
Abstract
Whole-genome and segmental duplications coupled with sequence and functional diversification are responsible for gene family expansion, and morphological and adaptive diversity. Although broad contours of such processes are understood, detailed investigations on regulatory elements, such as miRNA-transcription factor modules, especially in non-model crop plants with complex genomes, are few. The present study was performed to understand evolutionary history of MIR159 family, and changes in the miRNA-binding site (MBS) of the targets MYB33, MYB65, and MYB101 that may affect post-transcriptional gene silencing. We established orthology and paralogy between members of MIR159 family by reconstructing the phylogeny based on 240 precursor sequences sampled across green plants. An unambiguous paralogous relationship between MIR159A and MIR159B was observed only in Brassicaceae which prompted us to analyze the origin of this paralogy. Comparative micro-synteny of ca. 100 kb genomic segments surrounding MIR159A, MIR159B, and MIR159C loci across 15 genomes of Brassicaceae revealed segmental duplication that occurred in the common ancestor of Brassicaceae to be responsible for origin of MIR159A–MIR159B paralogy; extensive gene loss and rearrangements were also encountered. The impact of polyploidy was revealed when the three sub-genomes—least fractionated (LF), moderately fractionated (MF1), and most fractionated (MF2) sub-genomes of Brassica and Camelina sativa—were analyzed. Extensive gene loss was observed among sub-genomes of Brassica, whereas those in Camelina were largely conserved. Analysis of the target MYBs revealed the complete loss of MYB33 homologs in a Brassica lineage-specific manner. Our findings suggest that mature miR159a/b /c are capable of targeting MYB65 across Brassicaceae, MYB33 in all species except Brassica, and MYB101 only in Arabidopsis thaliana. Comparative analysis of the mature miRNA sequence and the miRNA-binding site (MBS) in MYB33, MYB65, and MYB101 showed the complexity of regulatory network that is dependent on strict sequence complementarity potentially leading to regulatory diversity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Segmental duplication and recurrent whole-genome duplications (WGD) have played a major role in plant morphological and adaptive diversity (Jiao et al. 2014; Vanneste et al. 2014; Dodsworth et al. 2015; Song and Chen 2015; Panchy et al. 2016; Cheng et al. 2018). Comparative genomics has now emerged as a powerful approach for understanding evolutionary processes on a genome-wide scale including the impact of polyploidization across various taxonomic hierarchies, and its impact on regulatory genes and elements (Ghircuta and Moret 2014; Chaney et al. 2016). In addition, it permits the study of impact of polyploidy on plant development and adaptation, and clues towards identification of orthologs that have implication in plant improvement programs (Peer et al. 2017).
Brassicaceae is a large family with high morphological diversity, and several members are highly economically valued. Six species of Brassica—three allo-tetraploids B. juncea (AABB), B. napus (AACC), and B. carinata (BBCC)—have formed as a result of pairwise interspecific hybridization between three diploid parents B. rapa (AA), B. nigra (BB), and B. oleracea (CC) (Nagaharu 1935; Warwick et al. 2009; Rakow 2004; Cheng et al. 2013). Several members of the family, including ancestral Brassica and Camelina sativa, are known to have the experienced genome triplication, followed by gene losses resulting in three distinct sub-genomes, designated as least fractionated (LF), moderately fractionated (MF1), and most fractionated (MF2) (Lysak et al. 2005, 2007; Wang et al. 2011a, b; Cheng et al. 2012; Kagale et al. 2014; Liu et al. 2014). Brassicaceae also has known histories of genome duplication as paleo-polyploidy (A. thaliana), meso-polyploidy (B. rapa/B. oleracea), and neo-polyploidy (B. napus; C. sativa). Brassicaceae has, thus, also been considered as a model family to analyze the effect of polyploidization and whole-genome duplications.
