
Abstract
Circular RNA (circRNA) is a class of non-coding RNA (ncRNA) that plays an important regulatory role in various biological processes of the organisms and has a major function in muscle growth and development. However, its molecular mechanisms of how it regulates pork quality remain unclear at present. In this study, we compared the longissimus dorsi (LD) muscle expression profiles of Queshan Black (QS) and Large White (LW) pigs to explore the role of circRNAs in meat quality using transcriptome sequencing. A total of 62 differentially expressed circRNAs (DECs), including 46 up- and 16 down-regulated, 39 differentially expressed miRNAs (DEmiRNAs), including 21 up- and 18 down-regulated and 404 differentially expressed mRNAs (DEMs), including 174 up- and 230 down-regulated were identified, and most circRNAs were composed of exons. Our results indicated that the DEC parent genes and DEMs were enriched in the positive regulation of fast-twitch skeletal muscle fiber contraction, relaxation of skeletal muscle, regulation of myoblast proliferation, AMPK signaling pathway, Wnt and Jak-STAT signaling pathway. Furthermore, circSETBP1/ssc-miR-149/PIK3CD and circGUCY2C/ssc-miR-425-3p/CFL1 were selected by constructing the competitive endogenous RNA (ceRNA) regulatory network, circSETBP1, circGUCY2C, PIK3CD and CFL1 had low expression level in QS, while ssc-miR-149 and ssc-miR-425-3p had higher expression level than LW, our analysis revealed that circSETBP1, circGUCY2C, ssc-miR-149, ssc-miR-425-3p, PIK3CD and CFL1 were associated with lipid regulation, cell proliferation and differentiation, so the two ceRNAs regulatory networks may play an important role in regulating intramuscular fat (IMF) deposition, thereby affecting pork quality. In conclusion, we described the gene regulation by the circRNA–miRNA–mRNA ceRNA networks by comparing QS and LW pigs LD muscle transcriptome, and the two new circRNA-associated ceRNA regulatory networks that could help to elucidate the formation mechanism of pork quality. The results provide a theoretical basis for further understanding the genetic mechanism of meat quality formation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The pork quality mainly includes intramuscular fat (IMF) content, dripping loss, tenderness, shear force, meat color, 24 h-pH postmortem and marbling, with these being important economic traits for the pig industry. The high quality and stability of meat products are important goals for the animal husbandry production. Fat is mainly stored subcutaneous, visceral or as IMF in mammals. The IMF content is the decisive factor to determine the meat quality and has a positive effect on the sensory quality traits, while very-low-fat content may lead to poorly flavor (Hocquette et al. 2010). A considerable number of studies have demonstrated the correlation between such as the slaughter weight, meat quality, carcass weight and age of the pig (Latorre et al. 2008; Lanferdini et al. 2018). The animals used in this study were Queshan Black (QS) and Large White (LW) pigs. As a domestic pig breed with a long history and its larger population being located in the Queshan County of Henan Province, QS is a typical fatty pig with high IMF content and excellent meat quality (Li et al. 2019). Due to the high feed conversion rate, rapid muscle growth, and high lean meat rate of the LW, it is a well-known and widely distributed typical western lean pig breed in all countries with developed pig industry in the world. Great differences in the IMF content have been reported between indigenous Chinese and western breeds of pig due to the different feeding methods and genetic background (Shen et al. 2016; Xing et al. 2019). Therefore, these two breeds are quite considerable value for the study of IMF in pigs. It is noteworthy that pigs can be used for biomedical research because they anatomically and physiologically resemble humans. Therefore, it has great significance to study IMF with these two breeds and understanding the fat deposition mechanism in pig muscles will not only help to unravel the molecular mechanism that affects pork quality but also provide a theoretical basis for pig breeding.
RNA-Seq can comprehensively and quickly obtain the sequence and expression information of almost all transcripts in a particular cell or tissue in a certain state, including mRNAs and non-coding RNAs (ncRNAs) (Trapnell et al. 2010). Compared to mRNA, circRNA is a class of ncRNA that can form a covalently closed continuous loop without 5′–3′ polarity and a poly(A) tail recently discovered (Hsu and Coca-Prados 1979), which is generated through back-splicing. Most circRNAs are conserved among species and resistant to RNase R, they are thus more stable than linear RNAs because of their cyclic structure (Qu et al. 2015). Several types of circRNAs have been found since the development of high-throughput sequencing technology, including circular RNA genome, circular RNA intron, circular RNA processing intermediate, circular non-coding RNA and circular RNA spliced exons (Lasda and Parker 2014). CircRNA is a relatively new discovery, and growing evidence shows that circRNA plays a key role in regulating meat quality. It has been reported that circEch1 may be a potential target for promoting muscle development and induce skeletal muscle regeneration and myoblast differentiation (Huang et al. 2021). It was reported that circArhgap5-2 is rich in human conserved circRNAs, and circTshz2-1 is highly expressed in mouse adipose tissue, while circTshz2-1 and circArhgap5-2 are shown to be essential for adipogenesis (Arcinas et al. 2019).
