
Abstract
Christianson syndrome (OMIM 300243), caused by mutations in the X-linked SLC9A6 gene, is characterized by severe global developmental delay and intellectual disability, developmental regression, epilepsy, microcephaly and impaired ocular movements. It shares many common features with Angelman syndrome. Carrier females have been described as having learning difficulties with mild to moderate intellectual disability, behavioural issues and psychiatric illnesses. There is little literature on the carrier female phenotype of Christianson syndrome. We describe a large extended family with three affected males, four carrier females, one presumed carrier female and one obligate carrier female with a c.190G>T, p.E64X mutation known to cause a premature stop codon in SLC9A6. We characterize and expand the clinical phenotype of female SLC9A6 mutation carriers by comparing our described family with female carriers previously discussed in the literature. In particular, we highlight the neurodevelopmental and psychiatric phenotypes observed in our family and previous reports.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Christianson et al. (1999) first described a five-generation, Caucasian South African family with multiple affected males presenting with profound mental retardation, epilepsy, mild craniofacial dysmorphism, bilateral ophthalmoplegia, truncal ataxia and limited life expectancy. This syndrome had many characteristic features overlapping with Angelman syndrome (OMIM105830), including developmental delay, acquired microcephaly and happy demeanour. The syndrome, now referred to as Christianson syndrome (CS), was found to be caused by mutations in SLC9A6 (Gilfillan et al. 2008). The SLC9A6 gene encodes for the sodium/hydrogen exchanger NHE6. NHE6 is highly expressed in the brain, and is thought to participate in the targeting of intracellular vesicles and recycling of synaptic vesicles (Garbern et al. 2010). The Na+/H+ exchanger gene family has been linked to autism and neurologic disease (Kondapalli et al. 2014).
Descriptions of female carriers of SLC9A6 mutations have been infrequent. Reported phenotypic findings of carrier females include learning problems, mild to moderate intellectual disability, behavioural problems and psychiatric illnesses (Schroer et al. 2010; Pescosolido et al. 2014).
We present a large extended family of Caucasian background with three affected males with CS, four confirmed SLC9A6 mutation carrier females, one obligate carrier female and one presumed carrier female. We characterize the carrier females in our family based on clinical features, developmental history and psychiatric disorders, and compare them to the available literature on other SLC9A6 carrier females.
Materials and methods
All male and female family members reported in this study were clinically assessed by at least one medical geneticist. A detailed chart review was also undertaken for consultation reports from other specialists, including developmental, neurological and psychiatric assessments, where available.
Genomic microarray analysis was performed using the 4 × 180 K Oligonucleotide Array (Agilent Technologies). Testing for Fragile X syndrome was performed by PCR amplification using the Asuragen AmplideX FMR1 PCR kit to detect CGG repeat size in the 5′ UTR of the FMR1 gene, and by PCR amplification and Southern blot analysis with methylation-sensitive enzymes to detect CCG repeat size and methylation status of the FMR2 gene. Testing for Prader–Willi syndrome was performed by multiplex ligation-dependent probe amplification to determine the methylation status at the SNRPN locus and gene dosage of the SNRPN gene. Testing for mutations in the NF1 and SPRED1 genes associated with neurofibromatosis type 1 (NF1) and Legius syndrome, respectively, was performed by Sanger sequencing.
Sequencing of SLC9A6 was performed as part of an X-linked mental retardation next-generation sequencing (NGS) panel (Ambry Genetics, CA, USA). In brief, genomic DNA was isolated from the patient’s specimen using a standardized kit and quantified by agarose gel electrophoresis. Sequencing was carried out using PCR and NGS using primer pairs designed to target the 81 genes on the panel. Additional Sanger sequencing was performed for any regions missing coverage and regions with sequence similarity elsewhere in the genome. Targeted Sanger sequencing was performed in additional family members after an SLC9A6 mutation was detected in the index case.
Results
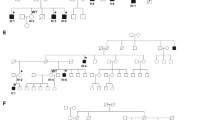
A disease-causing mutation was found in exon 1 of the SLC9A6 gene (c.190G>T, p.E64X). The G to T substitution at nucleotide position 190 causes an amino acid change from glutamic acid to a stop codon within exon 1. The mutation was first detected in individual IV–IV (Fig. 1).

