
Abstract
Through linkage and tagSNP-based association studies in 100 Dominican Republic (DR) families, we previously identified ANLN and AOAH (7p14.3) as candidate genes for carotid intima-media thickness at bifurcation (bIMT). Introns, exons, and flanking regions of ANLN and AOAH were re-sequenced in 151 individuals from nine families with evidence for linkage at 7p14.3. For common variants [CV, minor allele frequency (MAF) ≥5 %], single variant-based analysis was performed. For rare variants (RV, MAF < 5 %), gene-based analysis aggregating all RVs within a gene was performed. CV analysis revealed the strongest signal at rs3815483 (P = 0.0003) in ANLN and rs60023210 (P = 0.00005) in AOAH. In ANLN, RV analysis found suggestive evidence for association with exonic RVs (P = 0.08), and in particular non-synonymous RVs (P = 0.04) but not with all RVs (P = 0.15). The variant alleles of all non-synonymous RVs segregated with the major allele of rs3815483 and were associated with lower bIMT while a novel synonymous RV segregated with the minor allele of rs3815483 and was associated with greater bIMT. Additional analysis in 561 DR individuals found suggestive evidence for association with all ANLN non-synonymous RVs (P = 0.08). In AOAH, no evidence for association with RVs was detected. Instead, conditional analysis revealed that multiple independent intronic CVs are associated with bIMT in addition to rs60023210. We demonstrate the utility of using family-based studies to evaluate the contribution of RVs. Our data suggest two modes of genetic architecture underlying the linkage and association at ANLN (multiple exonic RVs) and AOAH (multiple intronic CVs with uncharacterized functions).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genome-wide association studies (GWAS) and organized consortium efforts have identified multiple susceptibility loci for vascular diseases, such as coronary artery disease and stroke, the leading causes of death and morbidity in the United States (CARDIoGRAMplusC4D Consortium et al. 2013; Bis et al. 2011). The associated single nucleotide polymorphisms (SNPs), however, explain only a small portion of the heritability estimated for these disabling diseases (Makinen et al. 2014). Given the extreme complexity of genetic and environmental contributions to vascular diseases, evaluation of the precursor phenotypes may reduce phenotypic heterogeneity and increase statistical power in genetic studies. As a result, this approach may facilitate identifying loci that were missed in previous GWAS focusing on the clinical manifestation of the diseases. Carotid intima-media thickness (cIMT) is an excellent phenotype for this approach because (1) cIMT is a subclinical measure of atherosclerosis, a leading cause of vascular diseases; (2) cIMT is a noninvasive, relatively inexpensive, highly reproducible measurement; (3) cIMT is a quantitative measure which provides additional statistical power; and (4) cIMT has a strong genetic contribution with estimated heritability ranging from 0.3 to 0.6 in various populations (Lange et al. 2002; Xiang et al. 2002; Fox et al. 2003; Juo et al. 2004).
Several large-scale genetic studies of cIMT have been reported. The first GWAS on cIMT was conducted in the Framingham Heart Study with no SNP meeting criteria for genome-wide significance, probably due to the relatively small sample size (O’Donnell et al. 2007). Subsequently, the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium organized a meta-analysis of 16 GWAS on cIMT (Bis et al. 2011). The combined meta-analysis from two stages included 42,484 participants of European ancestry. In the analysis of common cIMT, three independent loci, ZHX2, APOC1, and PINX, met the genome-wide significant criteria (P < 5 × 10−8). In addition, suggestive evidence for association was found with SLC17A4 (P = 7.8 × 10−8). No SNP was found to be genome-wide significant for association with internal cIMT. A large-scale candidate gene study using Illumina 200 K CardioMetabochip to interrogate hundreds of candidate genes simultaneously found that rs4888378 in the BCAR1-CFDP1-TMEM170A locus was associated with cIMT as well as coronary artery disease risk in multiple European cohorts (Gertow et al. 2012). In 2013, the first GWAS on a non-European population was described in Mexican Americans without any SNP reaching genome-wide significance (Melton et al. 2013).
In contrast to the above cohort studies, we have pursued a family-based approach combining linkage and association to map genetic determinants of cIMT in Caribbean Hispanics, an understudied population. The family-based design is more robust against population stratification and is less confounded by gene–environment interactions as family members are more likely to have similar environmental exposures. Indeed, our group has had notable success in using this approach to map susceptibility genes for cIMT. First, we mapped a quantitative trait locus (QTL) for cIMT at bifurcation (bIMT) to chromosome 7p using 100 extended Dominican Republic (DR) families in a genome-wide linkage study (Sacco et al. 2009). We then identified ANLN and AOAH as candidate genes in a peak-wide association study using tagSNPs to survey common variants within the QTL (Wang et al. 2012). The association between bIMT and these two genes was highly significant in 53 families with positive family-specific LOD scores (P = 0.0004 and 0.0002, respectively, for the top variant in each gene) but not in the 47 families with negative family-specific LOD scores (P = 0.13 and 0.41, respectively). This finding suggests that the genetic effects of these two genes are restricted to a specific subset of the families.
In previous studies, rare variants (RVs) have been understudied. With the rapid growth of next generation sequencing (NGS) technology, there are increasing interests in examining the contributions of RVs to complex diseases, such as vascular diseases. However, due to the extreme low frequency of RVs in the population, case–control studies require large sample sizes to achieve sufficient statistical power. Since related individuals will share RVs, a family-based study is a more powerful method to evaluate the contribution of RVs to the phenotype of interest. In an attempt to fully delineate the genetic architecture underlying the association at ANLN and AOAH, we carried out a targeted re-sequencing study to survey the genomic regions of the two genes, including exons, introns, and flanking regions, in nine DR families from the original linkage analysis that have a family-specific LOD score greater than 0.1 at the chromosome 7p QTL.