Recent studies found that circRNA acting as competing endogenous RNA (ceRNA) was widely involved in the regulation of meat quality, regulating cell proliferation, differentiation, apoptosis, and regulation of IMF deposition. The definition of ceRNA is based on a hypothesis initially introduced which suggests a new mechanism of mutual regulation of RNA at the post-transcriptional level. CircRNAs as ceRNAs, possess miRNA-binding sites and can serve as natural miRNA inhibitors to regulate gene expression (Salmena et al. 2011). It was found that circ-PLXNA1 could regulate CTNNB1 by adsorbing miR-214, and then affect the adipocytes differentiation of duck (Wang et al. 2020). The circINSR can regulate the proliferation and apoptosis of myoblasts and preadipocytes by negatively regulating miR-15/16 (Shen et al. 2020). It was found that bta-circ-03789-1 and bta-circ-05453-1 may act as miRNA sponges to regulate IGF1R and further affect muscle growth and development (Yan et al. 2020). It has been reported that lipid metabolism may be regulated through the circRNA-0000660-miR-693-IGFBP1 (Chen et al. 2020a). CircFUT10 can function as let-7c sponge to modulate the expression of PPARGC1B to promote the adipocyte proliferation and inhibit the cell differentiation (Jiang et al. 2020). CircSAMD4A acts as a miR-138-5p sponge to increase the expression of EZH2 and regulate the differentiation of preadipocytes (Liu et al. 2020). CircLCLAT1, circFNDC3AL, circCLEC19A and circARMH1 may affect fat formation by acting as miRNA sponges, regulating PPARγ and fatty acid metabolism-related pathways consequently (Zhang et al. 2020).
It is well known that great differences exist in the fat deposition, IMF content and meat quality between indigenous Chinese and western breeds of pig, but the underlying molecular mechanisms are not fully understood currently. Hence QS and LW were selected to study the meat quality differences of indigenous Chinese and western breeds of pig in our study. The LD muscles were collected from about weighing 100 kg QS and LW pigs for transcriptome sequencing. The circRNAs expression was systematically studied, additionally the interactions between circRNAs and miRNAs were predicted. CircRNAs and mRNAs related to fat formation or lipid metabolism were identified. Furthermore, the molecular mechanisms of differentially expressed circRNAs (DECs) and differentially expressed mRNAs (DEMs) regulating fat deposition were studied through functional enrichment and interaction network analysis. These results provide the theoretical basis for further analysis in the regulatory network of fat deposition in pigs.
Materials and methods
Experimental design and sample collection
In present study, both QS and LW were provided by Henan Fenghua Breeding Share Limited Company and kept feeding under the same conditions. Healthy individuals weighing about 100 kg were randomly selected according to the standard procedure. 24 h of fasting and humanely slaughtered, then the LD muscle at the sixth to seventh rib of the right side of the carcass was collected, immediately frozen in liquid nitrogen and stored at -80 ℃ until RNA isolation.
RNA isolation, library preparation, and sequencing
Total RNA was isolated using TRIzol reagent (15596026, Thermo Fisher Scientific, USA) according to the manufacturer's instructions. The concentration and quality of RNA were assessed using NanoDrop One/OneC spectrophotometer (Thermo Fisher Science, USA). We used 1% agarose gel to monitor RNA degradation and contamination, and the RNA Nano 6000 analysis kit of BioAnalyzer 2100 system (Agilent Technologies, California) was used to evaluate the RNA integrity number (RIN), such that only RNA with an RIN value larger than seven was used for sequencing. A NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (NEB, USA) was used to construct miRNA library following the manufacturer’s instructions. Finally, we used the Illumina Hiseq 2500 platform to generate 50 bp single-end reads (Illumina Inc., San Diego, CA, USA).
The ribosomal RNA was removed using the Epicentre Ribozero™ rRNA Removal Kit (Epicentre, USA), and the rRNA free residue was cleaned up by ethanol precipitation. Subsequently, the circRNA and mRNA sequencing library was constructed using the NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s recommendations. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia, San Diego, CA, USA) according to the manufacturer’s instructions. After the clusters were generated, the library was sequenced on an Illumina Hiseq 2500 platform (Illumina Inc., San Diego, CA, USA).
Quality control
Raw data (raw reads) in fastq format were firstly processed through in-house Perl scripts. In this step, clean data (clean reads) were obtained by removing reads containing ploy-N, with the 5’ adapter contaminants, the 3’ adapter or the insert tag, and the ones containing poly A/T/G/C and low-quality reads from raw data. At the same time, the Q20, Q30 and GC contents of the raw data were calculated. All the downstream analyses were based on the clean, high-quality data.
CircRNAs identification
After comparing the clean readout sequence with the pig reference genome, circRNAs were detected and identified using the find_circ and CIRI2 tools (version 1.2). The Circos software (version 0.62-1) was used to construct the Circos figure. Finally, the candidate circRNA with junction reads greater than or equal to 2 was used as the identified circRNA. The expression level of circRNA was reflected by mapped back-splicing junction Reads Per Million mapped reads (RPM).
MiRNA identification
The characteristics of the hairpin structure of the miRNA precursor can be used to predict novel miRNAs. The miREvo (Wen et al. 2012) and miRDeep2 (Friedlander et al. 2012) software applications were integrated to predict novel miRNAs by exploring the secondary structure, Dicer cleavage site and minimum free energy of the small RNA tags that were unannotated in the former steps.
Quantification expression level, differentially expressed and pathway analysis
The differentially expressed circRNAs (DECs), miRNAs (DEmiRNAs) and mRNAs (DEMs) were screened using the DESeq2 R package (version 1.10.1) using as threshold a q-value of 0.05 and an absolute log2(Foldchange) of 1, log2(Foldchange) of 1.45 for circRNA. The DAVID tool was used for performing Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed for the DECs (DECs parent genes) and DEMs to interpret their biological functions using p-value threshold of 0.05 to infer significantly enriched terms (Li et al. 2014).
