
Abstract
Mendelian mutations are the most medically actionable variants in the human genome and have always played a central role in its functional annotation. Despite the relative ease with which Mendelian mutations are identified compared to other classes of variants, the pace of their discovery has until recently been slow. However, recent technological advances in genomic sequencing have made the prospect of identifying all genes that can harbor Mendelian mutations an achievable near-term goal. The many lessons learned from previous discoveries of Mendelian mutations should inform future studies as I will discuss in this review. Also discussed are some of the challenges that will gain more prominence as we approach the last phase of the effort to map all Mendelian genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent advances in the study of the human variome have unraveled a remarkable degree of genomic diversity. Perhaps most surprising was the discovery of a tremendous degree of individual-level variation with many single nucleotide variants (SNVs) that are “private” to each individual genome (Kim et al. 2009; Lupski et al. 2010). Clearly, at least some of these private variants must be contributing to the biological processes that make each individual unique. The daunting challenge of establishing a link between these variants and the various phenotypic traits of the individuals who harbor them becomes more conspicuous when one realizes how little we know about the phenotypic influence of even the common variants that are shared by large sections of the human population. In the absence of a reasonable understanding of how variants of all classes influence the phenotype, the promise of personalized medicine may seem as a distant prospect.
Personalized medicine is not an all or none phenomenon, however. Rather, the delivery of personalized medicine should be viewed as an evolving and iterative process that builds on every discovery related to genotype/phenotype correlation and need not wait until such correlation, or lack thereof, is established for every variant. Indeed, the latter appears highly improbable in view of the virtually unlimited diversity of the human genome ensured by the introduction of 1.2 × 10−8 mutation per nucleotide per generation (Kong et al. 2012). Efforts to establish genotype/phenotype correlation for variants in the human genome should prioritize the medical relevance of such correlations. For example, some variants have been found to influence the relative abundance or spatial/temporal expression genes in ways that have profound implications on our understanding of the human lineage, but with very little apparent medical value (Indjeian et al. 2016). More relevant to personalized medicine are links to physiological processes that are perturbed in disease states. While most variants have a limited capacity to exert influence on such processes, the influence of some is so profound that their mere presence can reliably predict a measurable change that is readily detectable phenotypically and these are referred to as Mendelian variants, or more commonly mutations because the phenotype is usually a disease state (Mendelian diseases). Although this definition of Mendelian mutations overlooks important exceptions, it offers a compelling degree of practicality, so it will be used throughout this review.
The case for prioritizing the study of Mendelian mutations over other classes of variants is robust (Antonarakis and Beckmann 2006). The often straightforward link between these variants and disease states is much easier to study than variants that merely modify the risk, sometimes to an infinitesimal degree. The predictability of this link is also instrumental to making key medical and reproductive decisions. While some may argue that Mendelian diseases are rare, and that variants that influence the risk of more common diseases should receive more attention, one should bear in mind that the dichotomy of Mendelian vs common diseases is artificial (Antonarakis et al. 2010). Not only do Mendelian variants offer insight into the biology of common diseases, but there is also an increasing appreciation of the direct contribution of Mendelian variants to the risk of common diseases (Alkuraya 2015; Alsalem et al. 2013a; Cirulli and Goldstein 2010). Furthermore, Mendelian genes are enriched for druggability, and many of the best-selling drugs for common diseases target the protein products of Mendelian genes (Brinkman et al. 2006).
Given the great medical relevance of Mendelian mutations in rare and common diseases, this review will be dedicated to the issues related to how this special class of variants is identified. After a brief historical overview, the new wave of Mendelian mutations discovery made possible by next-generation sequencing is discussed. Important lessons will be drawn from past discoveries to inform a discussion about some of the remaining challenges in the path toward annotating all Mendelian genes.
Historical perspective
Identifying the one variant that causes a Mendelian phenotype from the billions of nucleotides in the human genome was particularly challenging when the blueprint of the human genome did not even exist. Clues were necessary to narrow the search space, and these came in different forms. Sickle cell disease is arguably the first “molecular disease” since its molecular basis was identified in 1949 (Pauling et al. 1949). This level of molecular understanding, including the abundance of the protein in a quasi-pure form, was critical to the identification of the causative mutation once the gene was cloned in 1977 (Marotta et al. 1977). A similar “forward genetics” approach was applied in several other disorders, the molecular basis of which was sufficiently understood to direct the search for mutations in the genes with relevant physiological function. Because these represent a minority of diseases, “reverse genetics” in the form of positional mapping provided the much needed alternative clue in the search for Mendelian mutations.