Extended pedigree of family with three affected males with Christianson syndrome, four SLC9A6 mutation carrier females, one assumed carrier (IV–XI) and one obligate carrier (II–VI)
Clinical report
Males affected with CS
Patient IV–IV (Index Case)
IV–IV is a 21-year-old male with global developmental delay, seizure disorder with onset at 1.5 years of age and autism spectrum disorder diagnosed at 15 years of age. Genetics assessment at 18 years of age showed a head circumference of 55 cm (47th percentile, −0.07 SD). He had a history of serious infection with influenza B myositis and encephalitis.
Patient IV–V
IV–V is an 18-year-old male with early childhood microcephaly, hearing loss, global developmental delay and seizure disorder. He was able to achieve some developmental milestones, including walking at 3 years of age. He suffered acquired brain damage at 5 years of age with accidental self-strangulation with subsequent regression in all spheres and is severely delayed. Genetics assessment at 16 years of age showed a head circumference of 51.5 cm (1st percentile, −2.42 SD).
Patient IV–X
IV–X is a 12-year-old male with severe developmental delay and seizures beginning at under 1 year of age. At genetics assessment at 10.5 years of age, he had a head circumference of 52 cm (21st percentile, −0.79 SD). He also has multiple neurocutaneous stigmata, including café-au-lait spots, extensive inguinal and axillary freckling, and neurofibromas. He was molecularly confirmed to be affected with NF1 with heterozygous deletion of exons 10–30 of the NF1 gene.
Female SLC9A6 mutation carriers
Table 1 summarizes the neurodevelopmental, psychiatric and neurologic findings of the female SLC9A6 mutation carriers in this family, along with their genetic testing results.
Patient II–VI
II–VI presented at 60 years of age with an akinetic right arm with apraxia, cortical sensory loss, worsening emotional lability, myoclonus and slow saccades with saccadic dysmetria. Initial neurologic assessment showed impaired fine motor control, spasticity in the upper extremities bilaterally, along with marked saccadic smooth pursuit. Vertical gaze was intact. Two years later, prior to her death, she had marked supranuclear gaze palsy and paralysis of the right side. Neurological opinion favoured a taupathy that fit within the corticobasal degeneration syndrome (CBDS) and progressive supranuclear palsy spectrum, with findings most compatible with CBDS. She had cognitive testing with Mini Mental Status Exam (Folstein et al. 1975) score of 22/30 indicating early cognitive decline, and Behaviour Neurology Assessment-Short Form (Darvesh et al. 2005) score of 48/114 with deficits in all cognitive domains. Initial MRI showed diffuse cortical and cerebellar atrophy. A second MRI with contrast showed a striking asymmetric pattern of atrophy, most prominent in the left frontal and parietal cortex, in keeping with CBDS. She had progression of the disease over time and multiple falls, and passed away at 64 years of age due to aspiration pneumonia. She did not undergo testing for the familial SLC9A6 mutation, but is an obligate carrier based on the test results of her offspring.
Patient III–III
III–III is a 38-year-old female with anxiety and depression treated with citalopram, learning difficulties and a history of self-cutting behaviour. She was able to complete grade 9 at an appropriate age but did not complete grade 12 until she was 38 years of age. At psychiatric assessment at age 38 years, she had moderate psychomotor retardation, was coherent and logical with slow mentation, and was cognitively grossly intact. She was diagnosed with major depressive disorder, anxiety disorder with symptoms of generalized anxiety disorder and panic disorder, along with psychosis not otherwise specified (NOS), possibly related to her underlying genetic condition. At genetics assessment at 38 years of age, she was apparently normocephalic, non-dysmorphic and had no obvious neurologic deficits.