Construction of the circRNA–miRNA–mRNA network
The RNA connectivity in the ceRNA network was defined as the number of co-expressed targeted miRNAs. Thus, ceRNAs with the highest connectivity were regarded as hub genes, which are more essential in biological networks. To identify possible interactions between miRNAs and mRNAs and circRNAs, the interactions between miRNAs and DEMs and DECs were analyzed using miRanda software (Pasquinelli 2012). The circRNA-miRNA-mRNA network was constructed by assembling all the above-identified co-expression competing triplets and visualized using Cytoscape (version 3.7.1, http://cytoscape.org/).
Validation of gene expression in RNA-seq
Approximately 1 μg of each RNA sample was used the PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (Code No. RR047A, Takara, Beijing, China) to convert the total RNA to cDNA, with random hexamers (for mRNA and circRNA) and Bulge-Loop U6 qPCR Primer Set for miRNAs (RN: R10031.8, RiboBio, Guangzhou, China) according to the manufacturer’s instructions. Next, qRT-PCR was performed using TB Green® Premix Ex Taq™ II (Code No. RR820A, Takara, Beijing, China) on the CFX96 Real Time PCR Detection System (Thermo Fisher Scientific, USA). The reaction program was as follows: denaturation at 95 °C for 3 min, followed by 45 cycles at 95 °C for 10 s and 60 °C for 30 s. Meanwhile, GAPDH (glyceraldehyde-3-phosphate dehydrogenase, for mRNA and circRNA) and U6 (for miRNA) were used as the normalization controls, and all reactions were carried out in triplicates. The 2−ΔΔCT method was used to calculate the relative gene expression levels.
Data accessibility
All raw data of high-throughput sequencing have been deposited to the National Genomics Data Center (NGDC, https://bigd.big.ac.cn) with the dataset accession number CRA005025.
Results
Predictions and properties of circRNAs in porcine LD muscles
The find_circ (Memczak et al. 2013) and CIRI2 (Gao et al. 2018) tools were used to screen circRNA. The results of circRNA identified by the two software applications were merged and intersected according to the position of circRNAs on the chromosome to minimize high false positive. Then, the junction reads of the candidate circRNA were filtered. QS and LW LD muscles generated 935,996,886 raw reads, while 887,486,586 clean reads were obtained (QS and LW obtained 446,859,590 and 440,626,996 clean reads, respectively) after quality control (Table 1). In addition, 7608 circRNAs were detected on all chromosomes (Fig. 1a). It could be found that circRNAs were distributed across all chromosomes, including sex chromosomes, while the number of the ones aligned on chromosome 1 was relatively large. The length of the revealed circRNAs ranged between 186 and 95,950 bp (Fig. 1b). Therefore, it was found that the length of circRNAs greatly varied. In addition, it is known that circRNA can be derived from exon or intron splicing. Our results revealed that most circRNAs consisted of exons (91.47%), while less than 9% of the circRNAs consisted of introns and intergenic areas (Fig. 1c). The reads density of each sample was compared for each chromosome in the genome, and the Circos software application was used to map the distribution of reads on each chromosome. The first 10 contigs or scaffolds were displayed in Fig. 1d.

source statistics and d density distribution on the chromosomes of circRNAs
Predictions and properties of circRNAs in porcine LD muscles. a Chromosomes, b length distribution, c
Differentially expressed analysis of circRNAs, miRNAs and mRNAs between QS and LW
Analysis for DECs, DEmiRNAs and DEMs, we found that 7287 (95.78%) circRNAs, 300 (83.10%) miRNAs and 8734 (86.42%) mRNAs were shared by both QS and LW pigs (Fig. 2a–c). Compared with the LW, there were 62 circRNAs (including 46 up- and 16 down-regulated), 39 miRNAs (including 21 up- and 18 down-regulated) and 404 mRNAs (including 174 up- and 230 down-regulated) that were differentially expressed in the QS (Fig. 2d–f and Table S1–S3).

Differentially expressed analysis of circRNAs, miRNAs and mRNAs between QS and LW. a The specific and shared circRNAs, b miRNAs and c mRNAs between QS and LW. d The differentially expressed circRNAs, e miRNAs and f mRNAs between QS and LW. The red points indicate the up-regulated circRNAs, miRNA or mRNA, the green points indicate the down-regulated ones, and the black points indicate those equally expressed. The vertical dotted lines represent |log2(Foldchange) |= 1, |log2(Foldchange)|= 1.45 for circRNA, and the horizontal dotted lines indicate q-value = 0.05
DECs parent genes and DEMs functional enrichment analysis
After obtaining the DECs parent genes and DEMs between QS and LW, GO and KEGG function and pathway enrichment analyses were performed, and 20 significantly enriched terms were found. The enriched GO terms for the DECs parent genes mainly included positive regulation of epidermal growth factor-activated receptor activity, positive regulation of ubiquitin-dependent protein catabolic process, positive regulation of ubiquitin-protein transferase activity, ubiquitin-dependent protein catabolic process, epidermal growth factor receptor signaling pathway, phosphatidylinositol 5-phosphate metabolic process, regulation of cell migration involved in sprouting angiogenesis, relaxation of skeletal muscle, regulation of NADP metabolic process and regulation of myoblast proliferation (Fig. 3a, Table S4). In addition, the results of the KEGG pathway enrichment analysis (Fig. 3b, Table S5) showed that the DECs parent genes were significantly enriched in Hippo signaling, Tight junction, Ubiquitin mediated proteolysis, Apoptosis, and AMPK signaling pathways.