Materials and methods
Participating subjects
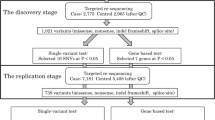
The design of our Family Study of Stroke Risk and Carotid Atherosclerosis has been described in detail previously (Sacco et al. 2009, 2007). Among the 100 enrolled Dominican families, 9 families with a family-specific LOD score greater than 0.1 at the chromosome 7p QTL for bifurcation bIMT were selected for this study (Table 1). In brief, the family study was derived from the Northern Manhattan Study (NOMAS) (Sacco et al. 2004). NOMAS participants had never been diagnosed with a stroke, were at least 40 years of age, and resided for at least 3 months in a household with a telephone in Northern Manhattan. Probands for the family study were selected from Caribbean Hispanic members of NOMAS with high risk for cardiovascular disease. Families were enrolled if the proband was able to provide a family history, obtain the family members’ permission for the research staff to contact them, and had at least three first-degree relatives able to participate. The probands were initially identified in Northern Manhattan and family members were enrolled in New York at Columbia University as well as in the Dominican Republic at the Clinicas Corazones Unidos in Santo Domingo. In total, 561 DR subjects in the population-based NOMAS who have bIMT measured and are not probands in the family study were used as an independent dataset. Demographic, socioeconomic, and risk factor data were collected through direct interview based on the NOMAS instruments (Elkind et al. 2006). All subjects provided informed consent and the study was approved by the Institutional Review Boards of Columbia University, University of Miami, the National Bioethics Committee, and the Independent Ethics Committee of Instituto Oncologico Regional del Cibao in the DR.
Carotid and associated risk factor measurements
All family members had high-resolution B-mode ultrasound measurement of cIMT. Carotid ultrasound was performed according to the standard scanning and reading protocols by a trained and certified sonologist as detailed previously (Rundek et al. 2002). IMT measurements were performed outside the areas of plaque as recommended by consensus documents (Touboul et al. 2007). IMT was measured using an automated computerized edge tracking software M’Ath (Intelligence in Medical Technologies, Inc., Paris, France) from the recorded ultrasound clips which improves precision and reduces variance of the measurements. (Sacco et al. 2004). The IMT protocols yield measurements of the distance between lumen-intima and media-adventitia ultrasound echoes. Bifurcation cIMT (bIMT) was defined as the near and the far wall IMT of the carotid bifurcation beginning at the tip of the flow divider and extending 10 mm proximal to the flow divider tip. Our bIMT measurements have excellent consistency: inter-reader reliability between two readers was demonstrated with a mean absolute difference in IMT of 0.11 ± 0.09 mm, variation coefficient 5.5 %, correlation coefficient 0.87, and the percent error 6.7 %; intra-reader mean absolute IMT difference was 0.07 ± 0.04 mm, variation coefficient 5.4 %, correlation coefficient 0.94, and the percent error 5.6 % (Sacco et al. 2004).
Sequencing and variant annotation
Introns, exons, and promoters of ANLN and AOAH were captured with a customized Agilent SureSelect Enrichment kit targeting 7 Mb sequence. DNA libraries with 24-sample barcoding were sequenced on Illumina HiSeq 2000 with pair-end sequencing at 100 base pair reading length. On average, each DNA sample generated about 15 million raw reads. The raw sequencing reads were aligned to the human reference sequence hg19 with the Burrows–Wheeler Aligner (BWA) (Li and Durbin 2010). About 14 million reads per sample passed filtering and were aligned to the target regions. The read duplication rate is 2.4 % on average, suggesting good library complexity. These reads yielded high-depth sequencing data: the mean target coverage is ~ 100X with a minimum of 68X. These high-depth sequencing data allowed us to make reliable genotype calls for downstream statistical analyses. At a depth of 8, 97 % of the target sequences were covered. Subsequent variant calling was done with the Genome Analysis ToolKit (GATK). Potential functions of variants were annotated using ANNOVAR (http://annovar.openbioinformatics.org/), SeattleSeq (http://snp.gs.washington.edu/SeattleSeqAnnotation138/index.jsp), and RegulomeDB (Boyle et al. 2012). In brief, we used three programs to annotate variants: ANNOVAR (gene-based annotation), SeattleSeq, and RegulomeDB. These programs, for a large part, complement each other on annotation features. The ANNOVAR annotates whether a variant hit exons, introns, UTRs, non-coding RNAs (ncRNAs), or intergenic regions, and the name of gene (if the variant is within the gene) or neighbor genes (if the variant is in intergenic regions). SeattleSeq provides additional information on the conservation among species, amino acid changes for coding variants, and potential damage caused by the amino acid changes. In contrast, RegulomeDB has an emphasis on annotating non-coding variants for regulatory potential. It integrates a large collection of genome-wide regulatory information (mainly produced by ENCODE) to predict functional impact of variants, in addition to manual curation of literature sources on low-throughput but high-quality assays, such as luciferase reporter and EMSA. The genome-wide information includes (1) e-QTLs; (2) chromatin state information; and (3) important regulatory factor binding sites. RegulomeDB scores variants into six categories 1–6, with category 1 and 2 having sub-category (1a–1f and 2a–2c). Lower scores indicate stronger evidence for a variant to be located in a functional region. All novel exonic variants were validated with Sanger sequencing.
Quality control
Quality control was conducted at both variant and sample levels. At the variant level, during Variant Quality Score Recalibration (VQSR) process, a VQSLOD score was assigned to each variant. Variants with VQSLOD <−4 were removed. Within each individual sample, variants with a depth <4 or phred-like (PL) score <100 were set as missing. Variants with call rate <75 % were also removed from further analysis. At the sample level, concordance between the sequencing data and genotypes from the previous peak-wide association study was assessed for each sample. Two samples with low concordance (<95 %) were removed. For the remaining samples, pedigree structure was confirmed using the graphical relationship representation software. Mendelian error checking was performed and Mendelian errors were set to missing for all the variants called using PLATO (Grady et al. 2010).