The power of positional mapping is that it does not require prior knowledge of the physiology/biochemistry of the disease in question, since it merely highlights a candidate region in the genome where the mutation is likely to reside. However, because the “critical locus” often contained many genes, prioritizing the most likely candidate gene within the locus was a practical necessity to avoid the costly and cumbersome brute-force Sanger sequencing of all the genes therein. The prioritization scheme only rarely assigned the causal gene the top score, which reflects the often surprising nature of the genes underlying Mendelian disorders and the opportunity for novel biological discoveries in the field of Mendelian genetics. Another major limitation of positional mapping is the requirement for sufficient meiotic events to identify the critical locus, often necessitating the procurement of large pedigrees. While this requirement could be fulfilled through the study of special populations in the case of recessive diseases, e.g., those with high rates of consanguinity, or dominant phenotypes that permit the study of multiple generations because their effect on reproductive fitness is limited, it was clear that this approach does not offer the throughput and generalizability needed to map all Mendelian genes and that alternative strategies were acutely needed.
Why was next-generation sequencing revolutionary in Mendelian genetics?
The ability to sequence many segments of DNA simultaneously was a distinct departure from the established norm of DNA sequencing, and represented a technological advance that abruptly made the sequencing of entire genomes an affordable endeavor. By essentially eliminating the need for the above-mentioned “clues”, genomic sequencing radically changed the kind of samples required to discover Mendelian mutations, from large pedigrees to simplex cases. A related advantage is that genomic sequencing can be applied to any phenotype including lethal unborn phenotypes that cannot be characterized otherwise, as well as dominant viable phenotypes with extremely limited reproductive fitness such as severe intellectual disability (Shamseldin et al. 2012b, c; Vissers et al. 2010). Also related to the agnostic nature of genomic sequencing is its power to obviate the historical requirement for strictly homogeneous phenotypes to map Mendelian genes, although this may still be helpful in defining the core features of a particular syndrome. One particularly unique advantage of genomic sequencing compared to old approaches in the discovery of Mendelian genes is in the area of postzygotic (somatic) mutations with resulting mosaicism (Lupski 2013). Prior to the era of genomic sequencing, the discovery of mosaic mutations in a novel disease gene was nearly impossible, because the challenge in identifying low-level mosaicism in accessible tissues by Sanger sequencing was an added layer of difficulty. The identification of somatic FGFR3 mutations in epidermal nevi, one of the very few examples prior to genomic sequencing, is an illustrative example where FGFR3 would not have been specifically pursued had there been no phenotypic overlap between epidermal nevus and acanthosis nigricans that accompanies germline FGFR3 mutations (Hafner et al. 2006). Finally, the affordability of next-generation sequencing ushered in an era of democratizing Mendelian gene discovery. No longer is it the case that large and specialized research laboratories are the only places equipped to identify novel Mendelian genes, because the wider base of clinicians who interact with these patients are increasingly capable of reporting novel gene discoveries made possible by the use of clinical genomic sequencing (Might and Wilsey 2014). This is critically important to the goal of identifying all Mendelian genes because while only a subset of patients is recruited in research programs, the majority are seen by clinicians.
Lessons learned from past discoveries
In this section, I will attempt to draw lessons from personal as well as the collective experience of the Mendelian genetics community in the discovery of Mendelian disorders. These lessons have the potential to inform future discovery efforts. Obviously, there is a clear bias in the literature to only publish success stories even though much can be learned from previous failures. Even success stories are typically presented in a way that often overlooks important mishaps that can provide valuable insights. This section, therefore, is dedicated to share some of these valuable “behind the scenes” scenarios.
Positional mapping remains an important tool in the era of genomic sequencing
Interpreting the large number of variants revealed by genomic sequencing remains a daunting task, especially when one considers the very large number of private or very rare variants (see “Introduction”), but can be simplified by focusing the search on a fraction of the genome highlighted by positional mapping (Alkuraya 2012, 2013). We have previously published on the identification of ISCA2, RTTN, GOLGA2 and UNC80 mutations as novel causes of Mendelian diseases in individuals in whom clinical exome, and in some cases clinical genome sequencing, failed to identify the likely cause despite full coverage of the respective gene, most likely due to the challenge of interpretation (Alazami et al. 2015; Shamseldin et al. 2015a, b, 2016). In all these examples, combining autozygosity mapping with exome sequencing was key to the successful identification of the likely causal mutation. This advantage is not limited to increasing efficiency, but extends to highlighting classes of variants that many not be conspicuous otherwise. For example, intronic mutations that are not in the consensus ±1/2 position are very difficult to interpret and can go unnoticed when analyzing a very large number of variants throughout the genome. However, when a single locus is being analyzed, more in-depth analysis of the variants allows even less conventional intronic mutations to come into focus, especially when no compelling coding mutations are identified. The identification of the RTTN as a novel cause of microcephalic primordial dwarfism based on a mutation in the -8 position was only possible through positional mapping of a family initially undiagnosed by clinical exome sequencing (Shamseldin et al. 2015a). Similarly, a mutation in the -24 position in COG6 was found to cause a novel syndrome of intellectual disability and hypohidrosis with the help of positional mapping, and clinical exome sequencing did not identify this mutation even after it was published because it was filtered out as a deep intronic mutation (Shaheen et al. 2013b; Yavarna et al. 2015). Finally, clinical exome sequencing failed to identify the causal variant in CTU2, most likely because it was synonymous and it was only through combining the three families with the novel phenotype of microcephaly, ambiguous genitalia, renal agenesis and polydactyly that the CTU2 variant appeared as a likely candidate and its effect on splicing was confirmed (Shaheen et al. 2015).