Patient III–VIII
III–VIII is a 34-year-old female with a history of learning difficulties in school, seizure disorder and psychiatric diagnoses. Her seizure disorder began with grand mal seizures in childhood that have evolved to occasional focal seizures around the eyes. These were described to be triggered by smells or spices. Psychiatric assessment at 33 years of age showed her to be cognitively intact with average intelligence, coherent and logical. She was given DSM-IV diagnoses of dysthymic disorder (with query for bipolar disorder type II), anxiety disorder NOS with symptoms of generalized anxiety disorder, panic disorder and post-traumatic stress disorder-like symptoms, some cluster B personality traits, impulsivity, interpersonal difficulties and affect dysregulation. She was found to be moderately impaired with a Global Assessment of Functioning (GAF) (Jones et al. 1995) score of 55. At genetics assessment at 34 years of age, she was apparently normocephalic, non-dysmorphic and had no obvious neurologic deficits. She had normal microarray testing.
Patient III–X
III–X is a 49-year-old female with a history of learning difficulties in school. She required extra help in all subjects and recently completed grade 12 at the age of 38. She had normal early development. She had behavioural issues in early childhood presenting as nocturnal enuresis. She was diagnosed with depression in her 40s, which has been treated with citalopram and quetiapine. Psychiatric assessment at 45 years of age showed intact cognition but poor memory and concentration. She was given DSM-IV diagnoses of chronic major depression (most likely in the phase of dysthymia), panic attacks and agoraphobia. She had a GAF (Jones et al. 1995) score of 52. At genetics assessment at 48 years of age, she was normocephalic with a head circumference of 54 cm (38th percentile, −0.30 SD), non-dysmorphic and had a normal neurologic examination. She had normal microarray and Fragile X syndrome testing.
Patient IV–VI
IV–VI is a 16-year-old female with developmental delays starting at 17 months of age in cognitive and fine motor skills. She had episodes of selective mutism that began at 4 years of age, along with speech difficulties. She has received speech therapy since 4 years of age. Because of difficulties in traditional school arrangements, she has required special classes with smaller numbers of students. She has been diagnosed with attention deficit hyperactivity disorder (ADHD), separation anxiety, oppositional defiant disorder (ODD) manifesting as violent tantrums and physical aggression, and possible schizophrenia. At genetics assessment at 13.5 years of age, she was normocephalic with a head circumference of 52.6 cm (15th percentile, −1.04 SD) and had a normal neurologic examination. She has multiple café-au-lait spots, and inguinal and axillary freckling, giving her a clinical diagnosis of NF1. She had normal ophthalmological assessments and a reportedly normal brain MRI. She had normal microarray and Fragile X syndrome testing, as well as normal testing for the NF1 and SPRED1 genes.
Patient IV–XI
IV–XI is a 9-year-old female with delays in speech development. Babbling began at 15 months and her first comprehensible words appeared at 3 years of age. At age 7 years, she was in grade 1 with extra help required for speech and language. Neuropsychological testing demonstrated good math skills and very poor language skills. Behavioural issues started at 1.5 to 2 years of age with temper tantrums. She has been diagnosed with ADHD and ODD, and a possible mood disorder. She has ongoing sleep difficulties and self-injurious behaviours, including head-banging and self-induced vomiting. At genetics assessment at 7.5 years of age, she had a head circumference of 54.7 cm (>97th percentile, +2.41 SD), with normal neurologic examination. She had normal microarray analysis, methylation studies for Prader–Willi syndrome and Fragile X syndrome testing. The family declined to have IV–XI undergo targeted mutation testing for the familial SLC9A6 mutation.
Discussion
CS was first described in 1999 in a single family with an initial cohort of 16 affected males and 10 carrier females over five generations. The initial ten carrier females (with eight obligate carriers and two carriers confirmed by linkage analysis) were briefly described with a wide clinical spectrum, ranging from mentally and physically normal in six females to two females who were mildly affected with behavioural problems and aggression during childhood (Christianson et al. 1999). The female SLC9A6 mutation carrier phenotype has since been expanded to include learning difficulties in school, speech and language delay, intellectual disability ranging from mild to moderate, and behavioural issues, most notably aggression (Christianson et al. 1999; Schroer et al. 2010; Pescosolido et al. 2014). In general, the female phenotype shows no dysmorphic features, no microcephaly [one described patient with microcephaly (−3 SD) (Pescosolido et al. 2014)] and no seizures.