Genotyping
To validate the RV finding in additional samples, the Illumina Exome Core chip with custom content was used to genotype additional families and NOMAS individuals as validation. This approach provides an overview of potentially functional variants at low MAF at a relative cheap cost per sample. The chip includes 240,000 pre-designed, commercially available exonic SNVs and 6000 exonic SNVs found during our re-sequencing effort in families. The selecting criteria of 6000 exonic SNVs are: (1) not included in the Exome Core chip; (2) passed sequencing QCs in at least three individuals; (3) cannot be effectively imputed using GWAS markers (imputation quality score <0.8); (4) has VQSLOD >−4; (5) has Illumina Infinium® design score >=0.6. The genotyping protocol and quality control have been described previously (Wang et al. 2012).
Statistical analysis
Bifurcation bIMT was natural log transformed to ensure a normal distribution as in the previous analyses (Sacco et al. 2009; Wang et al. 2012). An initial polygenic model was set up to include the same covariates as identified by a selection screen in the previous linkage and peak-wide association analyses: age, age2, sex, age × sex, pack years of smoking, waist hip ratio, and body mass index. We employed different analytical approaches to analyze common and rare variants. The same covariates were used in both the common and rare variant analysis. Common and rare variants were defined based on estimated frequencies from a population-based cohort of 600 Dominicans from NOMAS. The NOMAS sample was genotyped on the Affymetrix 6.0 genotyping array and then imputed using IMPUTE2 with the 1000 genomes phase 1, version 3 reference panel (Howie et al. 2009). Variants were classified as common if they had a minor allele frequency (MAF) ≥5 % in NOMAS Dominicans and classified as rare if they had MAF <5 % or could not be imputed efficiently (INFO ≤ 0.4) in NOMAS Dominicans.
For CVs, we tested each individual variant for association with bIMT using the quantitative transmission disequilibrium test (QTDT) in SOLAR (Almasy and Blangero 1998). The QTDT test was used for analyzing CVs because it was designed to evaluate association in the presence of linkage without inflating the type I error. In addition, the QTDT test is robust against population stratification and uses data from all available relatives in a pedigree, which makes it more powerful in analyzing data sets with missing parental data. The linkage disequilibrium (LD) between the CVs was estimated using the NOMAS Dominican sample. To determine if the effect of significant variants identified by QTDT were independent of each other, we performed a forward, stepwise QTDT conditional analysis. In short, a primary SNP was included as a covariate, and the QTDT analysis was carried out for all remaining SNPs. A likelihood ratio test was used to determine if any of the remaining SNPs in this conditioned analysis improved the fit of the model beyond the contribution of the primary SNP. The process was repeated until there was no additional SNP with a likelihood ratio test p value less than 0.01.
For RVs, we performed gene-based analyses using the Family SNP-set (Sequence) Kernel Association Test (Fam-SKAT) (Chen et al. 2013). Analyses were performed using three different filtering algorithms within each gene: all rare variants, exonic rare variants only, and non-synonymous rare variants only. For the NOMAS dataset, regular SKAT was used to evaluate non-synonymous rare variants in each gene for association with bIMT. To better visualize the distribution of these rare variants in relation to bIMT, both across and within families, we computed the residual bifurcation after adjusting for the associated risk factors (age, age2, sex, pack years of smoking, BMI, and WHR) using SAS. Haplotype phasing of exonic rare variants with the most strongly associated common variants was done with SimWalk2 (Sobel and Lange 1996).
Results
Overview of single nucleotide variants (SNVs) identified by re-sequencing
In total, we analyzed 149 individuals from the nine DR families with a family-specific LOD score >0.1 (ranging from 0.11 to 0.81), at the chromosome 7 QTL for bIMT (Table 1). We observed 1431 biallelic SNVs in this sample set. Of all the variants, 12.6 % (181 variants) were novel (i.e., not found in public databases) and 64.5 % (923 variants) were rare variants based on imputation in the NOMAS cohort (Table 2). The majority (97 %) of the variants were found in the introns and flanking regions. The genomic size (including 5 kb flanking regions on each side) for ANLN and AOAH were 74 and 222 kb, respectively. Consistent with a larger genomic size for AOAH, we found three times the number of novel intronic variants in AOAH and four times the number of total intronic variants in AOAH compared to ANLN. Despite a larger genomic size, AOAH had a smaller mRNA size (2559 bp) than ANLN (4780 bp). There was a 573-bp-long spliced non-coding RNA residing within a large intron of AOAH. Within the AOAH region, we observed 1 novel and 21 known exonic variants: 15 were located within the AOAH coding region, 4 were located within the AOAH untranslated regions (UTRs), and 3 were located within the non-coding RNA. Within ANLN, we observed 4 novel and 18 known exonic variants: 16 were located in the coding region and 6 were located in the UTRs.
Common variant analysis
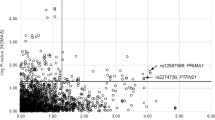
Single variant analysis was carried out for CVs. Figure 1 shows the regional association plot for CVs. In ANLN, the strongest association was found with rs3815483 (P = 0.0003), which was the top marker in the previous peak-wide association analysis (Wang et al. 2012). In AOAH, the strongest association was found at rs60023210 (P = 0.00005), which was in low LD (r 2 = 0.03; D′ = 0.25) with the top marker (rs2001600) identified in the previous study. Both SNPs remained significant (P < 0.05) after Bonferroni correction for CVs tested within each gene. There was limited LD between ANLN and AOAH, as evidenced by both the small r 2 values between CVs in ANLN and rs60023210 in AOAH, and the high recombination rate between the two genes (Fig. 1).

