
Abstract
Salinity severely affects crop yield by hindering nitrogen uptake and reducing plant growth. Plant growth-promoting bacteria (PGPB) are capable of providing cross-protection against biotic/abiotic stresses and facilitating plant growth. Genome-level knowledge of PGPB is necessary to translate the knowledge into a product as efficient biofertilizers and biocontrol agents. The current study aimed to isolate and characterize indigenous plant growth-promoting strains with the potential to promote plant growth under various stress conditions. In this regard, 72 bacterial strains were isolated from various saline-sodic soil/lakes; 19 exhibited multiple in vitro plant growth-promoting traits, including indole 3 acetic acid production, phosphate solubilization, siderophore synthesis, lytic enzymes production, biofilm formation, and antibacterial activities. To get an in-depth insight into genome composition and diversity, whole-genome sequence and genome mining of one promising Bacillus paralicheniformis strain ES-1 were performed. The strain ES-1 genome carries 12 biosynthetic gene clusters, at least six genomic islands, and four prophage regions. Genome mining identified plant growth-promoting conferring genes such as phosphate solubilization, nitrogen fixation, tryptophan production, siderophore, acetoin, butanediol, chitinase, hydrogen sulfate synthesis, chemotaxis, and motility. Comparative genome analysis indicates the region of genome plasticity which shapes the structure and function of B. paralicheniformis and plays a crucial role in habitat adaptation. The strain ES-1 has a relatively large accessory genome of 649 genes (~ 19%) and 180 unique genes. Overall, these results provide valuable insight into the bioactivity and genomic insight into B. paralicheniformis strain ES-1 with its potential use in sustainable agriculture.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Soil salinity is a major concern in agriculture, causing more than US$ 27 billion loss annually in crop production. Salinity causes a nutritional imbalance that adversely affects plant growth, development, and yield (Shrivastava and Kumar 2015). Bacterial strains that efficiently colonize the rhizosphere and promote plant growth directly or indirectly are known as plant growth-promoting bacteria (PGPB) (Ahemad and Kibret 2014). These strains carry plant growth-promoting traits such as the production of siderophores, nitrogenase, IAA, 1-aminocyclopropane 1-carboxylate (ACC) deaminase, hydrogen sulfate, and phosphate solubilization (Ahemad and Khan 2012; Glick 2012; Jahanian et al. 2012). Some bacterial strains hold additional habitat-specific plant growth-promoting traits such as salinity tolerance and biocontrol of plant pathogens and insects (Hynes et al. 2008). Their application in salinized agricultural soil is promising to support plant growth and tolerate higher salt concentrations. Plant growth-promoting bacteria are gaining more interest in agriculture applications due to their diverse biological activities. The use of particular strains for the induction of plant growth and suppression of phytopathogen provides a substitute for chemical fungicides and fertilizers (Basu et al. 2021).
Bacillus spp. are extensively investigated for inducing plant resistance against pathogens and plant growth development. In recent years, Bacillus strains are employed as an active ingredient in biofertilizer formulation. They colonize the rhizosphere, protect and induce root biomass and enhance plant potency (Gouda et al. 2018). B. paralicheniformis have synthesized several commercially important products such as antibiotics and enzymes. B. paralicheniformis strain MDJK30 was isolated from the rhizosphere and demonstrated to suppress the peony root rot disease (Wang et al. 2017). Next-generation sequencing technology has enabled whole-genome sequencing of bacteria and was recently employed to investigate the genome of several plant growth-promoting strains such as Pseudomonas and Bacillus spp. (Joshi and Chitanand 2020; Olanrewaju et al. 2021). Comprehensive analysis of whole-genome data and identification of genes that contribute to ecology and plant growth promotion will advance our understanding of molecular mechanisms and the development of plant growth-promoting-aided agriculture technology (Meena et al. 2017).
In the present study, one newly sequenced strain ES-1 showing in vitro plant growth-promoting traits and 14 B. paralicheniformis genomes available in the public databases (August 2021) were mined for antimicrobial metabolites BGCs and plant growth-promoting potentials. Furthermore, comparative genome analysis and the contribution of core and accessory genomes to secondary metabolites and plant growth promotion were analyzed. Our results revealed that the strain ES-1 core-genome harbor several secondary metabolite gene clusters coding for antimicrobial metabolites and plant growth-promoting genes majorly found in the accessory genome.
Materials and methods
Strain isolation and ecological features
Saline sodic soil and water samples (n = 14) were collected from various regions including salt mines Karak and Jhelum. The samples were collected in a sterile bottle aseptically to avoid ingress from exogenic bacterial contamination. The samples were sealed and transported in an ice box to the laboratory on the same day and stored at – 20 ℃. The pH, temperature, salinity, electrical conductivity, total dissolved solids (TDS), and global positioning system (GPS) coordinates of the sampling spot were recorded on the sampling sites. The strain ES-1 was isolated from salt mine sodic soil in Karak, Khyber Pakhtunkhwa, Pakistan. The Karak salt mines represent one of the largest reservoirs of salt in Pakistan, having an estimated reservoir of more than 10.5 billion tonnes (Sharif et al. 2007). The salt of Karak mines is light gray to dark gray and 98% pure. The Karak salt mine area consists of small to high rounded arid hills and is very hot in summer and cold in winter. The age of these salty rocks is predicted by geological horizons as 56–33.9 million years ago in the early Epoch era (Sharif et al. 2007).
A series of dilutions were prepared to start with 1 g of sample in 10 mL sterile 1 M NaCl solution. 100 µL from each dilution was spread on trypticase soy agar (Oxide, UK) supplemented with 5% NaCl. The plates were sealed and incubated at 37 ℃ for 5 days. The number of colonies were counted under a plate magnifier, and the colony-forming unit (CFU) per gram was calculated. The pure colonies were isolated via the sub-culturing technique and screened for plant growth-promoting potentials.
Antimicrobial activity assay
All the isolated bacterial strains (n = 72) were preliminarily screened for antimicrobial activity against a set of ATCC bacterial strains (Escherichia coli, Staphylococcus aureus, Listeria monocytogenes, and Agrobacterium radiobacter) and two locally isolated fungal pathogen; Aspergillus niger and Botrytis cinerea using the cross streaking method. Subsequently, the antimicrobial activity of strain ES-1 extract was confirmed via agar well diffusion assay (Iqbal et al. 2021a). Briefly, the strain was cultured in 500 mL tryptic soy broth (TSB) supplemented with 5% NaCl for 7 days at 37 ℃. The culture was extracted with 1:1 ethyl acetate and concentrated at reduced pressure and 60 ℃ in a rotary evaporator. The obtained extract was dissolved in 500 µL phosphate buffer solution and 100 µL was dispensed in an agar well plate for antibacterial activity. The agar plates were pre-inoculated with fresh bacterial indicator strains, comparable to 0.5 McFarland.
Antifungal activity was determined against phytopathogenic, A. niger MB-4 and B. cinerea KST-32 as performed earlier (Iqbal et al. 2021c). A Petri plate containing fungal strain MB-4 and KST-32 mycelial discs without treatment (ES-1 extract) were used as a control. The antifungal activity was calculated using the following formula.