Dominant and recessive mutations can tell different stories
It is well known that certain disorders can be caused by dominant and recessive mutations in the same gene, but these different classes of mutations are not commonly considered in the etiology of allelism (different phenotypes linked to the same gene). It is important, therefore, to consider genes as valid candidates for recessive phenotypes even if their dominant phenotypes appear very distinct and vice versa. For example, the phenotype associated with DNA2 heterozygous mutations (mitochondrial myopathy) is very different from homozygous loss of function, which causes Seckel syndrome (Ronchi et al. 2013; Shaheen et al. 2014a). When we identified homozygous loss of function mutations in ELOVL4 as a novel cause of a Sjogren–Larsson syndrome-like illness, this pathogenic mutation in the index was initially dismissed because heterozygous mutations in ELOVL4 are an established cause of Stargardt macular degeneration, a phenotype that lacks any overlap with that of the index (Aldahmesh et al. 2011b). This phenomenon can be explained on the basis of a different effect exerted by the heterozygous dominant negative or gain of function vs. homozygous loss of function, since haploinsufficiency would be expected to result in a phenotype in the carriers of the recessive loss of function mutations (Aldahmesh et al. 2011a).
Extreme forms of allelism
Some groups of disorders are known to display marked phenotypic variability, but these usually still fall within a broader phenotypic definition; ciliopathies are excellent examples. LMNA is a gene with probably the largest number of allelic disorders linked to recessive and dominant mutations in it (laminopathies) and the list of disorders was further expanded recently to include DAPJ (distal acroosteolysis, poikiloderma and joint stiffness) (Sewairi et al. 2016). The discovery of the DAPJ as a laminopathy was facilitated in part by its phenotypic overlap with other disorders in that group. However, it can be extremely challenging when the established phenotype caused by recessive missense mutations is very different from the one under investigation. Neu-Laxova syndrome (NLS) is a perinatally lethal disorder characterized by an extreme form of dysmorphism, so it was highly surprising when the underlying cause was found to be missense mutations in PHGDH, because biallelic missense mutations in the same gene are known to cause a serine deficiency disorder characterized by a relatively mild intellectual disability disorder with or without seizures and microcephaly (Shaheen et al. 2014b). It is conceivable that the PHGDH mutation could have been missed had it not been for the positional mapping advantage that confined the investigators’ search to a single locus. Allelism will inevitably account for a substantial number of “unsolved” Mendelian phenotypes that are listed in OMIM and presumed to be novel. It is interesting to observe how the classical use of the term “allelism” is being replaced by “phenotypic expansion”, because the rate of mutation discovery surpasses that at which distinct clinical phenotypes are established in the literature, as shown recently by a large cohort of apparently novel dysmorphology syndromes (Shaheen et al. 2015). Especially problematic is when the variability of phenotype leads to erroneous designation of affected members of the same family, thus complicating family-based segregation analysis. One extreme example can be seen in the founder C206Y mutation in THSD1, the single most common cause of recurrent lethal non-immune hydrops fetalis in Arabia (Shamseldin et al. 2015c). We initially dismissed this variant, because it was found to be homozygous in members of the family who appeared unaffected. Additional families clearly revealed that this mutation is not always lethal and some affected fetuses recover postnatally (Shamseldin et al. 2015c). Thus, even though embryonic lethality and apparently good health are strikingly different categories of phenotypes, they may still represent the phenotypic expression of the same allele.
Coverage matters
No available sequencing technique covers 100 % of the human genome. These inevitable gaps in coverage are a limitation that needs to be considered carefully when investigating the cause of a Mendelian disorder. This fact was beautifully illustrated in the study comparing the utility of genome vs exome sequencing in the setting of intellectual disability (Gilissen et al. 2014). The “missed” mutations by exome sequencing were not regulatory element mutations as had been assumed, but mostly complex genomic rearrangements and point mutations not adequately covered. Again, positional mapping can be very helpful in this regard, because meticulous examination of the coverage of one genomic interval is much easier when compared with the entire genome. For example, TMEM38B was initially missed as the candidate gene for an autosomal recessive form of osteogenesis imperfecta, but linking the disease to a single locus made it possible to uncover the small genomic deletion that disrupted the reading frame of that gene (Shaheen et al. 2012).