Table 1 summarizes the neurodevelopmental, psychiatric and neurologic findings of the females reported in our study compared to those previously reported in the literature. In our female cohort, there is an apparent overall decline in academic functioning in subsequent generations, along with more prevalent speech and language difficulties, and increased behavioural issues. In generation III, individuals III, VIII and X all reported learning issues in school, most prevalent in higher grades with difficulty in completing high school; III–III and III–X were able to complete high school-level courses as adults. No females in this third generation had issues with early development, speech or language. Their early behavioural concerns consisted of self-injurious behaviours (e.g. cutting) and nocturnal enuresis.
The female mutation carriers in the fourth generation of the family were all more severely affected after a period of normal early development. Subsequent developmental issues were generally noted to start between ages 2 and 4 years. Both IV–VI and IV–XI required speech therapy at an early age (3 and 5 years, respectively), and their learning difficulties began much earlier. With traditional classroom arrangements being too difficult, they have required special class arrangements since entering grade 1 and individualized education plans. Behavioural issues also began in early childhood, with ODD that manifested as aggression towards family, self-injurious behaviours and constant temper tantrums. The apparent progressively earlier presentation in subsequent generations has not been characterized before in the female phenotype. Modern surveillance with increased testing, vigilance, and awareness of early developmental and behavioural disorders may create an ascertainment bias when characterizing generational differences. Nevertheless, recognizing the potential for variable, and possibly more severe, expression in subsequent generations would be beneficial in assessment of large affected families to allow for earlier interventions.
In the family reported here, there was a higher frequency and variety of psychiatric disorders than previously described in the female SCL9A6 mutation carrier phenotype. The most common psychiatric disorders in female SLC9A6 mutation carriers in this family were mood, behavioural and anxiety disorders, including major depression, ADHD, ODD, generalized anxiety, panic disorder and agoraphobia. Several had symptoms suggestive of psychotic disorders. The prevalence of psychiatric disorders in adults with SLC9A6 mutations has not previously been described extensively in the literature. Riess et al. (2013) described one obligate carrier female who was healthy until 55 years, when she developed neurologic symptoms and depression. An ascertainment bias may be present in our family given the complexity of family dynamics and concomitant medical issues. However, there is a clear trend indicating that psychiatric manifestations are likely an underdiagnosed manifestation of SLC9A6 mutation carrier status that warrants closer attention. Furthermore, a similar generational phenomenon as seen with neurodevelopmental and behavioural disorders was also observed with psychiatric findings, with earlier and more severe psychiatric impairment present in subsequent generations of female mutation carriers. Those in the fourth generation showed much earlier ages of onset and a wider spectrum of psychiatric disorders, including mood and anxiety disorders, and more severe behavioural disorders, including ADHD, ODD and self-injurious behaviours.
Interestingly, Pescosolido et al. (2014) described a female SLC9A6 mutation carrier with a strikingly similar phenotype to patient IV–VI in our study. This individual (no age stated) was described to have moderate intellectual disability, speech and language delay, selective mutism, ODD, reactive attachment disorder and ADHD (Pescosolido et al. 2014). Unfortunately, there was no description of the SLC9A6 mutation for this patient to allow for potential genotype–phenotype correlations. Nevertheless, the similar clinical spectrums of these two patients underscore the emerging complex phenotypic picture of female SLC9A6 mutation carriers.
In our study, there were several confounding medical and genetic factors that may have influenced the ultimate phenotypes seen. One female (III–VIII) has a long-standing seizure disorder, the aetiology of which is unclear. No female mutation carriers have previously been reported with seizure disorders and it remains unclear whether this may be attributable to carrier status. Another female (IV–VI) met clinical criteria for a diagnosis of NF1 (OMIM 162200), with findings of multiple café-au-lait spots, and inguinal and axillary freckling on physical examination; molecular testing did not identify a pathogenic basis for her findings. Neurodevelopmental disorders, including learning difficulties, intellectual disability and autism spectrum disorders, are known to occur frequently in individuals with NF1 (Friedman 1998). Patient IV–XI did not undergo testing for the familial SLC9A6 mutation, though her clinical presentation, family history and other normal genetic test results strongly suggest that she is a carrier of the familial mutation.