In vitro plant growth-promoting ability and salinity tolerance
Initially, the strain ES-1 was tested for salinity tolerance using tryptic soy agar (TSA) medium supplemented with various concentrations of NaCl. Afterward, the strain was assessed for plant growth-promoting traits, including extracellular protease, cellulase, amylase and siderophore production, biofilm formation, phosphate solubilization, and 1-aminocyclopropane 1-carboxylic acid (ACC) deaminase. The biofilm formation was assessed using a colorimetric method as performed earlier by Djordjevic et al. (2002). The quantitative analysis of biofilm formation was assessed by destaining the glass walls with 95% ethanol and optical density (OD) was estimated at 595 nm. Fresh cultures (OD 600 nm = 0.8) of the isolates were used as inoculum and an uninoculated medium was used as a negative control. Siderophore synthesis was evaluated using colorimetric chrome azurol sulphonate (CAS) assay (Arora and Verma 2017) and OD was measured at 630 nm. The ACC deaminase activity was assessed using DF salt minimal medium modified with 3 mM ACC (Penrose and Glick 2003) and OD was measured at 540 nm. The strain ES-1 was screened for extracellular protease, cellulase, and amylase production as described earlier (Ji et al. 2014). Pre-sterilized skim milk, carboxymethyl cellulose, and starch was used as a substrate for protease, cellulase, and amylase production, respectively. Phosphate solubilization assay was performed as described earlier (Elhaissoufi et al. 2020) with minor modification. The strain ES-1 was spot inoculated on TSA medium containing insoluble tricalcium phosphate as a sole phosphate source. A clear halo zone of hydrolysis around the colony indicates positive results. The amylase, protease, cellulase, and phosphate-solubilizing potential was measured in mm and calculated as below.
All the plant growth-promoting assays were also evaluated with salinity stress supplemented with various concentrations of salt ranging from 0 to 4.27 M. Based on promising antagonistic and plant growth-promoting activities, strain ES-1 was subjected to whole-genome sequencing and subsequent genome analysis.
Whole-genome sequencing, assembly, and annotation
Genomic DNA was extracted from a fresh culture of strain ES-1 using DNA kit Pure link™ (Invitrogen, USA) according to manufacturer instructions. The purity and concentration of extracted gDNA was confirmed using NanoDrop and Qubit high sensitivity assay, respectively. Library of gDNA was prepared using Nextera XT library preparation kit (Illumina Inc SDCA, USA) and was sequenced using Hiseq Illumina 2500 platform with paired-end reads. The reads were trimmed using trimmomatic v 0.36 (Bolger et al. 2014) and de novo assembly was performed using SPAdes v 3.12 (Bankevich et al. 2012). Genome annotation was performed using Prokka (Prokka 2014) and PGAP v.4.10.
Genome mining
The biosynthetic gene clusters in the ES-1 genome were identified using the Antibiotic and Secondary Metabolite Analysis Accessory (AntiSMASH) online server (https://antismash.secondarymetabolites.org/). Moreover, both “known” and “unknown cluster” BLAST modules were selected to find similar clusters by genome comparison. Sequence similarities to known clusters and domain functions were predicted and annotated using BLASTp and Pfam analysis.
Identification of putative horizontal gene transfer (HGT) and prophages
GIs were predicted using an annotated whole-genome sequence of strain ES-1 as input to Islandviewer 4 online server (https://www.pathogenomics.sfu.ca/islandviewer/). Islandviewer 4 predicts GIs in the bacterial and archeal genome using three prediction methods: IslandPath-DIMOB, IslandPick, and SIGI-HMM. Additionally, a web-based PHASTER server (https://phaster.ca/) was used to identify prophage regions in the B. paralicheniformis ES-1 genome.
Comparative genome analysis
To date (August 2021), 87 genome assemblies of B. paralicheniformis genomes are available in public databases; however, most are incomplete or partial draft genomes. Therefore, only complete genomes or chromosomes (n = 15) were selected and used for comparative genome analysis. To assess genome-level diversity and similarities among B. paralicheniformis strains, high throughput average nucleotide identity (ANI) was conducted using orthoANI. The subsequent ANI matrices were visualized using a web-based heat mapper (http://www.heatmapper.ca/). In silico DNA–DNA hybridization (DDH) was performed for species boundaries delineation through genome-to-genome distance calculator (GGDC) version 2.1 (http://ggdc.dsmz.de/). The Bacterial Pan-Genome Analysis (BPGA) pipeline was used to evaluate genomic assortment and unique gene pool in B. paralicheniformis strains (Chaudhari et al. 2016). The core, accessory, and unique genes were extracted using the pan-genome extraction module. Phylogenetic analyses were conducted based on core proteins alignment using the presence/absence of binary pan gene matrix and the maximum likelihood tree was constructed based on the concatenated core proteins using MEGA X (Kumar et al. 2018). Cluster orthologous group (COGs) analyses were performed using orthoVenn with an inflation value of 1.5 and cutoff e-value of 1e−2 (https://orthovenn2.bioinfotoolkits.net/). Genome-wide analysis of orthologous clusters is important for understanding genome structure and gene/protein function. The information obtained from COGs comparison may serve as a foundation for taxonomic classification, thereby shedding light on the underlying mechanism of molecular evolution in genes/genomes. Furthermore, the ES-1 genome was mined for secondary metabolites gene clusters, and plant growth-promoting traits.
Results and discussion
Ecology, morphological characteristics, and antimicrobial activities
The sample temperature, pH, salinity, and TDS were recorded as 25 ℃, 7.85, 26%, and 37%, respectively (Supplementary file 2; Table S1). The sample revealed an average bacterial density of 2 × 102 ± 20 CFU/g. The strain ES-1 is a Gram-positive, rod-shaped bacterium and forms irregular dry colonies on tryptic soy agar. The antibacterial assay indicates that strain ES-1 produces broad-spectrum antibacterial metabolites, exhibiting promising antagonistic activity against E. coli, A. radiobacter, S. aureus, and L. monocytogenes (Fig. 1). B. paralicheniformis strain ES-1 extract also exhibited antifungal activity against A. niger MB-4 and B. cinerea KST-32 with inhibition of 32.5 and 39.25%, respectively (Fig. 1). In the earlier study, 35 halotolerant bacterial strains were isolated and tested against Fusarium culmorum. Out of 35, 3 strains exhibit antifungal activity by inhibiting mycelial growth in a dual culture plate assay (Albdaiwi et al. 2020).

Antimicrobial activities of B. paralicheniformis ES-1 extract against indicator strains; E. coli, S. aureus, L. monocytogenes, A. radiobacter, A. niger and B. cinerea
In vitro plant growth-promoting traits and salinity tolerance
Plant growth-promoting bacteria interact with plant roots directly by promoting the availability of essential nutrients such as phosphate and iron and indirectly by protecting plants against phytopathogens via competing with pathogens or producing hydrolytic enzymes (Oleńska et al. 2020). The results confirm that the strain ES-1 is capable of solubilizing phosphate, synthesizing siderophores, forming biofilm, and producing extracellular enzymes (Fig. 2). Salinity is an important abiotic stress change physicochemical properties of the soil that significantly affects bacterial and plant growth (Shrivastava and Kumar 2015). We found that the strain ES-1 is halotolerant and grows optimally up to 1.71 M salt concentration (Fig. 2). Recently, Reang et al., isolated several halotolerant bacterial isolates from coastal regions, showed in vitro plant growth-promoting traits and also optimally grow at 10–15% NaCl concentrations (Reang et al. 2022).

a Extracellular enzymes production and salinity tolerance of B. paralicheniformis strain ES-1. The enzymes activities were evaluated on a solid medium containing various concentrations of NaCl (indicated in various colors corresponding to the legend) and halozones were measured in mm. b Quantitative analysis of plant growth-promoting traits. The biofilm, siderophore, and ACC deaminase production were assessed using a colorimetric method, and optical densities (OD) were measured at 595, 630, and 540 nm, respectively. Results represent the mean values of triplicates experiments with standard deviations
Genome features
A total of 4,294,515 reads were obtained with a median insert size of 359 bp and 462.29 × mean coverage. The sequenced reads having a Phred score more than Q30 were subjected to de novo assembly. De novo assembly resulted in a high-quality draft genome of 4.47 Mbs having 47 contigs with N50 23,67,028 and L50 as one. The average GC content of the ES-1 genome is 45.73%. The draft genome annotation resulted in 4708 CDS and 94 RNAs (Fig. 3).