DECs parent genes and DEMs functional enrichment analysis. a GO biological processes and b KEGG analysis of DECs; c GO biological processes and d KEGG analysis of DEMs
GO enrichment analysis of DEMs showed that including the regulation of I-kappaB kinase/NF-kappaB signaling, anoikis, negative regulation of smooth muscle cell–matrix adhesion, intrinsic apoptotic signaling pathway in response to oxidative stress, negative regulation of NF-kappaB transcription factor activity, aging, phosphatidylinositol-mediated signaling, regulation of embryonic development, positive regulation of vascular associated smooth muscle cell migration and cell motility, were significantly enriched (Fig. 3c, Table S6). KEGG analysis showed that DEMs were significantly enriched in such as the Thyroid hormone signaling pathway, ECM-receptor interaction, NOD-like receptor signaling pathway, Cellular senescence, Rap1 signaling pathway, Th1 and Th2 cell differentiation, FoxO signaling pathway, Osteoclast differentiation, Insulin signaling pathway, Apoptosis, Regulation of actin cytoskeleton, Wnt signaling pathway, Th17 cell differentiation, Jak-STAT signaling pathway and others (Fig. 3d, Table S7).
Construction of a potential circRNA–miRNA–mRNA regulatory network
The ceRNA expression network of circRNA-miRNA-mRNA was constructed to determine the key circRNAs related to pork quality and regulation of cell proliferation and differentiation (Fig. 4a). The resulted co-expression network included 48 mRNAs, 9 miRNAs and 19 circRNAs. Each miRNA may be associated with multiple circRNAs or mRNAs. Using the CoGemiR tool (https://cogemir.tigem.it/) to analyze the conservation of miRNA, it was found that miR-149 and miR-425-3p were highly conserved in pig, human and mouse (Fig. 4b), and these two miRNAs play an important role in cell proliferation and differentiation. The analysis revealed that novel-circ-0017360 and novel-circ-0024588 (Fig. 4c) have miRNA-binding sites and can sponge ssc-miR-149 and ssc-miR-425-3p, respectively. Therefore, based on the above-mentioned results, two regulatory networks were reconstructed, namely novel-circ-0017360/ssc-miR-149/PIK3CD and novel-circ-0024588/ssc-miR-425-3p/CFL1, which represent potential regulators of IMF deposition, muscle growth and development.

Construction of potential circRNA-miRNA-mRNA regulatory network. a Analysis of the circRNA-miRNA-mRNA network. Rectangle nodes represent mRNAs, V nodes represent miRNAs, oval nodes represent circRNAs, red nodes represent the up-regulated, and green nodes represent the down-regulated transcripts in QS and LW; b conservation analysis of two miRNAs; c schematic diagram of circSETBP1 and circGUCY2C formation of SETBP1 and GUCY2C on chromosome. The gray arrows indicate specific junction sites
Validation of gene expression in RNA-seq
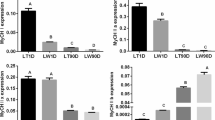
We selected two circRNAs with parent genes related to cell proliferation or adipose regulation. The parent genes were the Sus scrofa SET binding protein 1 (SETBP1) and Sus scrofa guanylate cyclase 2C (GUCY2C), and their corresponding circRNAs were novel-circ-0017360 and novel-circ-0024588, we designated them circSETBP1 and circGUCY2C based on their parent genes, respectively. The miRNAs adsorbed by these two circRNAs as well as their target genes were also validated, and it was found that the expression level of circSETBP1, circGUCY2C, PIK3CD and CFL1 in QS was lower than that in LW, while ssc-miR-149 and ssc-miR-425-3p were opposite (Fig. 5), which was consistent with the regulatory mechanism of ceRNAs, and the qRT-PCR results were in accordance with those of RNA-seq. The primers used in the real-time PCR amplification are listed in Table 2.

Validation of gene expression in RNA-seq. Verification of the accuracy of sequencing results by qRT-PCR. The data represent the Mean ± SEM from 3 biological replicates, and each measurement was repeated three times at least
Discussion
Meat quality is a key economic trait and an important concern for breeders. In our previous study, we found that QS with a higher IMF content have a better pork quality (Wang et al. 2019). In this study, we compared the expression profiles of mRNAs and ncRNAs in the LD muscles of QS and LW to identify the key factors affecting pork quality for the first time. In addition, we reconstructed the potential ceRNA regulatory network, providing a new perspective to study the molecular mechanisms of the pork quality.
CircRNAs can participate in a variety of biological processes through different mechanisms (Shen et al. 2020; Cen et al. 2021). At present, the studies of circRNAs conducted on domestic pigs are relatively few. In this study, a total of 7608 circRNAs were found to be shared from 7287 (95.78%), and there were 62 DECs in QS and LW were obtained.