Patient II–VI was not tested for the familial SLC9A6 mutation but could be determined to be an obligate carrier based on the results of testing in her offspring. Her distinct neurological findings were highly suggestive of a diagnosis of CBDS, which is one of the atypical parkinsonian disorders that can mimic the clinical presentation of classic Parkinson’s disease (Fogel et al. 2014). CBDS presents with distinct pathological findings of accumulation of 4R (four repeat) tau isoforms (Grijalvo-Perez and Litvan 2014). The classical clinical presentation is of asymmetric parkinsonism, ideomotor apraxia (inability to correctly use hand gestures and mime tool use), rigidity, myoclonus and dystonia (Grijalvo-Perez and Litvan 2014). Patient II–VI presented with an akinetic right arm with apraxia, cortical sensory loss, worsening emotional lability, myoclonus and slow saccades with saccadic dysmetria. CBDS has variable phenotypic presentation, which can make diagnosis difficult; an autopsy is needed for confirmation (Grijalvo-Perez and Litvan 2014).
Riess et al. (2013) described two obligate carrier females from their affected CS family. The grandmother of the index case, an obligate carrier, was healthy until 55 years when she developed neurologic symptoms and depression. Her brain imaging showed diffuse mild cerebral atrophy and she was given a diagnosis of Parkinson’s disease. Her mother was reported to have parkinsonism in her 70s and passed away at 82 years of age. Given the clinical similarity and age of onset of atypical parkinsonian disorders and Parkinson’s disease, distinguishing between the different etiologies is difficult without further clinical information or an autopsy of those affected. It is possible that these cases may represent onset of clinical findings of the female phenotype that presented at a later age. Interestingly, the neurologic findings in this family, as in our cohort, were also observed to worsen with earlier age of and more severe presentation in subsequent generations. Together, these findings represent a new aspect to the female SLC9A6 mutation carrier phenotype, with increasing evidence that females may be at risk for cognitive and neurologic decline as they age, with pathological and phenotypic features similar to CBDS or Parkinson’s disease.
There is increasing evidence to suggest that these neurodegenerative findings may reflect a tauopathic process associated with SLC9A6 functional impairment. Garbern et al. (2010) described two males with an in-frame 9-base pair deletion in SLC9A6 who were found to have pathologic findings of tau deposition very similar to CBDS. The two patients were reported to have profound mental retardation and severe language disturbance, similar to that seen in CS, but the dysmorphic features and severe microcephaly were not seen. They postulated that progressive neurodegeneration was caused in part by tau deposition, which contributed to the severe neurodevelopmental phenotype. Neuropathological examination showed widespread cerebral atrophy and marked neuronal loss. The cerebellar and brainstem white tracks showed diffuse glial tau pathology, while tau-positive inclusions were widespread in the cerebral cortex and hippocampus. Their observations suggested that the deposition of tau may have been mediated by the interaction with the mutant SLC9A6 protein. They postulated that mutations in SLC9A6 can cause aberrant splicing in the MAPT gene which encodes tau, leading to tau deposition.
Strømme et al. (2011) developed Slc9a6 knockout mouse models which showed that loss of function of Slc9a6 leads to altered endosomal–lysosomal function, and accumulation of GM2 gangliosides and cholesterol in select neuronal populations (including amygdala and hippocampus) similar to primary lysosomal storage diseases. They noted extensive Purkinje cell deterioration and axonal spheroid formation in the cerebellum, findings which were correlated with the cerebellar atrophy seen with human SLC9A6 mutations. The knockout mice also showed small increases of phosphorylated tau in soluble brain fractions which could be correlated with the behaviour of the knockout mice. Strømme et al. (2011) postulated that the specific areas involved could provide a unified explanation for the phenotypes seen. The findings of tau deposition as a downstream consequence of endosomal–lysosomal dysfunction (Strømme et al. 2011), along with the hypothesis that this deposition may be further provoked by the interaction of a mutant SLC9A6 protein (Garbern et al. 2010), raise interesting questions about the role of tau deposition in both the male and female phenotypes associated with SLC9A6 mutations.