Allele frequency can be deceiving
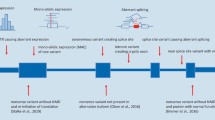
Screening 100 ethnically matched controls for a given allele to substantiate the claim of its pathogenicity, once a standard in human genetics literature, is clearly insufficient in view of the growing knowledge of the vast degree of private and rare variation in the human population. Many of the variants listed in HGMD as disease mutations are now clearly benign SNPs based on more recent compendiums of human variation made possible by large-scale sequencing projects (Group 2015). In general, a dominant allele should be absent in a variant database based on healthy controls or exceedingly rare to allow for reduced penetrance. An allele of high frequency, but only present in the heterozygous state, can still be disease causing in a recessive context depending on the prevalence of the disease in question. For example, the F508del mutation in CFTR has an MAF of >1 % in Caucasians and the Glu7Val mutation in HBB has an MAF of >12 % in Nigeria, and both are definitive disease-causing mutations despite their very high MAF because the disease (q 2) is very prevalent in the respective population, in agreement with the prediction of the Hardy–Weinberg equation. However, MAF may appear too high for the established q 2 (disease) frequency and yet the allele is disease causing, as revealed in a number of diseases caused by the dual presence of a common and rare allele in trans. This phenomenon was first described in thrombocytopenia-absent radius syndrome where the causative mutation is typically a loss of function mutation of RBM8A (usually a chromosomal deletion) in one allele and a regulatory SNP, usually in the 5′UTR of RBM8A, in trans (Albers et al. 2012). Indeed, the relatively high frequency of the regulatory SNP (>4 %) could only be reconciled with its involvement in such a rare disorder if one considers an unusual autosomal recessive inheritance where homozygosity for this SNP does not cause the disease, but rather its presence in trans with a loss of function allele in the same gene. Similarly, it has recently been found that Burn–McKeown syndrome, an oculo-oto-facial dysplasia, is caused by compound heterozygosity for a low-frequency SNP (MAF 0.76 %) in the promoter of TXNL4A in trans with a loss of function mutation in the same gene (Wieczorek et al. 2014). It is difficult to estimate the contribution of this unusual phenomenon to Mendelian diseases, but it seems prudent to consider this possibility when the causative mutation of a particular phenotype seems to defy the typical analytical pipeline for interpreting exome variants (Fig. 1).

Distilling all genomic variants into one or more causal variant requires iterative filtering informed by phenotypic data. While very helpful, this process requires flexibility to address the limitation of each filter
Animal models do not always recapitulate the human phenotype
This may seem too obvious to state in this review, but because researchers who evaluate variants in novel candidate genes often turn to published data on animal models, it is worth highlighting some of the limitations of this supportive line of evidence. The animal may truly lack the corresponding human phenotype despite being fully knocked out for the gene as in the knockout murine models of Hprt1, Ocrl1, Abcd1, Gla, Galt and Hexa, which were engineered to study classical Mendelian diseases in humans (Elsea and Lucas 2002). We had initially dismissed a missense variant in SBF1 in a family with a novel syndromic form of Charcot–Marie–Tooth, because the corresponding knockout mouse was reported to be normal neurologically, and it was only through the independent identification of SBF1 mutation in a Korean family with this phenotype that the link is now established in the literature (Alazami et al. 2014; Bohlega et al. 2011; Nakhro et al. 2013). An important consideration is that apparent lack of relevant phenotype in the animal model may simply reflect deficient phenotyping. For example, when we identified homozygous loss of function mutation CTSH as a novel cause of syndromic severe myopia, the knockout mouse had been described as having no visual involvement. However, subsequent collaboration and histological examination revealed marked elongation of the globe axis recapitulating the human phenotype (Aldahmesh et al. 2013; Bühling et al. 2011).