MicroRNAs are integral parts of regulatory networks involved in development, adaptive responses (Reinhart et al. 2002; Mallory and Vaucheret 2006; Jones-Rhoades et al. 2006; Luo et al. 2013; Comai et al. 2000; Jones-Rhoades et al. 2006), and genomic stability such as mediating responses to genomic shock experienced due to allo-polylploidization as in Arabidopsis suecica, [A. thaliana × (A) arenosa; Ha et al. 2009], and (B) juncea (B. rapa × B. nigra; Ghani et al. 2014).
In plants where sequence-dependent PTGS is the primary mode of interaction between miRNA and the cognate targets, understanding the comparative evolutionary history of microRNA and their targets is important to unravel their conservancy (Comai et al. 2000; Jones-Rhoades et al. 2006; Nozawa et al. 2012).
Previous reports suggest that MIR159 and MIR319 are descendants from a common ancestor (Li et al. 2011). Subsequent to their origin, differences in their mature miRNA sequences and expression domains led to functional specialization (Palatnik et al. 2007). It was also shown that mature miR and miR* region from MIR159 is conserved across land plants, and has more specialized target spectrum than miR319 in A. thaliana (Palatnik et al. 2007; Li et al. 2011). Homologs of MIR159 have been detected across land plants (Palatnik et al. 2007; Li et al. 2011). In Arabidopsis thaliana, MIR159 is a three member gene family with their mature products differing by a single nucleotide. The three targets of miR159–MYB33, MYB65, and MYB101—have been reported to promote floral induction (Achard et al. 2004), vegetative to reproductive transition and anther development (Allen et al. 2007; Millar and Gubler 2005; Alonso-Peral et al. 2012), male-specific cytokinesis (Liu et al. 2017), programmed cell death (PCD), seed germination (Alonso-Peral et al. 2010), leaf morphology, and various abiotic stresses (Li et al. 2016). Similar functions demonstrated in the other species such as Hordeum vulgare (Murray et al. 2003), Oryza sativa (Aya et al. 2009), Lolium temulentum (Woodger et al. 2003), and Fragaria vesca (Csukasi et al. 2012) indicate an evolutionarily conserved regulatory role. In spite of the stated importance, the impact of polyploidization on evolution of MIR159 family and their targets remains unexplored. We, therefore, analyzed in detail the evolutionary history of MIR159 family, and evaluated the impact on the components of the regulatory module MYB33, MYB65, and MYB101.
In the present endeavor, we employed comparative genomics to trace the origin of paralogy of MIR159A–MIR159B, investigated the impact of polyploidy, and co-evolution of miRNA-MBS in target based on strict sequence complementarity that has the potential to alter regulatory network leading to regulatory diversity. We began by estimating and reconstructing the phylogenetic relationship among homologs of MIR159 based on the precursor sequences across entire green plants to establish orthology and paralogy. The analysis led to the identification of Brassicaceae specific paralogy of MIR159A–MIR159B. The origin of MIR159A–MIR159B paralogy in Brassicaceae was an outcome of segmental duplication which was established through synteny-based comparative genomics between homologous and homoeologous segments harboring MIR159A, MIR159B and MIR159C. Impact of polyploidization was fully revealed when genome fractionation analysis was performed. Comparative analysis of mature miRNA of MIR159A, MIR159B, MIR159C, and the microRNA-binding site (MBS) in the putative targets—MYB33, MYB65, and MYB101—demonstrated that the target spectrum and the MBS in the targets known thus far are variously altered and revealed the intricacies of sequence based PTGS interaction between miR159 and target MYBs—MYB33, MYB65, and MYB101 in Brassicaceae. In conclusion, our study demonstrates the utility of comparative genomics to understand that polyploidy can impact regulatory interactions that are dependent on strict Watson–Crick pairing as in the case of miRNA-transcription factors, with a potential to generate regulatory diversity.