Circular genome map of strain ES-1 in comparison with the reference B. paralicheniformis Bac84 (two outermost circles) genome. The 3rd and 4th circles represent GC skew deviation from average, 5th and 6th circle indicate genome location of GIs and prophages, respectively
Putative GIs and prophages
GIs are regions in a genome that has evidence of horizontal gene transfer (HGT). GIs might be involved in important functions such as symbiosis or pathogenesis. HGT is also considered a vital factor that drives adaptation and evolution. The previous study demonstrated that B. japonicum acquired genes via HGT which leads to improved symbiotic N2 fixation capability as compared to other related strains (Itakura et al. 2009). Similarly, Richard and his colleagues analyzed the dynamic of genomic evolution in Streptococcus species and determined that eight GIs were attained during early evolution, consequently assisting a superior adaptation to a specific habitat (Richards et al. 2014). Herein, we determined five HGT events in B. paralicheniformis strain ES-1 (Fig. 3). The acquired genes were found to be associated with some important enzymes synthesis and stress-related proteins such as cross-over junction endoribonuclease (ruvC), Response regulator aspartate phosphatase A (rapA), Tyrosine recombinase (xerC), NADPH-dependent 7-cyano-7-deazaguanine reductase (queF), Putative hydrolase (ydeN), protease synthase and sporulation protein (paiB), cold shock protein (cspC), Spermidine/spermine N(1)-acetyltransferase (paiA), Phthiocerol/phenolphthiocerol synthesis polyketide synthase type I (ppsC), Adaptive-response sensory-kinase (sasA), Metallopeptidase (immA), Nisin biosynthesis protein (nisB/C), ICEBs1 excisionase (xis) and Metallopeptidase (immA) (Supplementary file 1; Table S1). The higher proportion of important enzymes and stress-related genes in GIs at divergence events propose their role in the adaptation/fitness of the strain ES-1 in the extreme environment. Several genes in GIs code for hypothetical proteins with unknown functions (supplementary file 1; Table S1).
Phage-mediated recombination is also important for environmental bacteria to exchange genetic material, which leads to numerous beneficial properties such as evolution, adaptation, and acquisition of antibiotic resistance/producing genes. In the present study, a total of seven prophage regions were identified in the ES-1 genome and classified as two intact, four incomplete, and one questionable (Table 1). The intact prophages (score 100 and 110%) is 38.2 and 35.3 kb in size with 43.24 and 47.16% GC content. These regions are composed of 57 and 46 coding sequences, of which 13 and 7 are phage hit proteins, respectively. The four incomplete prophages are 18.1, 13.3, 12.2, and 19.6 kb in size with 42.37, 39.96, 46.53, and 36.44% GC content, respectively. On the other hand, the questionable prophage is 26.7 kb in size with 40.70% GC content. The questionable prophage region comprises 28 coding sequences, of which 18 are phage-hit proteins and nine are hypothetical proteins (Supplementary file 1; Table S2).
Taxonomic classification
Pairwise genome similarity and distance were calculated against all (n = 15) available complete genome sequences of B. paralicheniformis strains. All ANI values were found above the threshold (< 95%) (Richter and Rosselló-Móra 2009) and a minimum ANI percentage (98.6) was noted between strain CBMAI 1303 and Bac48 (Fig. 4A). The heatmap generated using ANI values illustrates the four optimal clades in the dendrogram. Clade A and B are composed of two and five strains, respectively. Clades C and D consist of four strains with the highest ANI values (< 99%). The newly sequenced strain ES-1 lies in clade C, exhibiting the highest similarity with strain SUBG0010 (99.28%) followed by strain Bac84 and Bac48 (Fig. 4A). The strain Bac84 is a representative genome for B. paralicheniformis species in RefSeq. Database (https://www.ncbi.nlm.nih.gov/refseq/). In addition, in silico DDH values are above the threshold, confirming that all the strains belong to the same B. paralicheniformis species (Supplementary file 1; Table S3). The DDH value threshold > 70% was previously recommended for species delineation (On et al. 2017). Herein, the DDH values support the results of ANI since the recommended DDH threshold corresponds well to the ANI cutoff values.

a Whole-genome comparison of B. paralicheniformis strains. The cells in the heatmap correspond to the level of similarities on the scale of 98–100, where 98% similarity is depicted in green and 100 illustrated in blue color. The dendrogram was constructed based on ANI percentage and corresponds to the ANI values between the understudy strains. b Phylogenetic tree based on concatenated 3344 core proteins using maximum likelihood method. The values on each branch represent the estimated time of divergence. The tree was constructed using MEGA-X and edited in iTOL (https://itol.embl.de/)
Core pan-genome analysis of B. paralicheniformis
Core-pan genome analyses were conducted to identify unique genomic features and genomic diversity among B. paralicheniformis strains. To avoid incorrect inference from incomplete genomes, only complete genomes were considered (Supplementary file 2; Table S2). The pan-genome pool was generated by considering all genes found in all (n = 15) genomes and dividing them into core, accessory, and unique genes. The genes were clustered in the same group when the BLASTp coverage was more than 75%, with a cutoff value less than 1e−8. A total of 14,857 gene clusters (Pan-genome) were generated in 15 strains. Among these, 3344 genes are common in all strains and make the core genome. Based on 3344 concatenated core genome proteins, the maximum likelihood tree grouped 15 strains into 4 distinct clades with high confidence (Fig. 4B). The strain ES-1 cluster with SUBG0010, that was isolated from the rhizosphere, and Bac48 and Bac84, isolated from red sea sediments.
The power fit equation and exponential equation curve show that the pan-genome of B. paralicheniformis has the exponent >0 i.e., b = 0.117371 and d = − 0.0126432, respectively, suggesting an open pan-genome (Tettelin et al. 2008). The core versus pan-genome plot illustrates that the number of accessory and unique genes increases while the number of core genes slightly decreases with the addition of each genome (Fig. 5A). These results indicate that the pan-genome window is still open for expansion. A large number of accessory (10,654 genes) and unique genes (915) (Fig. 5B) might be due to the specialization of each B. paralicheniformis strain and correspond to their geographical location. Protein sequences of core, accessory, and unique genes were determined and established their KEGG identities. Interestingly, majority of the secondary metabolite gene clusters identified in ES-1 genome are associated with the core genome. The accessory genome which includes genes that are only found in a few genomes is majorly associated with amino acid and carbohydrate metabolism, emphasizing habitat-specific changes in the strain. Sun et al., reported that the diversity of amino acid and carbohydrate metabolism enhances the genetic fitness of Bifidobacterium to adapt in a particular habitat (Sun et al. 2015) which is desirable in sustainable agriculture applications. In addition, the genes that belong to the unique pan-genome are mostly involved in replication and repair, energy metabolism, and membrane transport (Fig. 5C). The detail of core, accessory, and unique genes present in each of B. paralicheniformis genomes are given in the Supplementary file 1; Table S4.