To further explore the differences between the regulation networks of pork quality from the QS and LW, we performed GO and KEGG pathway enrichment analyses for DECs parent genes. From the GO analysis, we found that DECs parent genes participated in the cellular response to glucose starvation, glycolysis from storage polysaccharide through glucose-1-phosphate in the current study. Previous studies have shown that glucagon and insulin are often considered to be anti-regulatory hormones that play a key role by maintaining glucose homeostasis in animals, regulates lipid synthesis and promotes triglyceride storage in adipocytes (Aronoff et al. 2004). The circRNAs associated with these items, all of which were expressed at higher expression level in QS than in LW and might be involved in the regulation of meat quality. MTMR3 (novel-circ-0007785 parent gene) can regulate cell proliferation and cell cycle progression, in muscle development significantly promoted the proliferation of skeletal muscle satellite cells but inhibited differentiation (Reiss et al. 1986; Hao et al. 2016), novel-circ-0007785 may have an important role in affecting pork quality. EIF2AK3 (novel-circ-0021263 parent gene), related to phenotypes relevant to the function of the digestive system and fat metabolism. PFKM (novel-circ-0025017) was significantly different between Chinese indigenous pig breed and Western pig breeds, and significantly associated with pork quality. Meanwhile, in the GO analyses, circRNA parent genes were also participated in regulation of phosphoprotein phosphatase activity, relaxation of skeletal muscle, regulation of myoblast proliferation. It is well known that the muscle fiber types have an impact on meat quality (Joo et al. 2013). Fat deposition is one of the major factors affecting the quality of the meat. The KEGG analysis implicated Tight junction, AMPK signaling pathway, MAPK signaling pathway. Previous studies have shown that processes related to cell junctions may interact with pathways associated with lipid metabolism, primarily through MAPK activity which affects the deposition of IMF (Cui et al. 2012).
The GO analysis of DEMs revealed significant enrichment in post-transcriptional regulation of gene expression, peptidyl-tyrosine sulfation, protein phosphorylation and negative regulation of amyloid fibril formation, glucose transmembrane transport, especially protein phosphorylation, which is the most prevalent and important post-translational modification of proteins in biology. Many studies showed that protein phosphorylation has important effects on the meat quality (Li et al. 2021b; Doumit and Bates 2000). It was previously reported that the activity of glycolytic enzymes in the LD muscles of pigs was affected by auto-phosphorylation. Thus, protein phosphorylation may improve the muscle glycolytic reaction, and protein phosphorylation may reduce the adverse effects on meat color and tenderness due to protein hydrolysis reaction after slaughtering by improving the muscle glycolytic reaction (Sale et al. 1987; Reiss et al. 1986). There were 20 DEMs, among which 8 and 12 were highly expressed in QS and LW, respectively, which may affect pork quality by affecting protein phosphorylation to different degrees. IMF is inextricably linked to all aspects of meat quality, especially the flavor, tenderness, and juiciness of meat. The amount of its content is also an important factor affecting the assessment of meat marbling (Fernandez et al. 1999a, b), so IMF content plays an important role in meat quality. In the present KEGG analysis, the Thyroid hormone signaling pathway, Osteoclast differentiation, Insulin signaling pathway and Regulation of actin cytoskeleton were significantly enriched, especially including the Wnt signaling pathway (Song et al. 2014) and Jak-STAT signaling pathway (Yao et al. 2019), which are important pathways related to adipogenesis. We also found that pathways associated with cell junctions (ECM-receptor interaction, Focal adhesion, Regulation of actin cytoskeleton) may influence the IMF formation in a pattern related to lipid metabolism (Cui et al. 2012).These results suggest that the differences in meat quality between QS and LW are not only associated with the IMF deposition but also lipid deposition and are jointly regulated by mRNAs and circRNAs. The results from GO and KEGG of DECs parent genes and DEMs showed that it is not only mRNA that affects the pork quality, but circRNA also regulates the deposition of IMF, which in turn affects the pork quality.
At present, more and more studies have shown that circRNAs can play role in regulating the transcription and expression of their parent genes (Siede et al. 2017). However, circRNAs may positively or negatively regulate the expression of their parent genes (Xu et al. 2020). Sus scrofa SET binding protein 1 (SETBP1) (Piazza et al. 2013; Chen et al. 2020b) and guanylate cyclase 2C (GUCY2C) (Folgueira et al. 2020; Rodriguez et al. 2016) have roles in cell proliferation or lipolysis, and serve as the parent genes of circSETBP1 and circGUCY2C, respectively. They may play a decisive role in the function of circRNAs. Thus, circSETBP1 and circGUCY2C may have an impact on cell proliferation and IMF deposition, which in turn affects meat quality. Previous studies have shown that circRNA can rescue the inhibitory effect on mRNA by adsorbing miRNA (Tang et al. 2021). Based on the ceRNA hypothesis that circRNA can sponge miRNA, our results found that circSETBP1 and circGUCY2C could sponge ssc-miR-149 and ssc-miR-425-3p, respectively. Many studies have shown that miR-149 (Mohamed et al. 2014; Jiao et al. 2018; Lu et al. 2018; Peng et al. 2020) and miR-425-3p play an important role in regulating muscle growth and development (Li et al. 2021a). Our analysis revealed that PIK3CD and CFL1 were targets of ssc-miR-149 and ssc-miR-425-3p, respectively. It has been shown that PIK3CD was associated with lipid metabolism and IMF deposition (Cui et al. 2012), CFL1 could regulate adipocyte differentiation (Kamal et al. 2013). Furthermore, our analysis results were consistent with the ceRNA expression pattern, circRNA (circSETBP1, circGUCY2C) and mRNA (PIK3CD, CFL1) had low expression level in QS, while miRNA (ssc-miR-149, ssc-miR-425-3p) expression level was higher than that in LW. Thus, circSETBP1/ssc-miR-149/PIK3CD and circGUCY2C/ssc-miR-425-3p/CFL1 may affect the deposition of IMF by regulating the proliferation and differentiation of adipocytes, thereby regulating the pork quality.