Materials and methods
Identification of homologues
Homologues from green plants were identified through BLASTN using precursor sequences of the three members of MIR159—MIR159A, MIR159B, and MIR159C from A. thaliana genome MIR159A/At1g73687 (184 bp); MIR159B/At1g18075 (196 bp); MIR159C/At2g46255 (225 bp) retrieved from miRBase (http://www.mirbase.org; Griffiths-Jones et al. 2006, 2007); Kozomara and Griffiths-Jones 2010, 2013) and used as query. BLASTN was performed at BRAD (http://brassicadb.org; Cheng et al. 2011) and Phytozome (https://phytozome.jgi.doe.gov/pz/portal.html#; Goodstein et al. 2011) databases using the default sets of parameters (program = BLASTN; expect = 10; description = 100; alignment = 50). Database search across miRBase was used for the complete retrieval of sequences across Viridiplantae. The data set reported by Li et al. (2011) on miR159/319 evolution was utilized to retrieve the sequences of miR159 of some of the species. Homologs of MYB33 (At5g06100), MYB65 (At3g11440), and MYB101 (At2g32460) across Brassicaceae were identified using A. thaliana CDS from TAIR Database as query to perform BLASTN at BRAD using the default parameters as described above. Mature miRNA and target-binding site sequences were identified based on miRBase (v21) and published literature and compared.
Phylogenetic reconstruction
Phylogenetic relationships among homologs of MIR159A/MIR159B/MIR159C were estimated using stem-loop precursor sequences from green plants; and using CDS for homologs of MYB33/MYB65/MYB101 from Brassicaceae. Sequences were aligned using Multiple Sequence Alignment using the default settings on MAFFT (http://www.ebi.ac.uk/Tools/msa/mafft/; Katoh and Standley 2013; Katoh et al. 2017). Alignments were saved in .NEXUS format and then subjected to BEAUti for 1,000,000 generations using GTR substitution model (Base frequencies = Estimated; Site Heterogeneity model = Gamma; no. of Gamma categories = 4, Yang96 model, and Yule process) to generate .xml file (Drummond et al. 2012; Bouckaert et al. 2014). This .xml file was then subjected to BEAST v1.8.4. The .tre file generated by BEAST was then annotated through TreeAnnotator using tree cut-off as 250 (Drummond et al. 2012; Bouckaert et al. 2014). The “TREE” file was visualized and manually edited using FigTree v4.3.1 (http://tree.bio.ed.ac.uk/software/figtree/).
Synteny across chromosomal segments and sub-genome fractionation across Brassicaceae
100 kb segments (flanking 50 kb upstream and downstream) harboring MIR159A, MIR159B, and MIR159C were retrieved from 14 genomes of Brassicaceae. Data set of B. rapa, B. nigra, B. oleracea, B. juncea, B. napus, Arabidopsis lyrata, Capsella rubella, Sisymbrium irio, Thellungiella halophila, Thellungiella salsuginea, Aethionema arabicum, and C. sativa was retrieved from BRAD; data sets for Capsella grandiflora and Boechera stricta were obtained from Phytozome. To perform genome fractionation, genomic segments of Brassica and Camelina genome that have lost homologs of MIR159A, MIR159B, and MIR159C were retrieved using A. thaliana homologs of protein-coding genes flanking the MIRNA genes on either side, and selecting the list from “Syntenic Gene” portal (http://brassicadb.org/brad/searchSyntenytPCK.php) of BRAD including all the sub-genomes. As the BRAD portal accepts only protein-coding genes as query, we employed At1g73680 (upstream of MIR159A), At1g18070 (upstream of MIR159B), and At2g46250 (upstream of MIR159C) as query.
Genomic segments were used as input for global alignment using AVID and gVISTA tool (http://genome.lbl.gov/cgi-bin/GenomeVista) against the A. thaliana genome (March 2004/2009 release) using default settings (rank VISTA threshold = 0.5) and conserved regions were visualized using VISTA (Bray et al. 2003; Frazer et al. 2004). The 100 kb segments from each species were then subjected to ab initio gene prediction through FGENESH (http://www.softberry.com) using gene model of A. thaliana (for (A) lyrata and Capsella species) and (B) rapa (rest of the species) as template using default gene finding parameters. Protein sequences obtained by FGENESH prediction tool were used for the BLASTP analysis using BLAST2GO against the nr database at NCBI using E-value = 1.0E−3 and number of BLAST hits = 20, word size = 6; low complexity filter = on; HSP length cut-off = 33; blast description annotator = on. A comparative list was prepared based on the identification of orthologs of A. thaliana, and annotated genes from BLAST2GO analysis. Synteny diagrams were manually constructed to depict gene conservation, loss, and duplications.