a Mathematical modeling of B. paralicheniformis genomes estimating the size of core and pan-genome. b Bar graph representing the number of unique genes. c KEGG distribution of representative protein sequences in the core, accessory, and unique pan-genome across 15 B. paralicheniformis strains
Orthologous clusters are clusters of genes originated by vertical descent from a single gene in the last common ancestor. Comparative orthologous cluster analysis in different genomes provides insight into gene structure, function, and molecular mechanism of evolution in genes/genome (Kristensen et al. 2011). Here, we performed a comparative orthologous cluster analysis which identified a total of 4756 clusters, 3548 orthologous clusters (found in at least two strains), and 1205 single-copy gene clusters in 15 B. paralicheniformis. The highest number of singletons were observed in strain RSC-3 (480) followed by the newly sequenced strain ES-1 (241) (Supplementary file 2; Table S3). Previously orthologous cluster analyses of Paenibacillus polymyxa were conducted. A total of 6052 families were prioritized and 3650 are shared by five strains (Xu et al. 2017). Similarly, proteome comparison of B. firmus strain I-1582 with five other related strains identified a total of 5371 clusters, 5211 orthologous clusters, and 3116 singletons (Susič et al. 2020). The low number of clusters in the current study are probably due to the small genome size of B. paralicheniformis as compared to Bacillus firmus. The orthologous clusters identified in ES-1 were compared with four B. paralicheniformis strain isolated from soil (Fig. 6a) as well as four strains originated from non-soil source (Fig. 6b). The soil-originated strains sharing 3767 clusters (core proteome) representing 79.20% repertoire of the proteins, as compared to B. paralicheniformis strains isolated from other source which shared 3737 (78.57%) clusters (Fig. 6a, b). These results indicate that the core-proteome is similar, regardless of the source of isolation. The newly sequenced strain ES-1 shared 37 and 33 clusters with strain Bac48 and SUBG0010, respectively. The strains Bac48 and SUBG0010 were earlier characterized as promising antimicrobial-exhibiting strains and were suggested to be a better candidate for biocontrol (Al-Amoudi et al. 2016; Bhatt et al. 2018). Recently, an in silico metabolic network was constructed for strain Bac48 and revealed twice as many secreted proteins as compared to model strain B. subtilis 168 (Othoum et al. 2019). These results suggest that these three strains shared particular modules of metabolites that have evolved in these strains due to the unique niche adaptation.

Genomic diversity in Bacillus paralicheniformis strains. a The strain ES-1 was compared with the strains that originated from soil. b The strain ES-1 was compared with the non-soil strains. Each strain is represented with an oval shape with a unique color and the number of orthologous proteins shared by all strains are mentioned in the center
Genome mining
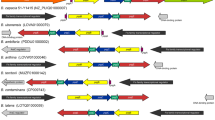
The AntiSMASH analysis identified 12 secondary metabolites biosynthetic gene clusters in the ES-1 genome (Fig. 7). These include four NRPS encoding for fengycin, bacitracin, bacillibactin, and lichenysin. Fengycin is a cyclic lipopeptide exhibiting promising antifungal activity. Bacitracin is a polypeptide that disrupts peptidoglycan synthesis of Gram-negative bacteria. Bacillibactin is an iron siderophore that enhances ES-1 capability to scavenge iron from the surrounding. Furthermore, the strain ES-1 harbor 4 BGCs coding for ribosomal synthesized post-translational modified peptides (RiPPs), one each for siderophore, terpene, type 3 polyketides (T3PK), and cyclodipeptides (CDPS). The identified BGCs were compared with known clusters and 6 BGCs showed homology with known gene clusters from other Bacillus species such as thiopeptide exhibited 7% similarity with butirosin A (BGC0000693) from B. circulans, fengycin showed 93% similarity with BGC0001095 from B. velezensis and bacitracin showed 100% similarity with BGC0000310 from B. licheniformis. Similarly, bacillibactin, lichenysin, and lanthipeptide revealed 53%, 100%, and 18% homology with BGC0000309 (B. subtilis str. 168), BGC0000381 (B. licheniformis DSM 13), and BGC0000506 (B. subtilis subsp. spizizenii), respectively. On the other hand, six clusters showed no similarity with known gene clusters, so-called orphan clusters. Comparative BGC analyses with other B. paralicheniformis genomes revealed that strain SUBG0010 carries highest (n = 13) and Bac48 harbor the lowest (n = 10) number of BGCs (Fig. 7). Overall, the B. paralichenifromis genomes carries a similar pattern of BGCs. However, a strain-specific hybrid BGC was identified in Bac48 genome as previously reported (Othoum et al. 2018). The genome mining data suggest that strain ES-1 potentially produces numerous antibacterial metabolites that correspond to the in vitro antibacterial results.

Comparative analysis of secondary metabolites biosynthetic gene clusters in B. paralicheniformis strain. The bar graph shows the consistent distribution of BGCs across the species. The number of BGCs in each strain is color-coded as per legend
Orphan BGCs in ES-1
Siderophore
Siderophore received great attention due to its application in agriculture. A previous study demonstrated that siderophore-producing Pseudomonas fluorescens play a vital role in controlling phytopathogens and also promote plant growth. (Cornelis and Matthijs 2007). Recent studies also support the siderophore theory of biological control by plant growth-promoting bacteria. Recently, Brevibacillus brevis was isolated from rhizosphere soil to produce a siderophore with potent antimicrobial activity (Sheng et al. 2020). One siderophore encoding gene cluster identified in strain ES-1 did not show similarity with a known cluster. The siderophore BGC is located in contig 1 of the ES-1 genome and contains 10 modules. The BLASTp results indicate 2 core biosynthetic genes, IucA and IucC, 6 additional biosynthetic genes including aminotransferase, aminotransferase class-III, Decarboxylase (pyridoxal-dependent), putative siderophore biosynthesis protein, lysine/ornithine N-monooxygenase, short-chain dehydrogenase/reductase SDR and 2 others genes. Siderophore coding BGCs were previously identified in plant growth-promoting B. subtilis and B. velezensis isolated from rhizosphere (Olanrewaju et al. 2021).
Terpene
Terpenes are an extremely diverse and largest class of natural products, widely used as herbicides, pharmaceuticals, biofuels, and flavoring. Previously, they have been mostly isolated from plants and fungi. However, the advances in genome technology and availability of bacterial genome sequences indicate that bacteria also harbor terpene synthase genes. Although, most of these terpenes synthase genes seem to be silent in the parent strains. Reddy et al. identified 22 terpenes genes in the bacterial genome and 15 were successfully expressed in E. coli (Reddy et al. 2020). Here, a terpene cyclase gene was identified as a core biosynthetic gene while 3 additional biosynthetic genes, beta-lactamase, aldehyde dehydrogenase, and GCN5-related N-acetyltransferase were identified in the ES-1 genome. Moreover, 13 “others” genes and one transport-related gene were identified.
Type III polyketide synthases (T3PKSs)
Type 3 polyketide synthases were earlier considered to be produced by only plants and fungi. However, the bacterial genome projects revealed that T3PKSs are widely distributed in bacterial genomes. Bacillus spp. are known to produce various biological active metabolites with polyketide group. In the earlier study, a new variant of antimicrobial aryl-crowned polyketide was isolated and characterized from a seaweed-associated B. subtilis strain MTCC 10403 (Chakraborty et al. 2018). The isolated polyketide exhibited promising activity against food-borne pathogens including E. coli. Furthermore, several Paenibacillus and Bacillus spp. are known to produce polyketide, antagonizing growth of human and phytopathogens (Olishevska et al. 2019). The current study identified T3PKS BGC in all B. paralicheniformis genomes including ES-1. The T3PKS cluster in ES-1 genome is composed of one core gene encoded for chalcone and stilbene synthase and three additional biosynthesis genes, isoprenylcysteine carboxyl methyltransferase (locus tag ctg1_1412, score: 100.9, e-value: 3.9e−29), acetyltransferase (ctg_1426, score: 49.6, e-value: 4.6e−13), and monogalactosyldiacylglycerol synthase (ctg_1428, score: 157.0, e-value: 4.8e−46) were identified in ES-1 genome. These findings suggest that T3PKS in strain ES-1 genome may encode important biological metabolites that could be involved in antimicrobial activities of B. paralicheniformis.