Regional association plot of common variants in ANLN and AOAH. Each point represents a common variants (with MAF >5 % in the NOMAS population) in ANLN and AOAH genomic region. The circles depict the P values of the single point QTDT analysis on a −log10 scale as a function of genomic position (NCBI Build 37). The most significant marker in the region (rs60023210, located within AOAH) is displayed as a purple diamond and the most significant marker located within ANLN (rs3815483) is labeled. The color of the circles indicates LD (estimated by r 2 values in the NOMAS cohort) between each SNP and rs600023210. The line displays the recombination rates estimated from Phase II HapMap. The structure of genes in the region is displayed at the bottom of the plot: each vertical bar represents an exon and horizontal arrow above each transcript depicts the transcription direction of the gene (color figure online)
To infer the functional impact of CVs, we used ANNOVAR, SeattleSeq, and RegulomeDB to annotate CVs with evidence for association (nominal P ≤ 0.05). Both rs3815483 and rs60023210 are intronic SNPs with no predicated function. In ANLN, three CVs with evidence for association are exonic variants: two were located within 3′UTR, and one was a synonymous variant. None of these exonic variants was in strong LD with rs3815483 (r 2 ≤ 0.005) and therefore was unlikely to account for the association observed at rs3815483. An intronic SNP rs3801306 in ANLN had a RegulomeDB score of 2b (transcription factor ARID3A binding in Hep2G cells; CTCF binding in K562 and HelaS3 cells), suggesting a regulatory potential. This SNP was moderately correlated with rs3815483 (r 2 = 0.43) and therefore could partially contribute to the association signal at rs3815483. In AOAH, no CV with evidence for association had an annotated functional impact.
Rare variant analysis
Gene-based tests were carried out to aggregate RVs in ANLN and AOAH separately. To investigate if RVs with functional implications were more likely to confer genetic association, RV analysis was done with three different filtering algorithms: all RVs, all exonic RVs, all non-synonymous RVs. These analyses revealed that exonic, especially non-synonymous RVs, in ANLN collectively contributed to bIMT (P = 0.04, Table 3). No evidence for association between RVs and bIMT was found for AOAH. In total, three non-synonymous RV in ANLN were observed in our data set: all were missense variants. Two of them, rs34285732 and rs138687533, have been observed only in African Americans in the 1000 genomes project (MAF = 1.5 and 0.2 %, respectively). The third missense variant was a novel one: Chr7_36463477. It was well conserved (PhastCon = 1, GERP = 4.6) and was predicted to be possibly damaging (Table 4). These missense RVs were carried within two families: F803 and F3630. In family F803, rs34285732 and rs138687533 occurred 3 times each in five individuals, with one compound heterozygote. Carriers of the rare allele of either rs34285732 or rs138687533 had lower average residual bIMT in F803; the compound heterozygote had the lowest residual bIMT (Fig. 2). In F3630, the novel Chr7_36463477 was observed only once, in individual 204, who had the lowest residual bIMT in the family (Fig. 2). All three rare missense variants segregated with the major allele of rs3815483. In addition, in F803, a novel synonymous RV (Chr7_36450217) occurred on the haplotype defined by the minor allele of rs3815483. Both the minor allele of Chr7_36450217 and rs3815483 were associated with higher residual bIMT (Fig. 3).

Correlation between three rare missense variants in ANLN and bIMT. In total, three rare missense variants were observed in two families (F803 and F3630). FAM-SKAT analysis suggested that the three missense variants collectively contribute to bIMT variations. To infer the direction of the association, we examined residual bIMT after adjusting for other risk factors (age, age squared, sex, and pack years of smoking, BMI, and WHR) in individuals carrying different numbers of the rare missense alleles. Twenty-six individuals from the two families were divided into three groups based on the number of rare allele they carry for the three missense variants: 20 individuals do not carry rare allele of any missense variants, 5 individuals carry one rare allele of any of the missense variants, and 1 individual carries a compound heterozygote of two rare alleles. The bars represent the mean residual bIMT values (after adjusting for age, age2, sex, pack years of smoking, BMI, and WHI) of each group and the error bars represent the standard error of residual bIMT values. The results suggest that the rare allele of the missense variants is associated with lower bIMT

Pedigree of family F803 depicting bIMT and a novel synonymous rare variant in ANLN. Residual bIMT values and the haplotype in ANLN defined by rs3815483 and a novel variant on chromosome 7 at base pair position 36,450,217 were displayed for each individual. Individuals with residual bifurcation bIMT value <0 are shaded black and individuals with residual bifurcation bIMT value ≥0 are shaded gray
Validation of rare variant analysis in additional datasets
To further test if RVs in ANLN and AOAH collectively contribute to bIMT variation, we used the Illumina Exome Core chip with custom content to genotype additional families and NOMAS individuals. With the custom content, the chip includes 29 exonic variants at low MAF in the two genes (14 in ANLN and 15 in AOAH). All exonic RVs or all non-synonymous RVs in each gene were analyzed in the validation families using FAM-SKAT and in the NOMAS using SKAT. In AOAH, no evidence for association was found in any analysis. In ANLN, a smaller number of RVs (5 exonic variants and one non-synonymous variant) were found in the validation families and gene-based tests did not reveal any significant results in this dataset. In the NOMAS cohort, however, suggestive evidence for association was found for exonic RVs (11 variants, P = 0.04), and non-synonymous RVs (six variants, P = 0.08) (Table 3), supporting the findings in the discovery family data set.