Results
Identification of homologues and phylogeny of MIR159
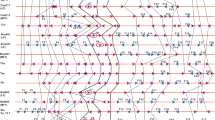
A total of 240 homologs of MIR159A, B, and C were identified from 84 species of green plants (1 family, 1 species of Bryophyte; 1 family, 1 species of Pteridophyte; 2 families, 6 species of Gymnosperms; 15 families, 76 species of Angiosperm) using A. thaliana precursor as query (Supplementary Table 1). We estimated the phylogenetic relationship between these homologs using GTR and doublet models of Bayesian method (Fig. 1). Phylogenetic reconstruction across plants shows two major clades—clade I comprising of MIR159C homologs and clade II with a mix of MIR159A/B homologs. Within both clade I and II, sequences from monocots and dicots form family-specific sub-clades. Clear distinction between MIR159A and MIR159B was visible only in the members of Brassicaceae. For the rest of the taxonomic groups, clusters with a mix of MIR159A + B were observed; within clade II, gymnosperms formed a separate distinct clade, and monocots formed a separate distinct group with a basal angiosperm—Amborella trichopoda. In core eudicots, the representative families (Fabaceae, Salicaceae, Solanaceae, and Rutaceae) formed independent and family-specific clusters of MIR159A/B.

Phylogenetic reconstruction of MIR159A, MIR159B, and MIR159C across green plants using GTR and doublet model of Bayesian method shows two major clades—clade I consisting of MIR159C homologs and clade II of MIR159A/MIR159B homologs. Within each clade, the majority of the homologs formed family-specific clusters. MIR159A/MIR159B were found to be mixed in angiosperms other than Brassicaceae, where a duplication causes origin of MIR159A–MIR159B paralogy (MIR159A—Blue; MIR159B—sea green). (Color figure online)
A total of 33 homologues of MIR159A, 18 of MIR159B, and 18 of MIR159C were identified across 15 Brassicaceae genomes (Table 1), and their orthology was confirmed through AVID/VISTA tool (data not shown) following Singh et al. (2017) before undertaking phylogeny, genome organization, and detailed synteny analysis. A separate phylogeny was reconstructed for members of Brassicaceae to understand the history of the MIR159 family in which also MIR159C formed a separate branch, and MIR159A and MIR159B form a distinct group with paralogous relationship (Supplementary Fig. 2). Within each group, clustering was based on genomic and sub-genomic affiliations–A genome homologs of Brapa_159a_A07-1 with homologs from A07-1 copies of B. napus (A genome counterpart from AC genome) and B. juncea (A genome counterpart from AB genome); B-genome homologs from B03-1 of B. juncea (B genome counterpart from AB genome) with scaffold 186 of B. nigra (B genome); and C genome homolog from C06-1 B. oleracea (C genome) with B. napus (C genome counterpart from AC genome).
Organization and synteny analysis of genomic segments encompassing MIR159A, MIR159B and MIR159C across Brassicaceae
To unravel the cause of paralogy, analyze the genomic organization of segment containing MIR159, and to understand the relationship between genome organization and evolutionary history, we identified homologs of A. thaliana MIR159A, MIR159B, and MIR159C from 14 genomes of Brassicaceae based on sequence similarity of the precursor region and retrieved a total of ca. 100 kb genomic segment, flanking 50 kb on either side of the precursor. Their homology was further validated using global alignment tool AVID/VISTA as previously reported (data not shown, available on request; Singh et al. 2017; Jain and Das 2016).