RiPP-like
RiPP-like are the unspecified ribosomally synthesized post-translationally modified peptides (RiPPs) products. In the present study, an unknown RiPP-like cluster was predicted in ES-1 genome located in contig 1. The RiPP-like cluster is composed of a core biosynthetic gene (345 nt) located on locus tag ctg1_1835 and Pfam hit results showed similarity with Bacteriocin class IId cyclical uberolysin-like (score: 40.5, e-value: 2.4e−10). Furthermore, 5 transport-related genes including 3 ABC transporter (ctg_1833, ctg1_1838 and ctg1_1839) and 2 binding protein-dependent transport system inner membrane component (ctg1_1840, ctg1_1841) were identified. Previously reported that members of bacteriocin class IId cyclical uberolysin-like are membrane-interacting peptides, and exhibit broad-spectrum antimicrobial activities (Wirawan et al. 2007).
Cyclodipeptides synthases (CDPSs)
CDPSs are a newly described family of peptidases that are responsible for the biosynthesis of various cyclopeptides, which act as a precursor for numerous natural products with important bioactivities (Gondry et al. 2009; Canu et al. 2020). CDPSs do not have a distinct structure; however, they still have a common architecture such as the rossmann fold domain. Initially, it was thought that NRPSs are responsible for CDPs biosynthesis until ribose-independent production was explored. The strain ES-1 genome harbor a unique CDPS BGC composed of core gene located on locus tag ctg2_293 and Pfam hit result (score: 270.4, e-value: 1.1e−80) revealed similarity with tRNA-dependent cyclodipeptide synthase. Moreover, five additional biosynthetic genes and two transport-related genes were identified. The two additional biosynthetic genes located upstream of the core gene, locus tag ctg_287 (1420 nt) revealed pfam hits with FGGY family of carbohydrate kinases, N-terminal domain (score: 104.9, e-value: 5e−30) and FGGY family of carbohydrate kinases, C-terminal domain (score: 100.2, e-value: 1.2e−28) and ctg_290 (2070 nt) showed pfam hit with Class II Aldolase and Adducin N-terminal domain (score: 91.2, e-value: 7.6e−26) and Enoyl/Acyl carrier protein reductase (score: 171.8, e-value: 1.7e−50). While the other 3 additional biosynthetic genes located downstream of the core gene locus tag; ctg2_294, ctg_297, ctg_298 exhibited pfam hits with Cytochrome P450 (score: 146.0, e-value: 1.4e−42), Amidase (score: 171.6, e-value: 2.8e−50) and Oxidoreductase family, NAD-binding Rossmann fold (score: 92.2, e-value: 3.9e−26). Previously, cyclic dipeptides were isolated from Bacillus sp. that antagonized growth of several microorganisms including fungal plant pathogens (Kumar et al. 2013). Similarly, B. amyloliquefaciens was reported to be secreting cyclic dipeptide that inhibits biofilm and virulence production in methicillin resistant Staphylococcus aureus (Gowrishankar et al. 2015).
Lassopeptide
Lassopeptides are important bacterial natural products that belong to a family of RiPPs with unique lasso-like structures. Lasso peptides have a broad spectrum of biological activities, including antimicrobial, antiviral and antimetastatic activities, while some act as enzyme inhibitors (Katahira et al. 1996). The biosynthetic machinery producing RiPPs contains a highly conserved lactam ring threaded by the carboxyl-terminal domain associated with post-translational modification of the precursor peptide, making them a perfect target for genome mining (Hetrick and Donk 2017). Genome mining of the ES-1 genome indicates a new putative macrolactam class II lasso peptide gene cluster with no similarity to the known gene cluster. The core biosynthetic genes are located at locus tag ctg2_713, ctg2_714 encoding for lasso peptide putative class II exhibiting Pfam hits with Asparagine synthase (score: 77.2, e-value: 1.8e−21) and transglutaminase-like superfamily (score: 55.2, e-value: 5.8e−15), respectively. One stand-alone RiPP recognition element (RRE) was also identified as a core biosynthetic gene located on locus tag ctg2_715. Recently, genome mining of Streptomyces humidus revealed a unique BGC for lasso peptide and heterologous expression identified a new peptide humidimycin with strong antimicrobial activities (Sánchez-Hidalgo et al. 2020).
Plant growth-promoting capabilities
B. paralicheniformis strain ES-1 genome was mined for plant growth-promoting genes that could be associated with their abilities to suppress phytopathogen, improve nutrient availability, and resist abiotic stress. The genome harbor genes associated with phosphate solubilization, protease, amylase, and cellulase synthesis, biofilm formation, and ACC deaminase production, which is in agreement with the in vitro plant growth-promoting assays (Supplementary file 2; Table S4). The ACC deaminase coding gene rimM was earlier reported in plant growth-promoting bacteria to function in reducing plant ethylene that inhibits root nodulation (Shah et al. 1998; Gupta et al. 2014). L-tryptophane (L-TRP) is an important residue required for normal plant growth/development and also acts as a precursor for plant growth regulators (Mustafa et al. 2018). It is generally supplied through seed priming or foliar spray. We identified a complete biosynthetic pathway of L-TRP in the strain ES-1 genome. The L-TRP pathway involved five enzymes encoded by seven genes (trpABCDEF). The last two steps in this pathway are catalyzed by a single enzyme tryptophan synthase which is composed of two (trpA and trpB) alpha and beta subunits. The enzyme anthranilate phosphoribosyltransferase catalyzes step 2 and converts anthranilate to phosphoribosyl anthranilate encoded by two genes trpD_1 and trpD_2 (Supplementary file 2; Table S4). Two volatile organic compounds, acetoin and 2,3-butanediol biosynthesis genes (ilvHB, alsD and budC) found in ES-1 genome which were reported to promote plant growth by increasing disease resistance (Han et al. 2006), stimulating root formation (Ryu et al. 2003) and drought tolerance (Cho et al. 2008). Furthermore, we identified genes (cysCJI) involved in the biosynthesis of hydrogen sulfide which is shown to promote plant growth and seed germination (Dooley et al. 2013). Two operons narHGX and nasABCDEF were determined and annotated as nitrate reductase and nitrate transporter, respectively. These clusters were previously found in plant growth-promoting B. subtilis strain MBI 600 and were predicted to be involved in nitrate reduction and nitrate transport (Samaras et al. 2021).
The chitinase coding gene was found which hydrolyzes the cell wall of pathogenic fungi and pests (Gupta et al. 2014; Loper et al. 2012). Besides these, the genes gabD and gabR were identified which are involved in the synthesis of disease/pest inhibiting gamma-aminobutyric acid (GABA) (Loper et al. 2012). The strain ES-1 genome also harbors speAGE and potAB operon which are associated with spermidine biosynthesis and transport system, respectively. Spermidine is crucial for plant cell viability and has been associated with lateral root expansion, phytopathogen confrontation, and mitigation of osmotic, oxidative, and acidic stress. Thus, spermidine synthesis by B. paralicheniformis ES-1 may establish additional biosynthetic machinery associated with increased plant growth. However, further wet-lab experiments are required to validate the direct involvement of spermidine produced by B. paralicheniformis ES-1 in plant growth promotion.
Root colonization and chemotaxis
Flagellar protein plays a vital role in the colonization of plant growth-promoting bacteria on plant roots. In strain ES-1, we found 20 genes associated with flagellar biosynthesis and assembly. These genes are mostly localized in 3 clusters, fliDEGJMSTPW, flhABHEF and flgBCGL, while gene hag encoding for flagellin and csrA encoding for a translational regulator found separately on locus tag 1 E1_02900 and E1_02901, respectively. Furthermore, the genes involved in chemotaxes such as methyl-accepting chemotaxis two-component systems protein-encoding genes like cheABCDRVWY, pomA, yoaH, and mcpBC were also found in ES-1 genome. The two-component system helps in signal recognition of exudate and adaptation to the environment. The presence of these genes indicates that strain ES-1 is capable of responding to a stimulus and consequently moving toward plant roots. These flagellar biosynthesis, assembly, and chemotaxis genes were previously identified in the plant growth-promoting Cronobacter muytjensii strain JZ38 genome (Eida et al. 2020). It was observed that plant growth-promoting genes are found in the accessory genome of ES-1, which implies an evolutionary process for adaptation in specific habitats, as suggested earlier (Iqbal et al. 2021b; Zhang et al. 2016).