In silico prediction algorithms are limited in sensitivity and specificity
In silico prediction algorithms can be very helpful in shortlisting variants, but caution is advised because the designation “likely benign” by these tools does not necessarily confirm the benign nature of the variant. The variant c.2233G>A in EZH1 would have been overlooked as disease causing because the alternative allele is the natural allele in monkeys so it was predicted to be “benign”, except that it was the second occurrence of the same de novo variant in patients with clinically confirmed Weaver syndrome (Al-Salem et al. 2013). An interesting recent paper shows that this phenomenon can be explained by a compensatory mutational mechanism as revealed by comparative genomic analysis (Jordan et al. 2015). Similarly, in silico prediction of splicing can also be misleading as we have shown in TMEM92, C21orf2 and DNA2 (Shaheen et al. 2014a, 2015; Shamseldin et al. 2015a). Not only is this especially problematic for deep intronic mutations as discussed above, but exonic splicing mutations that do not affect the terminal two base pairs can also very challenging. The FBXL4 exonic mutation we had originally identified as a novel candidate in mitochondrial encephalomyopathy replaced a very poorly conserved amino acid; however, it significantly affected splicing efficiency (Gai et al. 2013; Shamseldin et al. 2012a). Newer tools have been developed, but it is unlikely that 100 % specificity and sensitivity will be achieved. So careful examination of variants based on other criteria, including RTPCR in the case of suspected splicing mutations, is always advised.
The annotation of the reference genome is imperfect
There are various inconsistencies in gene naming, variant calling and localization and impact prediction that resulted from the use of a mixed collection of software versions and annotation database releases. Such inconsistencies can result in variants falsely classified as frameshift, e.g., two adjacent SNPs in the same codon are always coupled and their combined impact on the ORF is different from what is predicted if each SNP is evaluated independently. This was an important limitation to handle when attempting to catalog loss of function variants in the human genome (Alsalem et al. 2013b). Multiallelic sites (variable sites that can be occupied by up to three instead of one alternative alleles) can pose another challenge, since these may account for >6 % of the human genome and have the potential of masking the presence of pathogenic alleles when a more common allele is being called instead (Campbell et al. 2015). While these can be filtered out bioinformatically or even by Sanger sequencing in some cases, errors and gaps in defining exons and introns can be more consequential in a Mendelian gene discovery project. For example, our identification of EOGT as a novel gene for Adams–Oliver syndrome could not have been achieved without being guided by positional mapping data, because the causal mutation was presumably deep intronic when in fact it affected an unannotated exon leading to frameshift (Shaheen et al. 2013a).
Not every de novo mutation is disease causing
The power of trio-exome sequencing to reveal de novo variants cannot be overstated, and its relevance is particularly visible in the category of dominant and X-linked disorders with strong negative effect on the reproductive fitness (Veltman and Brunner 2012). The mutation rate in humans at 1.2 × 10−8 per nucleotide per generation predicts on average one de novo mutation per exome. Thus, while it may be tempting to assume the pathogenicity of such de novo events, probabilistic considerations are warranted. As elegantly illustrated by MacArthur et al., the finding of two independent de novo hits in TTN in two patients with intellectual disability is hardly a compelling evidence of causality, because the huge size of TTN makes it more likely to sustain de novo mutations, and intellectual disability is extremely heterogeneous and relatively common (MacArthur et al. 2014). While this may be an extreme example, the key message holds true, i.e., statistical support is needed to substantiate claims of causal links between de novo mutations in a novel disease gene and a particular phenotype.
Remaining challenges
The current rapid pace of disease gene discovery in Mendelian diseases suggests that virtually all Mendelian genes will be identified within the next few years. However, this process is likely to follow a tail end distribution where the last remaining genes are the most difficult to identify due to any or a combination of the factors listed above.
Mendelian diseases with an exceedingly low prevalence are certainly represented by the recent wave of successful mapping projects, but they are likely to be significantly enriched is the last phase of the race toward full mapping of all Mendelian genes. This has already highlighted the acute need for better exchange of data to overcome the increasingly apparent “n of one problem”, and matchmaking tools have been devised in response (Philippakis et al. 2015; Sobreira et al. 2015). The “wholesale” publication of novel candidates is another powerful solution to this problem and allows for a full and transparent matchmaking post-publication (Shaheen et al. 2015; Shamseldin et al. 2015a). Social media have also emerged lately as an important player, especially in participant-driven matchmaking (Chong et al. 2015; Lambertson et al. 2015).
Mosaic disorders are likely to become another frontier that assumes more prominence as more and more “easier” Mendelian gene mutations are identified. It is likely that mosaicism is underestimated as a cause of “unsolved” dominant disorders as exemplified by the study of “negative” Cornelia de Lange syndrome (Huisman et al. 2013). More challenging are disorders involving the CNS, because obtaining relevant tissue to evaluate mosaicism is often impractical. Fortunately, it has been demonstrated that ultradeep sequencing of blood-derived DNA can reveal the underlying mutation in a substantial proportion of these cases (Jamuar et al. 2014). Of note, mosaicism is not limited to dominant disorders and can also complicate the identification of recessive mutations as well (Anazi et al. 2014).