Conclusions
In summary, 62 DECs, 39 DEmiRNAs and 404 DEMs were found by analysis the LD muscle transcriptome of QS and LW, the result indicated that they were widely involved in the pathways related to lipid deposition, cell proliferation and differentiation. Furthermore, two ceRNA regulatory networks including circSETBP1/ssc-miR-149/PIK3CD and circGUCY2C/ssc-miR-425-3p/CFL1 were identified which may be related to meat quality. Taken together, these results may help to explain why indigenous Chinese pigs IMF deposition capacity better than the introduced and providing a theoretical basis for molecular breeding in pig.
References
Arcinas C, Tan W, Fang W, Desai TP, Teh DCS, Degirmenci U, Xu D, Foo R, Sun L (2019) Adipose circular RNAs exhibit dynamic regulation in obesity and functional role in adipogenesis. Nat Metab 1:16. https://doi.org/10.1038/s42255-019-0078-z
Aronoff SL, Berkowitz K, Shreiner B, Want L (2004) Glucose metabolism and regulation: beyond insulin and glucagon. Diabetes Spectr 17:183–190. https://doi.org/10.2337/diaspect.17.3.183
Cen J, Liang Y, Huang Y, Pan Y, Shu G, Zheng Z, Liao X, Zhou M, Chen D, Fang Y, Chen W, Luo J, Zhang J (2021) Circular RNA circSDHC serves as a sponge for miR-127-3p to promote the proliferation and metastasis of renal cell carcinoma via the CDKN3/E2F1 axis. Mol Cancer 20:1. https://doi.org/10.1186/s12943-021-01314-w
Chen Q, Liu M, Luo Y, Yu H, Zhang J, Li D, He Q (2020a) Maternal obesity alters circRNA expression and the potential role of mmu_circRNA_0000660 via sponging miR_693 in offspring liver at weaning age. Gene 731:1. https://doi.org/10.1016/j.gene.2020.144354
Chen Q, Liu M, Luo Y, Yu H, Zhang J, Li D, He Q (2020b) Downregulation of SETBP1 promoted non-small cell lung cancer progression by inducing cellular EMT and disordered immune status. Am J Transl Res 12:16
Cui HX, Liu RR, Zhao GP, Zheng MQ, Chen JL, Wen J (2012) Identification of differentially expressed genes and pathways for intramuscular fat deposition in pectoralis major tissues of fast-and slow-growing chickens. BMC Genomics 13:1. https://doi.org/10.1186/1471-2164-13-213
Doumit ME, Bates RO (2000) Regulation of pork water holding capacity, color and tenderness by protein phosphorylation. Pork Qual 63:6
Fernandez X, Monin G, Talmant A, Mourot J, Lebret B (1999a) Influence of intramuscular fat content on the quality of pig meat—1. Composition of the lipid fraction and sensory characteristics of m. longissimus lumborum. Meat Sci 53:7. https://doi.org/10.1016/s0309-1740(99)00037-6
Fernandez X, Monin G, Talmant A, Mourot J, Lebret B (1999b) Influence of intramuscular fat content on the quality of pig meat—2. Consumer acceptability of m. longissimus lumborum. Meat Sci 53:6. https://doi.org/10.1016/s0309-1740(99)00038-8
Folgueira C, Torres-Leal FL, Beiroa D, Pena-Leon V, Da Silva LN, Milbank E, Senra A, Al-Massadi O, Lopez M, Dieguez C, Seoane LM, Nogueiras R (2020) Oral pharmacological activation of hypothalamic guanylate cyclase 2C receptor stimulates brown fat thermogenesis to reduce body weight. Neuroendocrinology 110:13. https://doi.org/10.1159/000505972
Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N (2012) miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucl Acids Res 40:16. https://doi.org/10.1093/nar/gkr688
Gao Y, Zhang J, Zhao F (2018) Circular RNA identification based on multiple seed matching. Brief Bioinform 19:8. https://doi.org/10.1093/bib/bbx014
Hao F, Itoh T, Morita E, Shirahama-Noda K, Yoshimori T, Noda T (2016) The PtdIns3-phosphatase MTMR3 interacts with mTORC1 and suppresses its activity. FEBS Lett 590:13. https://doi.org/10.1002/1873-3468.12048
Hocquette JF, Gondret F, Baeza E, Medale F, Jurie C, Pethick DW (2010) Intramuscular fat content in meat-producing animals: development, genetic and nutritional control, and identification of putative markers. Anim Int J Anim Biosci 4:17. https://doi.org/10.1017/S1751731109991091
Hsu MT, Coca-Prados M (1979) Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature 280:2. https://doi.org/10.1038/280339a0
Huang K, Chen M, Zhong D, Luo X, Feng T, Song M, Chen Y, Wei X, Shi D, Liu Q, Li H (2021) Circular RNA profiling reveals an abundant circEch1 that promotes myogenesis and differentiation of bovine skeletal muscle. J Agric Food Chem 69:10. https://doi.org/10.1021/acs.jafc.0c06400
Jiang R, Li H, Yang J, Shen X, Song C, Yang Z, Wang X, Huang Y, Lan X, Lei C, Chen H (2020) circRNA profiling reveals an abundant circFUT10 that promotes adipocyte proliferation and inhibits adipocyte differentiation via sponging let-7. Mol Ther Nucl Acids 20:3. https://doi.org/10.1016/j.omtn.2020.03.011