As with many X-linked disorders, skewed X-inactivation may play a role in the clinical presentation of SLC9A6 mutations in female carriers. Sikora et al. (2016) created a heterozygous Slc9a6 knockout female mouse that showed the expression of the mutant Slc9a6 allele to follow the same neuroanatomic distribution and cell-specific patterns in hemizygous mutant males. The heterozygous knockout female mice developed progressive behavioural and neuropathological abnormalities that were similar to but milder than hemizygous male knockout mice. Similar to Strømme et al. (2011), they showed accumulation of GM2 gangliosides in the hippocampus and amygdala, along with Purkinje cell degeneration in the cerebellum. Using β-galactosidase as a marker for cellular expression of the mutant Slc9a6, they showed that X-inactivation was a likely random event in Purkinje cells. They concluded that further studies should be undertaken to determine neuroanatomic thresholds for behaviour pathologies (Sikora et al. 2016).
The emerging and expanding phenotypic spectrum of female SLC9A6 mutation carriers is reminiscent of the ever-expanding phenotypic spectrum associated with the Fragile X syndrome gene FMR1. Mutations in FMR1 are associated with Fragile X syndrome (OMIM 300624), while the premutation state can result in increased risk of Fragile X-associated tremor-ataxia syndrome (FXTAS) (OMIM 300623), as well as premature ovarian failure in females. The FMR1 narrative also began with the assumption that female carriers would have little to no susceptibility to the associated phenotypes. The less frequent, later onset and milder findings of FXTAS in female premutation carriers were thought to arise due to X-inactivation providing a protective mechanism, though this has not been fully proven (Grigsby et al. 2014). Further delving into the phenotype for female FMR1 premutation carriers has shown progressive cognitive impairment in executive functioning, working memory and information processing (Grigsby et al. 2014). Testing of neuropsychiatric functioning in females with a premutation has shown higher rates of depression and social anxiety (Kraan et al. 2013), as well as other psychiatric disorders, such as ADHD (Hunter et al. 2012). Loesch et al. (2015) showed that carriers of mid-sized FMR1 5′ UTR repeats (<100 CGG repeats) are at greatest risk for developing a psychiatric disorder. It is becoming clear that female FMR1 premutation carriers have a distinct clinical phenotype with progressive neurocognitive decline and increased risk for neuropsychiatric impairment. A similar pattern is now emerging with female SLC9A6 mutation carriers.
While the phenomenon of anticipation is most frequently associated with disorders involving expansion of triplet repeats, there are X-linked genetic syndromes not involving repeat expansions in which anticipation has been reported. X-linked dominant chondrodysplasia punctata (OMIM 302960) is a rare skeletal dysplasia that presents with stippled epiphyses, short stature and skin lesions. It is presumed lethal in males but presents variably in females. Females can be stillborn to mildly affected in adulthood. Traupe et al. (1992) described three families with variable presentations in affected family members with increasing severity in subsequent generations. They hypothesized a heritable unstable premutation found through exclusion mapping as the cause of this phenomenon. Sutphen et al. (1995) described a three-generation family affected with X-linked dominant chondrodysplasia punctata with increasing severity of presentation in subsequent generations. Skewed X-inactivation was hypothesized but not tested. Shirahama et al. (2003) described a family with marked variation and severity between mother and daughter which was attributed to confirmed skewed X-inactivation. Whether the observed increasing phenotypic severity can be attributable to variable skewed X-inactivation or the hypothesis of a heritable unstable premutation as proposed by Traupe et al. (1992) remains to be proven. Other X-linked conditions described with apparent anticipation attributable to skewed X-inactivation include dyskeratosis congenita (Devriendt et al. 1997), Wiskott–Aldrich Syndrome (Parolini et al. 1998) and X-linked myotubular myopathy (Tanner et al. 1999). Further studies, including in animal models, of these disorders are necessary to determine whether skewed X-inactivation provides an adequate explanation for the apparent anticipatory phenomenon seen in female carriers of non-triplet repeat X-linked disorders. Further longitudinal studies of human female SLC9A6 mutation carriers will also be essential to better characterize the neurodegenerative phenotype and determine the relative risk for developing these symptoms. X-inactivation studies may be of value for female mutation carriers to determine their risk for neurodegeneration. Neurologic and psychiatric assessments with periodic follow-up may be warranted to detect subtle neurologic findings or developing psychiatric illness.