Salt tolerance capabilities in ES-1
The strain ES-1 was isolated from the salt mine and can grow well on TSA supplemented with 0–1.71 M salt concentration (Fig. 2). Genome mining showed that the strain harbors several genes associated with salt tolerance. For instance, the genes betA and betB involved in glycine-betaine synthesis were identified in the ES-1 genome (Supplementary file 2; Table S5). These two genes are reported to be the most important genes associated with salt tolerance (Liu et al. 2016). Further, trehalose functions as an osmoprotectant under extreme conditions such as drought, high salt concentration, and osmotic stress (Duan et al. 2013). Garg and his colleagues previously reported that trehalose accumulates in plant and enhance systematic resistance to abiotic stresses (Garg et al. 2002). In bacteria, five trehalose biosynthesis pathways have been reported including ostsAB, treS, treT, treP, and treYZ (Paul et al. 2008). Here, in the strain ES-1 genome, treP biosynthesis pathway was identified. In addition, trehalose regulator (treR) and transport (sugA) genes were also identified in the ES-1 genome. Trehalose biosynthesis via treP biosynthesis is a single-step process where glucose are converted to trehalose by trehalose phosphorylase (treP). Subsequently, trehalose may be hydrolyzed via treA and produce two glucose molecules. This is a common pathway present in the microorganism and associated with adaptation and survival in extreme environments.
Furthermore, several osmoregulatory receptor genes were identified in the ES-1 genome (Supplementary file 2; Table S5). For instance, the kdpD is responsible for detecting hyperosmotic stress and triggering the expression of genes involved in the accumulation of solutes and cell wall synthesis (Möker et al. 2004). In response to high salt concentration, they may also trigger the expression of kdp operon, which encodes for a high-affinity K+ ions uptake system (Heermann and Jung 2010). Additionally, the genes encoding for transport systems such as Na+/H+ antiporter transport system for exporting Na+ and importing H+ and K+ transport system for K+ accumulation (Epstein 2003) were determined in the ES-1 genome to survive in hyperosmotic conditions ( Supplementary file 2; Table S5). The strain ES-1 genome also carries protein quality control system-related gene htpG, htpX, groL, groS, grpE, dnaK, and dnaJ, which are called molecular chaperons and play a vital role in stress response (Susin et al. 2006).
Conclusion
The current study sheds light on the genomic and biochemical characterization of a potential plant growth-promoting B. paralicheniformis strain ES-1 isolated from an unexplored salt mine in Karak, Pakistan. Among B. paralicheniformis species the strain MDJK30 was previously reported as potential PGPB and 11 secondary metabolites BGCs were identified (Wang et al. 2017) as compared to ES-1 where 12 BGCs were found. Furthermore, the current study revealed various features of B. paralicheniformis that are associated with their commensal lifestyle with plant roots such as open pan-genome, high diversity of transport, and the metabolism of amino acids and carbohydrates. The essential genomic and plant growth-promoting characteristics of strain ES-1 highlight the conjunction application of various microbes and imply the future research focus towards a holistic strategy for plant growth promotion. The complementarity of their antimicrobial activities and plant growth promotion will pave the way for their application as an alternative sustainable approach to enhance agriculture production under salt-stress conditions.
Data availability
The whole-genome sequence data of Bacillus paralicheniformis strain ES-1 has been deposited to National Centre for Biotechnology Information (NCBI) under accession number CP083398. The Bioproject and Biosample were submitted to NCBI and accession number PRJNA761002 and SAMN13510061, respectively.
References
Ahemad M, Khan MS (2012) Evaluation of plant-growth-promoting activities of rhizobacterium Pseudomonas putida under herbicide stress. Ann Microbiol 62:1531–1540. https://doi.org/10.1007/s13213-011-0407-2
Ahemad M, Kibret M (2014) Mechanisms and applications of plant growth promoting rhizobacteria: Current perspective. J King Saud Univ Sci 26:1–20. https://doi.org/10.1016/j.jksus.2013.05.001
Al-Amoudi S, Essack M, Simões MF, Bougouffa S, Soloviev I, Archer JAC et al (2016) Bioprospecting red sea coastal ecosystems for culturable microorganisms and their antimicrobial potential. Mar Drugs 14(9):165. https://doi.org/10.3390/md14090165
Albdaiwi RN, Khyami-Horani H, Ayad JY, Alananbeh KM, Al-Sayaydeh R (2020) Isolation and characterization of halotolerant plant growth promoting rhizobacteria from durum wheat (Triticum turgidum subsp. durum) cultivated in saline areas of the dead sea region. Oxid Med Cell Longev 10:1639. https://doi.org/10.3389/FMICB.2019.01639/BIBTEX
Arora NK, Verma M (2017) Modified microplate method for rapid and efficient estimation of siderophore produced by bacteria. 3 Biotech 7:381. https://doi.org/10.1007/s13205-017-1008-y
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS et al (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. https://doi.org/10.1089/cmb.2012.0021
Basu A, Prasad P, Das SN, Kalam S, Sayyed RZ, Reddy MS et al (2021) Plant growth promoting rhizobacteria (PGPR ) as green bioinoculants : recent developments, constraints, and prospects. Sustainability 13(3):1–20
Bhatt HD, Chudasama KS, Thaker VS (2018) Antimicrobial activity of Bacillus paralicheniformis SUBG0010 against plant pathogenic bacteria of mango. J Pharmacogn Phytochem 7:449–455
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Canu N, Moutiez M, Belin P, Gondry M (2020) Cyclodipeptide synthases: a promising biotechnological tool for the synthesis of diverse 2,5-diketopiperazines. Nat Prod Rep 37:312–321. https://doi.org/10.1039/C9NP00036D
Chakraborty K, Thilakan B, Kizhakkekalam VK (2018) Antibacterial aryl-crowned polyketide from Bacillus subtilis associated with seaweed Anthophycus longifolius. J Appl Microbiol 124:108–125. https://doi.org/10.1111/JAM.13627
Chaudhari NM, Gupta VK, Dutta C (2016) BPGA-an ultra-fast pan-genome analysis pipeline. Sci Rep 6:1–10. https://doi.org/10.1038/srep24373
Cho SM, Kang BR, Han SH, Anderson AJ, Park J-Y, Lee Y-H et al (2008) 2R,3R-butanediol, a bacterial volatile produced by Pseudomonas chlororaphis O6, is involved in induction of systemic tolerance to drought in Arabidopsis thaliana. Mol Plant Microbe Interact 21:1067–1075. https://doi.org/10.1094/MPMI-21-8-1067
Cornelis P, Matthijs S (2007) Pseudomonas Siderophores and their Biological Significance. In: Varma A, Chincholkar SB (eds) Microbial siderophores. Springer, Berlin, pp 193–203
Djordjevic D, Wiedmann M, McLandsborough LA (2002) Microtiter plate assay for assessment of Listeria monocytogenes biofilm formation. Appl Environ Microbiol 68:2950–2958. https://doi.org/10.1128/AEM.68.6.2950-2958.2002
