schutte BC (2012) Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am J Hum Genet 90:69–75
Niemann C, Watt FM (2002) Designer skin: lineage commitment in postnatal epidermis. Trends Cell Biol 12:185–192
Pagnan NAB, Visinoni ÁF (2014) Update on ectodermal dysplasias clinical classification. Am J Med Genet Part A 164:2415–2423
Prasad GL, Valverius EM, McDuffie E, Cooper H (1992) Complementary DNA cloning of a novel epithelial cell marker protein, HME1, that may be down-regulated in neoplastic mammary cells. Cell Growth Differ 3:507
Shaheen R, Ansari S, Alshammari MJ, Alkhalidi H, Alrukban H, Eyaid W, Alkuraya FS (2013) A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency. J Med Genet 50:431–436
Shamseldin HE, Maddirevula S, Faqeih E, Ibrahim N, Hashem M, Shaheen R, Alkuraya FS (2016) Increasing the sensitivity of clinical exome sequencing through improved filtration strategy. Genet Med. doi:10.1038/gim.2016.155
Truong AB, Kretz M, Ridky TW, Kimmel R, Khavari PA (2006) p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev 20:3185–3197
Westfall MD, Mays DJ, Sniezek JC, Pietenpol JA (2003) The ΔNp63α phosphoprotein binds the p21 and 14-3-3σ promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol Cell Biol 23:2264–2276
Wright JT, Grange DK, Richter MK (2014) Hypohidrotic ectodermal dysplasia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K (eds) GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2016
Xu D, Zhang Y (2012) Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80:1715–1735. doi:10.1002/prot.24065
Acknowledgements
We thank the study family for their enthusiastic participation. We also thank the Sequencing and Genotyping Core Facilities at KFSHRC for their technical help. This work was supported by KACST Grant 13-BIO1113-20 (FSA) and King Abdullah University of Science and Technology (KAUST) (STA).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding authors state that there is no conflict of interest.
Additional information
H. E. Shamseldin and O. Khalifa contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
439_2016_1741_MOESM1_ESM.pdf
Supplementary material 1 (PDF 429 kb) Figure S1. (A) Prediction of secondary structure (top) and disorder (bottom) for KDF1. F251 is identified by an asterisk. The grey underlined fragment was subjected to ab initio 3D structure prediction, resulting in the model shown in (B), where F521 is highlighted in green, and S218 is shown in yellow. The Quark TM score for the 3D model was 0.56 ± 0.08, and TM score of top 10 best models was 0.63 ± 0.07, indicating that the modeling procedure converged on highly similar models, and hence that the overall fold of the resulting model is close to reality
Rights and permissions
About this article

Cite this article
Shamseldin, H.E., Khalifa, O., Binamer, Y.M. et al. KDF1, encoding keratinocyte differentiation factor 1, is mutated in a multigenerational family with ectodermal dysplasia. Hum Genet 136, 99–105 (2017). https://doi.org/10.1007/s00439-016-1741-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1741-z