Population-rare, family-common variants
Some variants that were rare in the population were enriched in the nine DR families studied, providing statistical power to evaluate them individually in our family data set. To test the contribution of these variants to bIMT, we analyzed the population-rare (MAF <5 % or cannot be efficiently imputed in NOMAS), family-common (MAF >5 % in the family dataset and observed in more than one family) variants for association with bIMT using the single variant analysis QTDT. A total of 15 ANLN variants and 142 AOAH variants were included in this analysis (Supplementary Table). The most significant associations were found with rs6954831 in ANLN (P = 0.0006) and rs2727794 (P = 0.0003) in AOAH (Table 5) The estimated MAF of each of these SNPs in the NOMAS cohort is ~2 %. According to the 1000 genome data, rs6954831 is only polymorphic in Caucasians (MAF = 5 %) and rs2727794 is only polymorphic in African Americans (MAF = 16 %). We observed both variants in our admixed Hispanic population: rs6954831 is observed in five families with 26 carriers and rs2727794 is observed in all nine families with 43 carriers. Both variants are located within introns without predicated functional consequences.
To examine if multiple independent “family-common” variants in the chr7 QTL contribute to bIMT, we performed a stepwise conditional analysis using all “family-common” variants identified in the single variant analysis and starting with the most significant variant, i.e., rs60023210. This analysis revealed that rs2727794 (QTDT P = 0.0003 after conditioning on rs60023210, likelihood ratio P = 0.001) and rs11971903 (conditional QTDT P = 0.0004 after conditioning on rs60023210 and rs2727794, likelihood ratio P = 0.002) were independently associated with bIMT. After conditioning on rs60023210, rs2727794, and rs11971903, no more variants remained significant in the likelihood ratio test (P > 0.01). All three variants are intronic variants in AOAH with no obvious evidence for regulatory functions.
Discussion
We have previously identified ANLN and AOAH as candidate genes for bIMT in Dominicans using a tagSNP-based approach that focused on CVs in the human genome. In the current study, we sought to further characterize the genetic basis of bIMT variations at these loci using next generation sequencing (NGS) technology to re-sequence the genomic region of the two genes. The thorough examination of variants with a full spectrum of frequencies allowed us to evaluate whether RVs, which were not captured in previous studies, contribute to bIMT. In addition, by cataloging all SNVs in the genomic region of the two genes, this approach allowed us to systematically search for functional variants underlying the association at the top markers in previous studies.
In ANLN, the strongest evidence for association was found to rs3815483, the same top marker in the previous study (Wang et al. 2012). We found no evidence supporting a regulatory function for this intronic SNP. Interestingly, the RV analysis revealed that multiple rare exonic variants aggregated on haplotypes defined by rs3815483, which might explain some of the association. These rare exonic variants included three missense variants that were associated with lower bIMT, and one novel synonymous variant that was associated with higher bIMT. The functional importance of the novel synonymous variant needs to be further examined in future studies. Potential functions for a synonymous variant include affecting splicing and translation efficiency of RNA. Analysis in the population-based NOMAS samples supported that rare exonic variants in ANLN collectively contribute to bIMT variation. In addition to the rare exonic variants, we found that a relatively common intronic SNP, rs3801306 (MAF = 11 %) with functional potential was in modest LD with rs3815483. The sequence surrounding this SNP has been identified as a transcription factor binding site for ARID3A in Hep2G cells and for CTCF in K562 and HelaS3 cells. The regulatory potential of rs381306 and the LD between it and rs3815483 provides another possible mechanism underlying the association observed at rs3815483.
In AOAH, the strongest evidence for association was found to rs60023210, a different SNP than the top marker identified in the previous study (Wang et al. 2012). Unlike the genetic architecture in ANLN, we found no CVs or RVs with annotated functional effects that could account for the association signal at rs60023210 in AOAH. Instead, our analysis revealed multiple independent common intronic variants with no predicted function to be associated with bIMT. It is unclear how these CVs confer the genetic association with bIMT. It is possible that the sequence variations at these CVs are involved in regulatory mechanisms that have not been characterized.
Despite the success of GWAS in many complex diseases, it has been increasingly recognized that the identified variants in GWAS only account for a small fraction of the phenotypic variation, which is referred as the “missing heritability”. Given that GWAS is designed to interrogate CVs, the underrepresented RVs are believed to account for some of the “missing heritability” in GWAS (Eichler et al. 2010). For example, in this study, only one-third of variants observed have a MAF greater than 5 %, the targeted variants in GWAS. Indeed, many recent studies have focused on incorporating RVs in the genome-wide analysis using either exome-chip or whole exome sequencing (WES) (Ratnapriya et al. 2014; SIGMA Type 2 Diabetes Consortium et al. 2014). One limitation of these studies is the lack of coverage of the non-exonic variants. Herein, we have performed a targeted sequencing study using a custom capture design to re-sequence the genomic regions of candidate genes identified in previous studies, which allowed us to survey not only exonic variants but also non-exonic variants. Indeed, we found an intronic variant with regulatory potential that is in LD with the top marker, which provided a plausible mechanism for the genetic association observed in the previous study.
The CHARGE consortium has led to a substantial effort on targeted sequencing of candidate genes for bIMT and carotid plaque nominated in a previous GWAS meta-analysis (Bis et al. 2014). Over 370 kb of genomic region surrounding SLC17A4 (associated with bIMT) and PIK3CG (associated with plaque) was sequenced among 3669 participants with European ancestry that are part of the original GWAS conducted by CHARGE. The most significant CV association was found in CCDC71L (P = 0.0003 for bIMT and P = 0.0004 for plaque), which did not survive multiple testing correction. RV analysis found nominal evidence for association between all RVs in CCDC71L and bIMT (P = 0.05) but not plaque (P = 1.0). Interestingly, CCDC71L is upstream of PIK3CG, the gene associated with plaque but not bIMT in the original GWAS. Compared to the CHARGE-targeted sequencing study based on unrelated individuals, our study offers a unique advantage with the family-based study design. By selecting families that are genetically enriched for the trait, our family-based design provides additional power in evaluating the genetic contribution of RVs, which can be largely attributed to the enrichment of trait-related RVs within families. For example, the novel synonymous variant (Chr7_36450217), which would have an extremely low frequency in the general population, was carried by 6 individuals in family F803 and all of them had above average residual bIMT in the family. The multiple observations of the RV and segregation with the phenotype of interest within the family dramatically increased the chance to detect such genotype–phenotype correlation.