Jiao Y, Huang B, Chen Y, Hong G, Xu J, Hu C, Wang C (2018) Integrated analyses reveal overexpressed Notch1 promoting porcine satellite cells’ proliferation through regulating the cell cycle. Int J Mol Sci 19:1. https://doi.org/10.3390/ijms19010271
Joo ST, Kim GD, Hwang YH, Ryu YC (2013) Control of fresh meat quality through manipulation of muscle fiber characteristics. Meat Sci 95:828–836. https://doi.org/10.1016/j.meatsci.2013.04.044
Kamal AH, Kim WK, Cho K, Park A, Min JK, Han BS, Park SG, Lee SC, Bae KH (2013) Investigation of adipocyte proteome during the differentiation of brown preadipocytes. J Proteomics 94:10. https://doi.org/10.1016/j.jprot.2013.10.005
Lanferdini E, Andretta I, Fonseca LS, Moreira RHR, Cantarelli VS, Ferreira RA, Saraiva A, Abreu MLT (2018) Piglet birth weight, subsequent performance, carcass traits and pork quality: a meta-analytical study. Livest Sci 214:5. https://doi.org/10.1016/j.livsci.2018.05.019
Lasda E, Parker R (2014) Circular RNAs: diversity of form and function. RNA 20:1829–1842. https://doi.org/10.1261/rna.047126.114
Latorre MA, García-Belenguer E, Ariño L (2008) The effects of sex and slaughter weight on growth performance and carcass traits of pigs intended for dry-cured ham from Teruel (Spain). J Anim Sci 86:10. https://doi.org/10.2527/jas.2007-0764
Li A, Zhang J, Zhou Z (2014) PLEK a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinform 15:1. https://doi.org/10.1186/1471-2105-15-311
Li X, Qiao R, Ye J, Wang M, Zhang C, Lv G, Wang K, Li X, Han X (2019) Integrated miRNA and mRNA transcriptomes of spleen profiles between Yorkshire and Queshan black pigs. Gene 688:11. https://doi.org/10.1016/j.gene.2018.11.077
Li J, Tu J, Gao H, Tang L (2021a) MicroRNA-425-3p inhibits myocardial inflammation and cardiomyocyte apoptosis in mice with viral myocarditis through targeting TGF-beta1. Immun Inflamm Dis 9:11. https://doi.org/10.1002/iid3.392
Li X, Zhang D, Ren C, Bai Y, Ijaz M, Hou C, Chen L (2021b) Effects of protein posttranslational modifications on meat quality: a review. Compr Rev Food Sci Food Saf 20:43. https://doi.org/10.1111/1541-4337.12668
Liu Y, Liu H, Li Y, Mao R, Yang H, Zhang Y, Zhang Y, Guo P, Zhan D, Zhang T (2020) Circular RNA SAMD4A controls adipogenesis in obesity through the miR-138-5p/EZH2 axis. Theranostics 10:15. https://doi.org/10.7150/thno.42417
Lu M, Xu L, Wang M, Guo T, Luo F, Su N, Yi S, Chen T (2018) miR149 promotes the myocardial differentiation of mouse bone marrow stem cells by targeting Dab2. Mol Med Rep 17:8. https://doi.org/10.3892/mmr.2018.8903
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, le Noble F, Rajewsky N (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495:333–338. https://doi.org/10.1038/nature11928
Mohamed JS, Hajira A, Pardo PS, Boriek AM (2014) MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1alpha network in skeletal muscle. Diabetes 63:1546–1559. https://doi.org/10.2337/db13-1364
Pasquinelli AE (2012) MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 13:12. https://doi.org/10.1038/nrg3162
Peng W, Li T, Pi S, Huang L, Liu Y (2020) Suppression of circular RNA circDHCR24 alleviates aortic smooth muscle cell proliferation and migration by targeting miR-149-5p/MMP9 axis. Biochem Biophys Res Commun 529:7. https://doi.org/10.1016/j.bbrc.2020.06.067
Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, Antolini L, Mologni L, Donadoni C, Papaemmanuil E, Schnittger S, Kim DW, Boultwood J, Rossi F, Gaipa G, De Martini GP, di Celle PF, Jang HG, Fantin V, Bignell GR, Magistroni V, Haferlach T, Pogliani EM, Campbell PJ, Chase AJ, Tapper WJ, Cross NC, Gambacorti-Passerini C (2013) Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet 45:7. https://doi.org/10.1038/ng.2495
Qu S, Yang X, Li X, Wang J, Gao Y, Shang R, Sun W, Dou K, Li H (2015) Circular RNA: a new star of noncoding RNAs. Cancer Lett 365:8. https://doi.org/10.1016/j.canlet.2015.06.003
Reiss N, Kanety H, Schlessinger J (1986) Five enzymes of the glycolytic pathway serve as substrates for purified epidermal-growth-factor-receptor kinase. Biochem J 239:7. https://doi.org/10.1042/bj2390691
Rodriguez A, Gomez-Ambrosi J, Catalan V, Ezquerro S, Mendez-Gimenez L, Becerril S, Ibanez P, Vila N, Margall MA, Moncada R, Valenti V, Silva C, Salvador J, Fruhbeck G (2016) Guanylin and uroguanylin stimulate lipolysis in human visceral adipocytes. Int J Obes (lond) 40:11. https://doi.org/10.1038/ijo.2016.66
Sale EM, White MF, Kahn CR (1987) Phosphorylation of glycolytic and gluconeogenic enzymes by the insulin receptor kinase. J Cell Biochem 33:12. https://doi.org/10.1002/jcb.240330103
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell 146:6. https://doi.org/10.1016/j.cell.2011.07.014