Dooley FD, Nair SP, Ward PD (2013) Increased growth and germination success in plants following hydrogen sulfide administration. PLoS ONE 8:e62048
Duan J, Jiang W, Cheng Z, Heikkila JJ, Glick BR (2013) The complete genome sequence of the plant growth-promoting bacterium Pseudomonas sp. UW4. PLoS ONE 8:e58640
Eida AA, Bougouffa S, L’Haridon F, Alam I, Weisskopf L, Bajic VB et al (2020) Genome insights of the plant-growth promoting bacterium Cronobacter muytjensii JZ38 with volatile-mediated antagonistic activity against phytophthora infestans. Front Microbiol 11:369. https://doi.org/10.3389/fmicb.2020.00369
Elhaissoufi W, Khourchi S, Ibnyasser A, Ghoulam C, Rchiad Z, Zeroual Y et al (2020) Phosphate solubilizing rhizobacteria could have a stronger influence on wheat root traits and aboveground physiology than rhizosphere P solubilization. Front Plant Sci 11:979. https://doi.org/10.3389/FPLS.2020.00979/BIBTEX
Epstein W (2003) The roles and regulation of potassium in bacteria. Prog Nucleic Acid Res Mol Biol 75:293–320. https://doi.org/10.1016/s0079-6603(03)75008-9
Garg AK, Kim J-K, Owens TG, Ranwala AP, Choi Y, Kochian LV et al (2002) Trehalose accumulation in rice plants confers high tolerance levels to different abiotic stresses. Proc Natl Acad Sci U S A 99:15898–15903. https://doi.org/10.1073/pnas.252637799
Glick BR (2012) Plant growth-promoting bacteria: mechanisms and applications. Scientifica (cairo). https://doi.org/10.6064/2012/963401
Gondry M, Sauguet L, Belin P, Thai R, Amouroux R, Tellier C et al (2009) Cyclodipeptide synthases are a family of tRNA-dependent peptide bond-forming enzymes. Nat Chem Biol 5:414–420. https://doi.org/10.1038/nchembio.175
Gouda S, Kerry RG, Das G, Paramithiotis S, Shin H-S, Patra JK (2018) Revitalization of plant growth promoting rhizobacteria for sustainable development in agriculture. Microbiol Res 206:131–140. https://doi.org/10.1016/j.micres.2017.08.016
Gowrishankar S, Kamaladevi A, Ayyanar KS, Balamurugan K, Pandian SK (2015) Bacillus amyloliquefaciens-secreted cyclic dipeptide—cyclo(l-leucyl-l-prolyl) inhibits biofilm and virulence production in methicillin-resistant Staphylococcus aureus. RSC Adv 5:95788–95804. https://doi.org/10.1039/C5RA11641D
Gupta A, Gopal M, Thomas GV, Manikandan V, Gajewski J, Thomas G et al (2014) Whole genome sequencing and analysis of plant growth promoting bacteria isolated from the rhizosphere of plantation crops coconut, cocoa and arecanut. PLoS ONE 9:e104259
Han SH, Lee SJ, Moon JH, Park KH, Yang KY, Cho BH et al (2006) GacS-dependent production of 2R, 3R-butanediol by Pseudomonas chlororaphis O6 is a major determinant for eliciting systemic resistance against Erwinia carotovora but not against Pseudomonas syringae pv. tabaci in tobacco. Mol Plant Microbe Interact 19:924–930. https://doi.org/10.1094/MPMI-19-0924
Heermann R, Jung K (2010) The complexity of the “simple” two-component system KdpD/KdpE in Escherichia coli. FEMS Microbiol Lett 304:97–106. https://doi.org/10.1111/j.1574-6968.2010.01906.x
Hetrick KJ, van der Donk WA (2017) Ribosomally synthesized and post-translationally modified peptide natural product discovery in the genomic era. Curr Opin Chem Biol 38:36–44. https://doi.org/10.1016/j.cbpa.2017.02.005
Hynes RK, Leung GCY, Hirkala DLM, Nelson LM (2008) Isolation, selection, and characterization of beneficial rhizobacteria from pea, lentil, and chickpea grown in western Canada. Can J Microbiol 54:248–258. https://doi.org/10.1139/w08-008
Iqbal S, Vohra MS, Janjua HA (2021a) Whole-genome sequence and broad-spectrum antibacterial activity of Chryseobacterium cucumeris strain MW-6 isolated from the Arabian Sea. 3 Biotech 11(12):1–7. https://doi.org/10.1007/s13205-021-03039-5
Iqbal S, Vollmers J, Janjua HA (2021b) Genome mining and comparative genome analysis revealed niche-specific genome expansion in antibacterial Bacillus pumilus strain SF-4. Genes (basel) 12:1060–1079. https://doi.org/10.3390/genes12071060
Iqbal S, Ullah N, Janjua HA (2021c) In vitro evaluation and genome mining of Bacillus subtilis strain RS10 reveals its biocontrol and plant. Agriculture 11:1273. https://doi.org/10.3390/agriculture11121273
Itakura M, Saeki K, Omori H, Yokoyama T, Kaneko T, Tabata S et al (2009) Genomic comparison of Bradyrhizobium japonicum strains with different symbiotic nitrogen-fixing capabilities and other Bradyrhizobiaceae members. ISME J 3:326–339. https://doi.org/10.1038/ismej.2008.88
Jahanian A, Chaichi M, Rezaei K, Rezayazdi K, Khavazi K (2012) The effect of plant growth promoting rhizobacteria (PGPR) on germination and primary growth of artichoke (Cynara scolymus). Int J Agric Crop Sci 4(14):923–929
Ji SH, Gururani MA, Chun S-C (2014) Isolation and characterization of plant growth promoting endophytic diazotrophic bacteria from Korean rice cultivars. Microbiol Res 169:83–98. https://doi.org/10.1016/j.micres.2013.06.003
Joshi A, Chitanand M (2020) Complete genome sequence of plant growth promoting Pseudomonas aeruginosa AJ D 2 an isolate from monocropic cotton rhizosphere. Genomics 112:1318. https://doi.org/10.1016/J.YGENO.2019.07.022
Katahira R, Yamasaki M, Matsuda Y, Yoshida M (1996) MS-271, A novel inhibitor of calmodulin-activated myosin light chain kinase from Streptomyces sp.—II. Solution structure of MS-271: characteristic features of the ‘lasso’ structure. Bioorg Med Chem 4:121–129. https://doi.org/10.1016/0968-0896(95)00176-X
Kristensen DM, Wolf YI, Mushegian AR, Koonin EV (2011) Computational methods for Gene Orthology inference. Brief Bioinform 12:379–391. https://doi.org/10.1093/BIB/BBR030
Kumar N, Mohandas C, Nambisan B, Kumar DRS, Lankalapalli RS (2013) Isolation of proline-based cyclic dipeptides from Bacillus sp. N strain associated with rhabitid entomopathogenic nematode and its antimicrobial properties. World J Microbiol Biotechnol 29:355–364. https://doi.org/10.1007/S11274-012-1189-9
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Liu W, Wang Q, Hou J, Tu C, Luo Y, Christie P (2016) Whole genome analysis of halotolerant and alkalotolerant plant growth-promoting rhizobacterium Klebsiella sp D5A. Sci Rep 6:26710. https://doi.org/10.1038/srep26710
Loper JE, Hassan KA, Mavrodi DV, Davis EW II, Lim CK, Shaffer BT et al (2012) Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PLOS Genet 8:e1002784
Meena KK, Sorty AM, Bitla UM, Choudhary K, Gupta P, Pareek A et al (2017) Abiotic stress responses and microbe-mediated mitigation in plants: the omics strategies. Front Plant Sci 8:172. https://doi.org/10.3389/fpls.2017.00172
Möker N, Brocker M, Schaffer S, Krämer R, Morbach S, Bott M (2004) Deletion of the genes encoding the MtrA-MtrB two-component system of Corynebacterium glutamicum has a strong influence on cell morphology, antibiotics susceptibility and expression of genes involved in osmoprotection. Mol Microbiol 54:420–438. https://doi.org/10.1111/j.1365-2958.2004.04249.x