Genetic studies of complex diseases, including those on cardiovascular diseases, have been focused primarily on non-Hispanic whites. Our study of the Caribbean Hispanics provides essential data in this underrepresented population. As discussed above, the chr 7 QTL for bIMT identified in our linkage study was not covered in the re-sequencing study led by CHARGE. Therefore, we cannot evaluate the contribution of rare variants in the region in the Caucasian population and compare it to our findings.
Our next generation sequencing fine-mapping study supports the concept that both rare and common variants contribute to the genetic basis of subclinical atherosclerosis, as measured by bifurcation bIMT. Some of these variants have annotated functions, while others have no clear predicated function. Future studies are needed to validate and characterize the function of these variants in the context of atherogenesis.
References
Almasy L, Blangero J (1998) Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62:1198–1211
Bis JC, Kavousi M, Franceschini N, Isaacs A, Abecasis GR, Schminke U, Post WS, Smith AV, Cupples LA, Markus HS, Schmidt R, Huffman JE, Lehtimaki T, Baumert J, Munzel T, Heckbert SR, Dehghan A, North K, Oostra B, Bevan S, Stoegerer EM, Hayward C, Raitakari O, Meisinger C, Schillert A, Sanna S, Volzke H, Cheng YC, Thorsson B, Fox CS, Rice K, Rivadeneira F, Nambi V, Halperin E, Petrovic KE, Peltonen L, Wichmann HE, Schnabel RB, Dorr M, Parsa A, Aspelund T, Demissie S, Kathiresan S, Reilly MP, the CARDIoGRAM Consortium, Taylor K, Uitterlinden A, Couper DJ, Sitzer M, Kahonen M, Illig T, Wild PS, Orru M, Ludemann J, Shuldiner AR, Eiriksdottir G, White CC, Rotter JI, Hofman A, Seissler J, Zeller T, Usala G, Ernst F, Launer LJ, D’Agostino RBS, O’Leary DH, Ballantyne C, Thiery J, Ziegler A, Lakatta EG, Chilukoti RK, Harris TB, Wolf PA, Psaty BM, Polak JF, Li X, Rathmann W, Uda M, Boerwinkle E, Klopp N, Schmidt H, Wilson JF, Viikari J, Koenig W, Blankenberg S, Newman AB, Witteman J, Heiss G, Duijn CV, Scuteri A, Homuth G, Mitchell BD, Gudnason V, O’Donnell CJ (2011) Meta-analysis of genome-wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nat Genet. doi:10.1038/ng.920;10.1038/ng.920
Bis JC, White CC, Franceschini N, Brody J, Zhang X, Muzny D, Santibanez J, Gibbs R, Liu X, Lin H, Boerwinkle E, Psaty BM, North KE, Cupples LA, O’Donnell CJ, CHARGE Subclinical Atherosclerosis Working Group (2014) Sequencing of 2 subclinical atherosclerosis candidate regions in 3669 individuals: cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium Targeted Sequencing Study. Circ Cardiovasc Genet 7:359–364. doi:10.1161/CIRCGENETICS.113.000116
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, Cherry JM, Snyder M (2012) Annotation of functional variation in personal genomes using regulomeDB. Genome Res 22:1790–1797. doi:10.1101/gr.137323.112;10.1101/gr.137323.112
CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, Goldstein BA, Stirrups K, Konig IR, Cazier JB, Johansson A, Hall AS, Lee JY, Willer CJ, Chambers JC, Esko T, Folkersen L, Goel A, Grundberg E, Havulinna AS, Ho WK, Hopewell JC, Eriksson N, Kleber ME, Kristiansson K, Lundmark P, Lyytikainen LP, Rafelt S, Shungin D, Strawbridge RJ, Thorleifsson G, Tikkanen E, Van Zuydam N, Voight BF, Waite LL, Zhang W, Ziegler A, Absher D, Altshuler D, Balmforth AJ, Barroso I, Braund PS, Burgdorf C, Claudi-Boehm S, Cox D, Dimitriou M, Do R, DIAGRAM Consortium, CARDIOGENICS Consortium, Doney AS, El Mokhtari N, Eriksson P, Fischer K, Fontanillas P, Franco-Cereceda A, Gigante B, Groop L, Gustafsson S, Hager J, Hallmans G, Han BG, Hunt SE, Kang HM, Illig T, Kessler T, Knowles JW, Kolovou G, Kuusisto J, Langenberg C, Langford C, Leander K, Lokki ML, Lundmark A, Mccarthy MI, Meisinger C, Melander O, Mihailov E, Maouche S, Morris AD, Muller-Nurasyid M, Nikus K, Peden JF, Rayner NW, Rasheed A, Rosinger S, Rubin D, Rumpf MP, Schafer A, Sivananthan M, Song C, Stewart AF, Tan ST, Thorgeirsson G, van der Schoot CE, Wagner PJ, Wellcome Trust Case Control Consortium, Wells GA, Wild PS, Yang TP, Amouyel P, Arveiler D, Basart H, Boehnke M, Boerwinkle E, Brambilla P, Cambien F, Cupples AL, de Faire U, Dehghan A, Diemert P, Epstein SE, Evans A, Ferrario MM, Ferrieres J, Gauguier D, Go AS, Goodall AH, Gudnason V, Hazen SL, Holm H, Iribarren C, Jang Y, Kahonen M, Kee F, Kim HS, Klopp N, Koenig W, Kratzer W, Kuulasmaa K, Laakso M, Laaksonen R, Lee JY, Lind L, Ouwehand WH, Parish S, Park JE, Pedersen NL, Peters A, Quertermous T, Rader DJ, Salomaa V, Schadt E, Shah SH, Sinisalo J, Stark K, Stefansson K, Tregouet DA, Virtamo J, Wallentin L, Wareham N, Zimmermann ME, Nieminen MS, Hengstenberg C, Sandhu MS, Pastinen T, Syvanen AC, Hovingh GK, Dedoussis G, Franks PW, Lehtimaki T, Metspalu A, Zalloua PA, Siegbahn A, Schreiber S, Ripatti S, Blankenberg SS, Perola M, Clarke R, Boehm BO, O’Donnell C, Reilly MP, Marz W, Collins R, Kathiresan S, Hamsten A, Kooner JS, Thorsteinsdottir U, Danesh J, Palmer CN, Roberts R, Watkins H, Schunkert H, Samani NJ (2013) Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 45:25–33. doi:10.1038/ng.2480