The female SLC9A6 mutation carrier phenotypic spectrum encompasses learning difficulties, speech and language delay, mild to moderate intellectual disability, and behavioural issues. The results of this study provide further support for these phenotypes and expand the phenotype to suggest a significant risk for a variety of psychiatric disorders, including mood and anxiety disorders, ADHD, ODD, and disorders that can involve psychosis, such as bipolar disorder and schizophrenia. Some of these disorders had been previously infrequently reported. The most prominent behavioural phenotypes seen are aggression and self-injury. There is also increasing evidence of a neurodegenerative component to the female carrier phenotype, with this and other reports of CBDS and parkinsonian disorders, and the possibility of SLC9A6 mutations being involved in a tauopathic process. Finally, empiric evidence from this study and the literature suggests an anticipatory element, with worsening of the female carrier phenotype in subsequent generations. This would have important implications in the counselling and management of families in which multiple generations have been identified to carry a mutation in SLC9A6. It remains unclear whether the emerging findings constitute a distinct female phenotype for SLC9A6 mutations or whether they are more akin to the FMR1 premutation status. There is currently no known correlation between X-inactivation status and phenotype (Christianson et al. 1999; Gilfillan et al. 2008; Riess et al. 2013), but further studies on larger cohorts may aid in elucidating other pathomechanisms for the phenotypic findings. In the meantime, the growing body of evidence for a female phenotype supports that description of “affected males” and “carrier females” would be more accurately represented by describing individuals as simply being affected or not with a mutation in SLC9A6.
References
Christianson AL, Stevenson RE, van der Meyden CH, Pelser J, Theron FW, van Rensburg PL et al (1999) X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellaratrophy in a large South African kindred is localised to Xq24-q27. J Med Genet 36(10):759–766
Darvesh S, Leach L, Black SE, Kaplan E, Freedman M (2005) The behavioural neurology assessment. Can J Neurol Sci 32(2):167–177
Devriendt K, Matthijs G, Legius E, Schollen E, Blockmans D, van Greet C et al (1997) Skewed X-chromosome inactivation in female carriers of dyskeratosis congenital. Am J Hum Genet 60:581–587
Fichou Y, Bahi-Buisson N, Nectoux J, Chelly J, Héron D, Cuisset L, Bienvenu T (2009) Mutation in the SLC9A6 gene is not a frequent cause of sporadic Angelman-like syndrome. Eur J Hum Genet. 17(11):1378–1380
Fogel BL, Clark MC, Geshwind DH (2014) The neurogenetics of atypical Parkinsonian disorders. Semin Neurol 34(2):217–224
Folstein MF, Folstein SE, McHugh PR (1975) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12(3):189–198
Friedman JM (1998) [Updated 2014 Sep 4] Neurofibromatosis 1. In: Pagon RA, Adam MP, Ardinger HH et al (eds) GeneReviews® [Internet]. University of Washington, Seattle; 1993–2015. https://www.ncbi.nlm.nih.gov/books/NBK1109/. Accessed 05 January 2016
Garbern JY, Neumann M, Trojanowski JQ, Lee VM, Feldman G, Norris JW et al (2010) A mutation affecting the sodium/proton exchanger, SLC9A6, causes mental retardation with tau deposition. Brain. 133(Pt 5):1391–1402
Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K et al (2008) SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet 82(4):1003–1010
Grigsby J, Cornish K, Hocking D, Kraan C, Olichney JM, Rivera SM et al (2014) The cognitive neuropsychological phenotype of carriers of the FMR1 premutation. J Neurodev Disord. 6:28
Grijalvo-Perez AM, Litvan I (2014) Corticobasal degeneration. Semin Neurol. 34:160–173