One major question that is often raised is what percentage of unsolved Mendelian phenotypes is caused by regulatory mutations, i.e., mutations in UTR, promoters or enhancers? A complicating factor here is the presence of phenocopies, especially for non-specific phenotypes such as intellectual disability. Does the fact that more than one-third of cases with severe intellectual disability remain undiagnosed molecularly even after whole genome sequencing suggest that they harbor regulatory mutations that remain difficult to call by researchers even if they are sequenced, or do they perhaps represent non-Mendelian phenocopies? Unbiased analysis of large cohorts of phenotypes with tight linkage loci will be needed to answer this question for each class of Mendelian mutations (recessive, dominant and X-linked).
Conclusion
The reward of identifying causal mutations for Mendelian disorders is far from being purely academic. Each discovery can have medical implications and will leave a lasting imprint on the annotation of our genome. Because the stakes are high, no effort should be spared to accelerate these discoveries, and this review is a humble attempt in this regard to share experience and learn from past mistakes. The mapping of all Mendelian genes will inevitably inform research into common diseases. Going forward, we should expect more challenging classes of variants to be identified and these will require a greater level of collaboration within the Mendelian genetics community.
References
Alazami AM, Alzahrani F, Bohlega S, Alkuraya FS (2014) SET binding factor 1 (SBF1) mutation causes Charcot-Marie-Tooth disease type 4B3. Neurology 82:1665–1666
Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, Hijazi H, Alshammari M, Aldahmesh MA, Salih MA (2015) Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell reports 10:148–161
Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P (2012) Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 44:435–439
Aldahmesh MA, Khan AO, Mohamed J, Alkuraya FS (2011a) Novel recessive BFSP2 and PITX3 mutations: insights into mutational mechanisms from consanguineous populations. Genet Med 13:978–981
Aldahmesh MA, Mohamed JY, Alkuraya HS, Verma IC, Puri RD, Alaiya AA, Rizzo WB, Alkuraya FS (2011b) Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am J Hum Genet 89:745–750
Aldahmesh MA, Khan AO, Alkuraya H, Adly N, Anazi S, Al-Saleh AA, Mohamed JY, Hijazi H, Prabakaran S, Tacke M (2013) Mutations in LRPAP1 are associated with severe myopia in humans. Am J Hum Genet 93:313–320
Alkuraya FS (2012) Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr Protoc Hum Genet: 6.12. 1–6.12. 13
Alkuraya FS (2013) The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Hum Genet 132:1197–1211
Alkuraya FS (2015) Human knockout research: new horizons and opportunities. Trends Genet 31:108–115
Alsalem AB, Halees AS, Anazi S, Alshamekh S, Alkuraya FS (2013) Autozygome sequencing expands the horizon of human knockout research and provides novel insights into human phenotypic variation. PLoS Genet 9:e1004030
Al-Salem A, Alshammari MJ, Hassan H, Alazami AM, Alkuraya FS (2013) Weaver syndrome and defective cortical development: a rare association. Am J Med Genet Part A 161:225–227
Anazi S, Al-Sabban E, Alkuraya F (2014) Gonadal mosaicism as a rare cause of autosomal recessive inheritance. Clin Genet 85:278–281
Antonarakis SE, Beckmann JS (2006) Mendelian disorders deserve more attention. Nat Rev Genet 7:277–282
Antonarakis SE, Chakravarti A, Cohen JC, Hardy J (2010) Mendelian disorders and multifactorial traits: the big divide or one for all? Nat Rev Genet 11:380–384
Bohlega S, Alazami A, Cupler E, Al-Hindi H, Ibrahim E, Alkuraya F (2011) A novel syndromic form of sensory-motor polyneuropathy is linked to chromosome 22q13. 31–q13. 33. Clin Genet 79:193–195
Brinkman RR, Dubé M-P, Rouleau GA, Orr AC, Samuels ME (2006) Human monogenic disorders—a source of novel drug targets. Nat Rev Genet 7:249–260
Bühling F, Kouadio M, Chwieralski CE, Kern U, Hohlfeld JM, Klemm N, Friedrichs N, Roth W, Deussing JM, Peters C (2011) Gene targeting of the cysteine peptidase cathepsin H impairs lung surfactant in mice. PLoS One 6(10):e26247