Chen H, Meigs JB, Dupuis J (2013) Sequence kernel association test for quantitative traits in family samples. Genet Epidemiol 37:196–204. doi:10.1002/gepi.21703
Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH (2010) Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet 11:446–450. doi:10.1038/nrg2809
Elkind MS, Sciacca R, Boden-Albala B, Rundek T, Paik MC, Sacco RL (2006) Moderate alcohol consumption reduces risk of ischemic stroke: the Northern Manhattan Study. Stroke 37:13–19. doi:10.1161/01.STR.0000195048.86810.5b
Fox CS, Polak JF, Chazaro I, Cupples A, Wolf PA, D’Agostino RA, O’Donnell CJ, Framingham Heart Study (2003) Genetic and environmental contributions to atherosclerosis phenotypes in men and women: heritability of carotid intima-media thickness in the Framingham Heart Study. Stroke 34:397–401
Gertow K, Sennblad B, Strawbridge RJ, Ohrvik J, Zabaneh D, Shah S, Veglia F, Fava C, Kavousi M, McLachlan S, Kivimaki M, Bolton JL, Folkersen L, Gigante B, Leander K, Vikstrom M, Larsson M, Silveira A, Deanfield J, Voight BF, Fontanillas P, Sabater-Lleal M, Colombo GI, Kumari M, Langenberg C, Wareham NJ, Uitterlinden AG, Gabrielsen A, Hedin U, Franco-Cereceda A, Nyyssonen K, Rauramaa R, Tuomainen TP, Savonen K, Smit AJ, Giral P, Mannarino E, Robertson CM, Talmud PJ, Hedblad B, Hofman A, Erdmann J, Reilly MP, O’Donnell CJ, Farrall M, Clarke R, Franzosi MG, Seedorf U, Syvanen AC, Hansson GK, Eriksson P, Samani NJ, Watkins H, Price JF, Hingorani AD, Melander O, Witteman JC, Baldassarre D, Tremoli E, de Faire U, Humphries SE, Hamsten A (2012) Identification of the BCAR1-CFDP1-TMEM170A locus as a determinant of carotid intima-media thickness and coronary artery disease risk. Circ Cardiovasc Genet 5:656–665. doi:10.1161/CIRCGENETICS.112.963660
Grady BJ, Torstenson E, Dudek SM, Giles J, Sexton D, Ritchie MD (2010) Finding unique filter sets in PLATO: a precursor to efficient interaction analysis in GWAS data. Pac Symp Biocomput 15:315–326
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5:e1000529. doi:10.1371/journal.pgen.1000529
Juo SH, Lin HF, Rundek T, Sabala EA, Boden-Albala B, Park N, Lan MY, Sacco RL (2004) Genetic and environmental contributions to carotid intima-media thickness and obesity phenotypes in the Northern Manhattan Family Study. Stroke 35:2243–2247. doi:10.1161/01.STR.0000142132.20442.d8
Lange LA, Bowden DW, Langefeld CD, Wagenknecht LE, Carr JJ, Rich SS, Riley WA, Freedman BI (2002) Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke 33:1876–1881
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26:589–595. doi:10.1093/bioinformatics/btp698
Makinen VP, Civelek M, Meng Q, Zhang B, Zhu J, Levian C, Huan T, Segre AV, Ghosh S, Vivar J, Nikpay M, Stewart AF, Nelson CP, Willenborg C, Erdmann J, Blakenberg S, O’Donnell CJ, Marz W, Laaksonen R, Epstein SE, Kathiresan S, Shah SH, Hazen SL, Reilly MP, Lusis AJ, Samani NJ, Schunkert H, Quertermous T, McPherson R, Yang X, Assimes TL (2014) Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet 10:e1004502. doi:10.1371/journal.pgen.1004502
Melton PE, Carless MA, Curran JE, Dyer TD, Goring HH, Kent JW Jr, Drigalenko E, Johnson MP, Maccluer JW, Moses EK, Comuzzie AG, Mahaney MC, O’Leary DH, Blangero J, Almasy L (2013) Genetic architecture of carotid artery intima-media thickness in Mexican Americans. Circ Cardiovasc Genet 6:211–221. doi:10.1161/CIRCGENETICS.113.000079
O’Donnell CJ, Cupples LA, D’Agostino RB, Fox CS, Hoffmann U, Hwang SJ, Ingellson E, Liu C, Murabito JM, Polak JF, Wolf PA, Demissie S (2007) Genome-wide association study for subclinical atherosclerosis in major arterial territories in the NHLBI’s Framingham Heart Study. BMC Med Genet 8(Suppl 1):S4. doi:10.1186/1471-2350-8-S1-S4