Hunter JE, Epsten MP, Tinker SW, Abramowitz A, Sherman SL (2012) The FMR1 premutation and attention-deficit hyperactivity disorder (ADHD): evidence for complex inheritance. Behav Genet 42(3):415–422
Jones SH, Thornicroft G, Coffey M, Dunn G (1995) A brief mental health outcome scale-reliability and validity of the Global Assessment of Functioning (GAF). Br J Psychiatry. 166(5):654–659
Kondapalli KC, Prasad H, Rao R (2014) An inside job: how endosomal Na(+)/H(+) exchangers link to autism and neurological disease. Front Cell Neurosci 8:172. doi:10.3389/fncel.2014.00172
Kraan C, Hocking D, Bradshaw JL, Fielding J, Cohen J, Georgiou-Karistianis N et al (2013) Neurobehavioural evidence for the involvement of the FMR1 gene in female carriers of fragile X syndrome. Neurosci Biobehav Rev. 37(3):522–547
Loesch DZ, Bui MQ, Hammersley E, Schneider A, Storey E, Stimpson P et al (2015) Psychological status in female carriers of permutation FMR1 allele showing a complex relationship with the size of CGG expansion. Clin Genet 87:173–178
Parolini O, Ressmann G, Haas OA, Pawlowsky J, Gadner H, Knapp W et al (1998) X-linked Wiskott-Aldrich syndrome in a girl. N Eng J Med. 338:291–295
Pescosolido MF, Stein DM, Schmidt M, El Achkar CM, Sabbagh M, Rogg JM et al (2014) Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome. Ann Neurol. 76(4):581–593
Riess A, Rossier E, Krüger R, Dufke A, Beck-Woedl S, Horber V et al (2013) Novel SLC9A6 mutations in two families with Christianson syndrome. Clin Genet 83(6):596–597
Schroer RJ, Holden KR, Tarpey PS, Matheus MG, Griesemer DA, Friez MJ et al (2010) Natural history of Christianson syndrome. Am J Med Genet A. 152A(11):2775–2783
Shirahama S, Miyahara A, Kitoh H, Honda A, Kawase A, Yamada K et al (2003) Skewed X-chromosome inactivation causes intra-familial phenotypic variation of an EBP mutation in a family with X-linked dominant chondrodysplasia punctata. Hum Genet 112(1):78–83
Sikora J, Leddy J, Gulinello M, Walkley SU (2016) X-linked Christianson syndrome: heterozygous female Slc9a6 knockout mice develop mosaic neuropathological changes and related behavioral abnormalities. Dis Model Mech. 9(1):13–23. doi:10.1242/dmm.022780
Strømme P, Dobrenis K, Sillitoe RV, Gulinello M, Ali NF, Davidson C et al (2011) X-linked Angelman-like syndrome caused by Slc9a6 knockout in mice exhibits evidence of endosomal-lysosomal dysfunction. Brain. 134(Pt 11):3369–3383
Sutphen R, Amar MJ, Kousseff BG, Toomey KE (1995) XXY male with X-linked dominant chondrodysplasia punctate (Happle syndrome). Am J Med Genet 57:489–492
Tanner SM, Ørstavik KH, Kristiansen M, Lev D, Lerman-Sigie T, Sadeh M et al (1999) Skewed X-inactivation in a manifesting carrier of X-linked myotubular myopathy and in her non-manifesting mother. Hum Genet. 104:249–253
Traupe H, Muller D, Atherton D, Kalter DC, Cremers FP, van Oost BA et al (1992) Exclusion mapping of the X—linked dominant chondrodysplasia punctate/ichthyosis/cataract/short stature (Happle) syndrome: possible involvement of an unstable pre-mutation. Hum Genet 89:659–665
Zanni G, Barresi S, Cohen R, Specchio N, Basel-Vanagaite L, Valente EM et al (2014) A novel mutation in the endosomal Na+/H+ exchanger NHE6 (SLC9A6) causes Christianson syndrome with electrical status epilepticus during slow-wave sleep (ESES). Epilepsy Res. 108(4):811–815
Acknowledgments
The authors would like to thank the family and their contributions to this paper. On behalf of all authors, the corresponding author states that there is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sinajon, P., Verbaan, D. & So, J. The expanding phenotypic spectrum of female SLC9A6 mutation carriers: a case series and review of the literature. Hum Genet 135, 841–850 (2016). https://doi.org/10.1007/s00439-016-1675-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1675-5