Campbell IM, Gambin T, Jhangiani SN, Grove ML, Veeraraghavan N, Muzny DM, Shaw CA, Gibbs RA, Boerwinkle E, Yu F (2015) Multiallelic positions in the human genome: challenges for genetic analyses. Hum Mutat 37(3):231–234
Chong JX, Yu J-H, Lorentzen P, Park KM, Jamal SM, Tabor HK, Rauch A, Saenz MS, Boltshauser E, Patterson KE, Nickerson DA, Bamshad MJ (2015) Gene discovery for Mendelian conditions via social networking: de novo variants in KDM1A cause developmental delay and distinctive facial features. Genet Med. doi:10.1038/gim.2015.161
Cirulli ET, Goldstein DB (2010) Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet 11:415–425
Elsea SH, Lucas RE (2002) The mousetrap: what we can learn when the mouse model does not mimic the human disease. ILAR J 43:66–79
Gai X, Ghezzi D, Johnson MA, Biagosch CA, Shamseldin HE, Haack TB, Reyes A, Tsukikawa M, Sheldon CA, Srinivasan S (2013) Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet 93:482–495
Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, Kwint M, Janssen IM, Hoischen A, Schenck A (2014) Genome sequencing identifies major causes of severe intellectual disability. Nature 511(7509):344–347
Group SM (2015) Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome Biol 16:134
Hafner C, van Oers JM, Vogt T, Landthaler M, Stoehr R, Blaszyk H, Hofstaedter F, Zwarthoff EC, Hartmann A (2006) Mosaicism of activatingFGFR3 mutations in human skin causes epidermal nevi. J Clin Investig 116:2201
Huisman SA, Redeker EJ, Maas SM, Mannens MM, Hennekam RC (2013) High rate of mosaicism in individuals with Cornelia de Lange syndrome. J Med Genet 50:339–344
Indjeian VB, Kingman GA, Jones FC, Guenther CA, Grimwood J, Schmutz J, Myers RM, Kingsley DM (2016) Evolving new skeletal traits by cis-regulatory changes in bone morphogenetic proteins. Cell 164(1–2):45–56
Jamuar SS, Lam A-TN, Kircher M, D’Gama AM, Wang J, Barry BJ, Zhang X, Hill RS, Partlow JN, Rozzo A (2014) Somatic mutations in cerebral cortical malformations. N Engl J Med 371:733–743
Jordan DM, Frangakis SG, Golzio C, Cassa CA, Kurtzberg J, Davis EE, Sunyaev SR, Katsanis N (2015) Identification of cis-suppression of human disease mutations by comparative genomics. Nature 524:225–229
Kim J-I, Ju YS, Park H, Kim S, Lee S, Yi J-H, Mudge J, Miller NA, Hong D, Bell CJ (2009) A highly annotated whole-genome sequence of a Korean individual. Nature 460:1011–1015
Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A (2012) Rate of de novo mutations and the importance of father/’s age to disease risk. Nature 488:471–475
Lambertson KF, Damiani SA, Might M, Shelton R, Terry SF (2015) Participant-driven matchmaking in the genomic era. Hum Mutat 36:965–973
Lupski JR (2013) Genome mosaicism-one human, multiple genomes. Science 341:358–359
Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA (2010) Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. N Engl J Med 362:1181–1191
MacArthur D, Manolio T, Dimmock D, Rehm H, Shendure J, Abecasis G, Adams D, Altman R, Antonarakis S, Ashley E (2014) Guidelines for investigating causality of sequence variants in human disease. Nature 508:469–476
Marotta CA, Wilson JT, Forget BG, Weissman SM (1977) Human beta-globin messenger RNA. III. Nucleotide sequences derived from complementary DNA. J Biol Chem 252:5040–5053
Might M, Wilsey M (2014) The shifting model in clinical diagnostics: how next-generation sequencing and families are altering the way rare diseases are discovered, studied, and treated. Genet Med 16(10):736–737
Nakhro K, Park J-M, Hong YB, Park JH, Nam SH, Yoon BR, Yoo JH, Koo H, Jung S-C, Kim H-L (2013) SET binding factor 1 (SBF1) mutation causes Charcot-Marie-Tooth disease type 4B3. Neurology 81:165–173
Pauling L, Itano HA, Singer SJ, Wells IC (1949) Sickle cell anemia, a molecular disease. Science 110:543–548
Philippakis AA, Azzariti DR, Beltran S, Brookes AJ, Brownstein CA, Brudno M, Brunner HG, Buske OJ, Carey K, Doll C (2015) The matchmaker exchange: a platform for rare disease gene discovery. Hum Mutat 36:915–921
Ronchi D, Di Fonzo A, Lin W, Bordoni A, Liu C, Fassone E, Pagliarani S, Rizzuti M, Zheng L, Filosto M (2013) Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am J Hum Genet 92:293–300