Ratnapriya R, Zhan X, Fariss RN, Branham KE, Zipprer D, Chakarova CF, Sergeev YV, Campos MM, Othman M, Friedman JS, Maminishkis A, Waseem NH, Brooks M, Rajasimha HK, Edwards AO, Lotery A, Klein BE, Truitt BJ, Li B, Schaumberg DA, Morgan DJ, Morrison MA, Souied E, Tsironi EE, Grassmann F, Fishman GA, Silvestri G, Scholl HP, Kim IK, Ramke J, Tuo J, Merriam JE, Merriam JC, Park KH, Olson LM, Farrer LA, Johnson MP, Peachey NS, Lathrop M, Baron RV, Igo RP Jr, Klein R, Hagstrom SA, Kamatani Y, Martin TM, Jiang Y, Conley Y, Sahel JA, Zack DJ, Chan CC, Pericak-Vance MA, Jacobson SG, Gorin MB, Klein ML, Allikmets R, Iyengar SK, Weber BH, Haines JL, Leveillard T, Deangelis MM, Stambolian D, Weeks DE, Bhattacharya SS, Chew EY, Heckenlively JR, Abecasis GR, Swaroop A (2014) Rare and common variants in extracellular matrix gene Fibrillin 2 (FBN2) are associated with macular degeneration. Hum Mol Genet 23:5827–5837. doi:10.1093/hmg/ddu276
Rundek T, Elkind MS, Pittman J, Boden-Albala B, Martin S, Humphries SE, Juo SH, Sacco RL (2002) Carotid intima-media thickness is associated with allelic variants of stromelysin-1, interleukin-6, and hepatic lipase genes: the Northern Manhattan Prospective Cohort Study. Stroke 33:1420–1423
Sacco RL, Anand K, Lee HS, Boden-Albala B, Stabler S, Allen R, Paik MC (2004) Homocysteine and the risk of ischemic stroke in a triethnic cohort: the northern MAnhattan Study. Stroke 35:2263–2269. doi:10.1161/01.STR.0000142374.33919.92
Sacco RL, Sabala EA, Rundek T, Juo SH, Huang JS, DiTullio M, Homma S, Almonte K, Lithgow CG, Boden-Albala B (2007) Design of a family study among high-risk Caribbean Hispanics: the Northern Manhattan Family Study. Ethn Dis 17:351–357
Sacco RL, Blanton SH, Slifer S, Beecham A, Glover K, Gardener H, Wang L, Sabala E, Juo SH, Rundek T (2009) Heritability and linkage analysis for carotid intima-media thickness: the family study of stroke risk and carotid atherosclerosis. Stroke 40:2307–2312. doi:10.1161/STROKEAHA.109.554121
SIGMA Type 2 Diabetes Consortium, Estrada K, Aukrust I, Bjorkhaug L, Burtt NP, Mercader JM, Garcia-Ortiz H, Huerta-Chagoya A, Moreno-Macias H, Walford G, Flannick J, Williams AL, Gomez-Vazquez MJ, Fernandez-Lopez JC, Martinez-Hernandez A, Centeno-Cruz F, Mendoza-Caamal E, Revilla-Monsalve C, Islas-Andrade S, Cordova EJ, Soberon X, Gonzalez-Villalpando ME, Henderson E, Wilkens LR, Le Marchand L, Arellano-Campos O, Ordonez-Sanchez ML, Rodriguez-Torres M, Rodriguez-Guillen R, Riba L, Najmi LA, Jacobs SB, Fennell T, Gabriel S, Fontanillas P, Hanis CL, Lehman DM, Jenkinson CP, Abboud HE, Bell GI, Cortes ML, Boehnke M, Gonzalez-Villalpando C, Orozco L, Haiman CA, Tusie-Luna T, Aguilar-Salinas CA, Altshuler D, Njolstad PR, Florez JC, MacArthur DG (2014) Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 311:2305–2314. doi:10.1001/jama.2014.6511
Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores and marker-sharing statistics. Am J Hum Genet 58:1323–1337
Touboul PJ, Hennerici MG, Meairs S, Adams H, Amarenco P, Bornstein N, Csiba L, Desvarieux M, Ebrahim S, Fatar M, Hernandez Hernandez R, Jaff M, Kownator S, Prati P, Rundek T, Sitzer M, Schminke U, Tardif JC, Taylor A, Vicaut E, Woo KS, Zannad F, Zureik M (2007) Mannheim carotid intima-media thickness consensus (2004–2006). An update on behalf of the advisory board of the 3rd and 4th watching the risk symposium, 13th and 15th European Stroke Conferences, Mannheim, Germany, 2004, and Brussels, Belgium, 2006. Cerebrovasc Dis 23:75–80. doi:10.1159/000097034
Wang L, Beecham A, Zhuo D, Dong C, Blanton SH, Rundek T, Sacco RL (2012) Fine mapping study reveals novel candidate genes for carotid intima-media thickness in Dominican Republican families. Circ Cardiovasc Genet 5:234–241. doi:10.1161/CIRCGENETICS.111.961763
Xiang AH, Azen SP, Buchanan TA, Raffel LJ, Tan S, Cheng LS, Diaz J, Toscano E, Quinonnes M, Liu CR, Liu CH, Castellani LW, Hsueh WA, Rotter JI, Hodis HN (2002) Heritability of subclinical atherosclerosis in Latino families ascertained through a hypertensive parent. Arterioscler Thromb Vasc Biol 22:843–848
Acknowledgments
The authors are grateful to all the families and research staff who participated in the study. This research was supported by grants from the National Institute of Neurological Disorders and Stroke R01NS040807.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
None.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, L., Beecham, A., Dueker, N. et al. Sequencing of candidate genes in Dominican families implicates both rare exonic and common non-exonic variants for carotid intima-media thickness at bifurcation. Hum Genet 134, 1127–1138 (2015). https://doi.org/10.1007/s00439-015-1592-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-015-1592-z