Mustafa A, Imran M, Ashraf M, Mahmood K (2018) Perspectives of using L-tryptophan for improving productivity of agricultural crops: a review. Pedosphere 28:16–34. https://doi.org/10.1016/S1002-0160(18)60002-5
Olanrewaju OS, Ayilara MS, Ayangbenro AS, Babalola OO (2021) Genome mining of three plant growth-promoting Bacillus species from maize rhizosphere. Appl Biochem Biotechnol 193:3949–3969. https://doi.org/10.1007/S12010-021-03660-3/FIGURES/8
Oleńska E, Małek W, Wójcik M, Swiecicka I, Thijs S, Vangronsveld J (2020) Beneficial features of plant growth-promoting rhizobacteria for improving plant growth and health in challenging conditions: A methodical review. Sci Total Environ 743:140682. https://doi.org/10.1016/j.scitotenv.2020.140682
Olishevska S, Nickzad A, Déziel E (2019) Bacillus and Paenibacillus secreted polyketides and peptides involved in controlling human and plant pathogens. Appl Microbiol Biotechnol 103(3):1189–1215. https://doi.org/10.1007/S00253-018-9541-0
On SLW, Miller WG, Houf K, Fox JG, Vandamme P (2017) Minimal standards for describing new species belonging to the families Campylobacteraceae and Helicobacteraceae: Campylobacter, Arcobacter, Helicobacter and Wolinella spp. Int J Syst Evol Microbiol 67:5296–5311. https://doi.org/10.1099/ijsem.0.002255
Othoum G, Bougouffa S, Razali R, Bokhari A, Alamoudi S, Antunes A et al (2018) In silico exploration of Red Sea Bacillus genomes for natural product biosynthetic gene clusters. BMC Genomics 19:1–11. https://doi.org/10.1186/S12864-018-4796-5/FIGURES/5
Othoum G, Prigent S, Derouiche A, Shi L, Bokhari A, Alamoudi S et al (2019) Comparative genomics study reveals Red Sea Bacillus with characteristics associated with potential microbial cell factories (MCFs). Sci Rep 9:19254. https://doi.org/10.1038/s41598-019-55726-2
Paul MJ, Primavesi LF, Jhurreea D, Zhang Y (2008) Trehalose metabolism and signaling. Annu Rev Plant Biol 59:417–441. https://doi.org/10.1146/annurev.arplant.59.032607.092945
Penrose DM, Glick BR (2003) Methods for isolating and characterizing ACC deaminase-containing plant growth-promoting rhizobacteria. Physiol Plant 118:10–15. https://doi.org/10.1034/j.1399-3054.2003.00086.x
Prokka ST (2014) Rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. https://doi.org/10.1093/bioinformatics/btu153
Reang L, Bhatt S, Tomar RS, Joshi K, Padhiyar S, Vyas UM et al (2022) Plant growth promoting characteristics of halophilic and halotolerant bacteria isolated from coastal regions of Saurashtra Gujarat. Sci Rep 121(1):1–16. https://doi.org/10.1038/s41598-022-08151-x
Reddy GK, Leferink NGH, Umemura M, Ahmed ST, Breitling R, Scrutton NS et al (2020) Exploring novel bacterial terpene synthases. PLoS ONE 15:e0232220
Richards VP, Palmer SR, Bitar PDP, Qin X, Weinstock GM, Highlander SK et al (2014) Phylogenomics and the dynamic genome evolution of the genus Streptococcus. Genome Biol Evol 6:741–753. https://doi.org/10.1093/gbe/evu048
Richter M, Rosselló-Móra R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131. https://doi.org/10.1073/pnas.0906412106
Ryu C-M, Farag MA, Hu C-H, Reddy MS, Wei H-X, Paré PW et al (2003) Bacterial volatiles promote growth in Arabidopsis. Proc Natl Acad Sci 100:4927–4932. https://doi.org/10.1073/pnas.0730845100
Samaras A, Nikolaidis M, Antequera-Gómez ML, Cámara-Almirón J, Romero D, Moschakis T et al (2021) Whole genome sequencing and root colonization studies reveal novel insights in the biocontrol potential and growth promotion by Bacillus subtilis MBI 600 on cucumber. Front Microbiol 11:3437. https://doi.org/10.3389/fmicb.2020.600393
Sánchez-Hidalgo M, Martín J, Genilloud O (2020) Identification and heterologous expression of the biosynthetic gene cluster encoding the lasso peptide humidimycin, a caspofungin activity potentiator. Antibiotics 9(2):67. https://doi.org/10.3390/antibiotics9020067
Shah S, Li J, Moffatt BA, Glick BR (1998) Isolation and characterization of ACC deaminase genes from two different plant growth-promoting rhizobacteria. Can J Microbiol 44:833–843
Sharif QM, Hussain M, Hussain MT (2007) Chemical evaluation of major salt deposits of Pakistan. J Chem Soc Pakistan 29:569–574
Sheng MM, Jia HK, Zhang GY, Zeng LN, Zhang TT, Long YH et al (2020) Siderophore production by rhizosphere biological control bacteria Brevibacillus brevis GZDF3 of Pinellia ternata and its antifungal effects on Candida albicans. J Microbiol Biotechnol 30:689–699. https://doi.org/10.4014/jmb.1910.10066
Shrivastava P, Kumar R (2015) Soil salinity: a serious environmental issue and plant growth promoting bacteria as one of the tools for its alleviation. Saudi J Biol Sci 22:123–131. https://doi.org/10.1016/j.sjbs.2014.12.001
Sun Z, Zhang W, Guo C, Yang X, Liu W, Wu Y et al (2015) Comparative genomic analysis of 45 type strains of the genus Bifidobacterium: a snapshot of its genetic diversity and evolution. PLoS ONE 10:e0117912
Susič N, Janežič S, Rupnik M, Stare BG (2020) Whole genome sequencing and comparative genomics of two nematicidal Bacillus strains reveals a wide range of possible virulence factors. G3 10:881–890. https://doi.org/10.1534/G3.119.400716
Susin MF, Baldini RL, Gueiros-Filho F, Gomes SL (2006) GroES/GroEL and DnaK/DnaJ have distinct roles in stress responses and during cell cycle progression in Caulobacter crescentus. J Bacteriol 188:8044–8053. https://doi.org/10.1128/JB.00824-06
Tettelin H, Riley D, Cattuto C, Medini D (2008) Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol 11:472–477. https://doi.org/10.1016/j.mib.2008.09.006
Wang Y, Liu H, Liu K, Wang C, Ma H, Li Y et al (2017) Complete genome sequence of Bacillus paralicheniformis MDJK30, a plant growth-promoting rhizobacterium with antifungal activity. Genome Announc 5:e00577-e617. https://doi.org/10.1128/genomeA.00577-17
Wirawan RE, Swanson KM, Kleffmann T, Jack RW, Tagg JR (2007) Uberolysin: a novel cyclic bacteriocin produced by Streptococcus uberis. Microbiology 153:1619–1630. https://doi.org/10.1099/mic.0.2006/005967-0
Xu H, Qin S, Lan Y, Liu M, Cao X, Qiao D et al (2017) Comparative genomic analysis of Paenibacillus sp. SSG-1 and its closely related strains reveals the effect of glycometabolism on environmental adaptation. Sci Rep 7(1):1–11. https://doi.org/10.1038/s41598-017-06160-9
Zhang N, Yang D, Kendall JRA, Borriss R, Druzhinina IS, Kubicek CP et al (2016) Comparative genomic analysis of Bacillus amyloliquefaciens and Bacillus subtilis reveals evolutional traits for adaptation to plant-associated habitats. Front Microbiol 7:2039. https://doi.org/10.3389/fmicb.2016.02039
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Martine Collart.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Iqbal, S., Qasim, M., Rahman, H. et al. Genome mining, antimicrobial and plant growth-promoting potentials of halotolerant Bacillus paralicheniformis ES-1 isolated from salt mine. Mol Genet Genomics 298, 79–93 (2023). https://doi.org/10.1007/s00438-022-01964-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-022-01964-5