Sewairi W, Assiri A, Patel N, Alhashem A, Alkuraya FS (2016) Distal acroosteolysis, poikiloderma and joint stiffness: a novel laminopathy&quest. Eur J Hum Genet
Shaheen R, Alazami AM, Alshammari MJ, Faqeih E, Alhashmi N, Mousa N, Alsinani A, Ansari S, Alzahrani F, Al-Owain M (2012) Study of autosomal recessive osteogenesis imperfecta in Arabia reveals a novel locus defined by TMEM38B mutation. J Med Genet 49:630–635
Shaheen R, Aglan M, Keppler-Noreuil K, Faqeih E, Ansari S, Horton K, Ashour A, Zaki MS, Al-Zahrani F, Cueto-González AM (2013a) Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome. Am J Hum Genet 92:598–604
Shaheen R, Ansari S, Alshammari MJ, Alkhalidi H, Alrukban H, Eyaid W, Alkuraya FS (2013b) A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency. J Med Genet 50:431–436
Shaheen R, Faqeih E, Ansari S, Abdel-Salam G, Al-Hassnan ZN, Al-Shidi T, Alomar R, Sogaty S, Alkuraya FS (2014a) Genomic analysis of primordial dwarfism reveals novel disease genes. Genome Res 24:291–299
Shaheen R, Rahbeeni Z, Alhashem A, Faqeih E, Zhao Q, Xiong Y, Almoisheer A, Al-Qattan SM, Almadani HA, Al-Onazi N (2014b) Neu-Laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH. Am J Hum Genet 94:898–904
Shaheen R, Patel N, Shamseldin H, Alzahrani F, Al-Yamany R, Al A, Ewida N, Anazi S, Alnemer M, Elsheikh M, Alfaleh K, Alshammari M, Alhashem A, Alangari AA, Salih MA, Kircher M, Daza RM, Ibrahim N, Wakil SM, Alaqeel A, Altowaijri I, Shendure J, Al-Habib A, Faqieh E, Alkuraya FS (2015) Accelerating matchmaking of novel dysmorphology syndromes through clinical and genomic characterization of a large cohort. Genet Med. doi:10.1038/gim.2015.147
Shamseldin HE, Alshammari M, Al-Sheddi T, Salih MA, Alkhalidi H, Kentab A, Repetto GM, Hashem M, Alkuraya FS (2012a) Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J Med Genet 49:234–241
Shamseldin HE, Swaid A, Alkuraya FS (2012b) Lifting the lid on unborn lethal Mendelian phenotypes through exome sequencing. Genet Med 15:307–309
Shamseldin H, Alazami AM, Manning M, Hashem A, Caluseiu O, Tabarki B, Esplin E, Schelley S, Innes AM, Parboosingh JS (2015a) RTTN mutations cause primary microcephaly and primordial Dwarfism in humans. Am J Hum Genet 97:862–868
Shamseldin HE, Tulbah M, Kurdi W, Nemer M, Alsahan N, Al Mardawi E, Khalifa O, Hashem A, Kurdi A, Babay Z (2015b) Identification of embryonic lethal genes in humans by autozygosity mapping and exome sequencing in consanguineous families. Genome Biol 16:116
Shamseldin HE, Faqeih E, Alasmari A, Zaki MS, Gleeson JG, Alkuraya FS (2015b) Mutations in UNC80, Encoding Part of the UNC79-UNC80-NALCN Channel Complex, Cause Autosomal-Recessive Severe Infantile Encephalopathy. Am J Hum Genet
Shamseldin HE, Bennett AH, Alfadhel M, Gupta V, Alkuraya FS (2016) GOLGA2, encoding a master regulator of golgi apparatus, is mutated in a patient with a neuromuscular disorder. Hum Genet 135:245–251
Sobreira N, Schiettecatte F, Valle D, Hamosh A (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 36:928–930
Veltman JA, Brunner HG (2012) De novo mutations in human genetic disease. Nat Rev Genet 13:565–575
Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M (2010) A de novo paradigm for mental retardation. Nat Genet 42:1109–1112
Wieczorek D, Newman WG, Wieland T, Berulava T, Kaffe M, Falkenstein D, Beetz C, Graf E, Schwarzmayr T, Douzgou S (2014) Compound heterozygosity of low-frequency promoter deletions and rare loss-of-function mutations in TXNL4A causes Burn-McKeown syndrome. Am J Hum Genet 95:698–707
Yavarna T, Al-Dewik N, Al-Mureikhi M, Ali R, Al-Mesaifri F, Mahmoud L, Shahbeck N, Lakhani S, AlMulla M, Nawaz Z (2015) High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum Genet 134:967–980
Acknowledgments
I thank Ranad Shaheen and Hanan E Shamseldin for their critical review of this manuscript. FSA was supported by a grant from King Salman Center for Disability Research and a grant from King Abdulaziz City for Science and Technology (13-BIO1113-20).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest.
Rights and permissions
About this article

Cite this article
Alkuraya, F.S. Discovery of mutations for Mendelian disorders. Hum Genet 135, 615–623 (2016). https://doi.org/10.1007/s00439-016-1664-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-016-1664-8