Shen Y, Mao H, Huang M, Chen L, Chen J, Cai Z, Wang Y, Xu N (2016) Long noncoding RNA and mRNA expression profiles in the thyroid gland of two phenotypically extreme pig breeds using Ribo-Zero RNA sequencing. Genes (basel) 7:1. https://doi.org/10.3390/genes7070034
Shen X, Tang J, Ru W, Zhang X, Huang Y, Lei C, Cao H, Lan X, Chen H (2020) CircINSR regulates fetal bovine muscle and fat development. Front Cell Dev Biol 8:1. https://doi.org/10.3389/fcell.2020.615638
Siede D, Rapti K, Gorska AA, Katus HA, Altmuller J, Boeckel JN, Meder B, Maack C, Volkers M, Muller OJ, Backs J, Dieterich C (2017) Identification of circular RNAs with host gene-independent expression in human model systems for cardiac differentiation and disease. J Mol Cell Cardiol 109:9. https://doi.org/10.1016/j.yjmcc.2017.06.015
Song K, Wang S, Mani M, Mani A (2014) Wnt signaling, de novo lipogenesis, adipogenesis and ectopic fat. Oncotarget 5:4. https://doi.org/10.18632/oncotarget.2769
Tang L, Xiong W, Zhang L, Wang D, Wang Y, Wu Y, Wei F, Mo Y, Hou X, Shi L, Xiong F, Zhang S, Gong Z, Liao Q, Xiang B, Zhang W, Zhou M, Li X, Li G, Guo C, Zeng Z (2021) CircSETD3 regulates MAPRE1 through miR-615-5p and miR-1538 sponges to promote migration and invasion in nasopharyngeal carcinoma. Oncogene 40:15. https://doi.org/10.1038/s41388-020-01531-5
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:5. https://doi.org/10.1038/nbt.1621
Wang MY, Zhang C, Xue YH, Ye JW, Han XL, Qiao RM, Li XL, Liu X, Wang XW, Wu SJ, Wang MM, Li XJ (2019) Comparative analysis of body size indexes, slaughter performance, meat quality and blood indicators between Yuxi Black pigs and Queshan Black pigs (Chinese). China Anim Husb Vet Med 46:10. https://doi.org/10.16431/j.cnki.1671-7236.2019.07.007
Wang L, Liang W, Wang S, Wang Z, Bai H, Jiang Y, Bi Y, Chen G, Chang G (2020) Circular RNA expression profiling reveals that circ-PLXNA1 functions in duck adipocyte differentiation. PLoS ONE 15:1. https://doi.org/10.1371/journal.pone.0236069
Wen M, Shen Y, Shi S, Tang T (2012) miREvo an integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform 13:1. https://doi.org/10.1186/1471-2105-13-140
Xing K, Wang K, Ao H, Chen S, Tan Z, Wang Y, Xitong Z, Yang T, Zhang F, Liu Y, Ni H, Sheng X, Qi X, Wang X, Guo Y, Wang C (2019) Comparative adipose transcriptome analysis digs out genes related to fat deposition in two pig breeds. Sci Rep 9:12925. https://doi.org/10.1038/s41598-019-49548-5
Xu X, Zhang J, Tian Y, Gao Y, Dong X, Chen W, Yuan X, Yin W, Xu J, Chen K, He C, Wei L (2020) CircRNA inhibits DNA damage repair by interacting with host gene. Mol Cancer 19:1. https://doi.org/10.1186/s12943-020-01246-x
Yan XM, Zhang Z, Meng Y, Li HB, Gao L, Luo D, Jiang H, Gao Y, Yuan B, Zhang JB (2020) Genome-wide identification and analysis of circular RNAs differentially expressed in the longissimus dorsi between Kazakh cattle and Xinjiang brown cattle. PeerJ 8:1. https://doi.org/10.7717/peerj.8646
Yao Y, Bi Z, Wu R, Zhao Y, Liu Y, Liu Q, Wang Y, Wang X (2019) METTL3 inhibits BMSC adipogenic differentiation by targeting the JAK1/STAT5/C/EBPbeta pathway via an m(6)A-YTHDF2-dependent manner. FASEB J off Publ Fed Am Soc Exp Biol 33:16. https://doi.org/10.1096/fj.201802644R
Zhang M, Han Y, Zhai Y, Ma X, An X, Zhang S, Li Z (2020) Integrative analysis of circRNAs, miRNAs, and mRNAs profiles to reveal ceRNAs networks in chicken intramuscular and abdominal adipogenesis. BMC Genomics 21:1. https://doi.org/10.1186/s12864-020-07000-3
Acknowledgements
This work was supported by Henan Key Research & Development Program (192102110067 and 202102110242), Pig Industry Technology System Innovation Team Project of Henan Province (S2012-06-G03), Grand Science and Technology Special Project in Tibet (212102110003), and the National Natural Science Foundation of China (32002142).
Author information
Authors and Affiliations
Contributions
Author contributions XH proposed conceptualization, methodology, wrote reviews and editing, administrated project; KQ performed verification and visualization, wrote original draft preparation; XH, YL and CL investigated; XH, XL and XL provided resources; KW and RQ performed supervision. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
All of the experiments involving animals were carried out in accordance with the guidelines for the care and use of experimental animals established by the Ministry of Science and Technology of the People’s Republic of China (Approval Number 2006–398).
Additional information
Communicated by Joan Cerdá.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Qi, K., Liu, Y., Li, C. et al. Construction of circRNA-related ceRNA networks in longissimus dorsi muscle of Queshan Black and Large White pigs. Mol Genet Genomics 297, 101–112 (2022). https://doi.org/10.1007/s00438-021-01836-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-021-01836-4