Each of the genomic segments of ca. 100 kb was subjected to FGENESH analysis followed by gene annotation using BLAST2GO, and the gene content was manually represented (Fig. 2, Supplementary Fig. 2A, B). We also computed overall conservation of A. thaliana homologs present in genomes and sub-genomes (Fig. 3), conservation of genes in each of the genomes and sub-genomes (Fig. 4; Tables 2, 3), and gene density (Supplementary Fig. 4). A comparison of the rate of gene conservation across genomes between the genomic segment harboring MIR159A, MIR159B, and MIR159C revealed that the 100 kb segment containing MIR159C was most conserved with as many as 11 genes conserved in more than 80% of the genomes. MIR159A and MIR159B homologous segments have only 3 and 5 genes, respectively, in more than 80% of the genomes MIR159A/MIR159B.

Micro-synteny analysis across different genomes of Brassicaceae. Synteny analysis across genomic regions (∼ 100 kb) flanking MIR159A showing duplication, losses, and rearrangement in Non-Brassica (a) and Brassica species (b). Protein-coding genes, and MIRNA in A. thaliana and their homologs are represented by different colors, and connected. Genes unique to a particular genomic segment are marked by black arrows; oval region marked as P in Brassica lineage (II) show Brassica juncea specific duplication of miR159A on Chromosome A02. Details of genes in each genome and alphabetical letters marking specific duplications and insertion are discussed in detail in text, and in Supplementary Table 2. (Color figure online)

Graphical representation showing percent conservation of A. thaliana homologs across different genomes of Brassicaceae. Only a few genes (marked by oval) across MIR159A (a, d), MIR159B (b, e) genomic segments are conserved in more than 80% of the genomes, except in MIR159C (c, f) genomic segment which shows the highest conservation among the three segments across Brassicaceae. (Color figure online)

a Graphical representation of number of genes shared across any two or all three sub-genomes (LF and MF1, MF1 and MF2, LF and MF2; LF, MF1, and MF2) harboring MIR159A, MIR159B, and MIR159C. The three sub-genomes of Camelina sativa share the maximum number of genes (orange bar) across all MIR159A, MIR159B, and MIR159C genomic segments. b Gene density (one gene per × kb) across different sub-genomes (LF, MF1, and MF2) harboring MIR159A, MIR159B and MIR159C of B. rapa, B. oleracea, B. napus A, B. napus C, and C. sativa (b). (Color figure online)
Synteny across genomic segments harboring MIR159A
We detected one homologue each of MIR159A in A. lyrata, (A) arabicum, (B) stricta, (C) rubella, C. grandiflora, S. irio, T. halophila, and T. salsuginea; in C. sativa, three copies were detected on chromosome 7, 9, and 16. Among the various Brassica genomes, three copies each of MIR159A were detected each in B. rapa (A genome), B. nigra (B genome), and B. oleracea (C genome); five copies in B. napus (AACC genome) and seven copies in B. juncea (AABB genome). Two copies of MIR159A were located on the same chromosome in B. juncea (ChrA02 within a distance of 11.096 kb; Fig. 2 marked P). A critical analysis of the genomic segments revealed that the 100 kb segments of Bol-C02 and Bnap-A07-1 are not fully sequenced and are represented by N’s; similarly, the genome sequence of Capsella grandiflora is not yet fully assembled, and the 100 kb genomic segment is present on two different scaffolds. To avoid ambiguity and prevent mis-interpretation, we omitted these three genomes for further synteny analysis leaving us with 29 genomic segments. For ease of representation, the output is divided into two figures, with 5 Brassica species (with 17 homoeologous segments representing 18 copies) and 9 non-Brassica species (representing 11 genomes) separately, with the A. thaliana genomic segment being common to both (Fig. 2a, b; Supplementary Table 2).
The 100 kb segment surrounding MIR159A locus in A. thaliana is known to contain 27 protein-coding genes (Fig. 2a, b). Gene prediction based on homology to (A) thaliana/(B) rapa through FGENESH followed by annotation using BLAST2GO revealed that the number of genes ranged from a low of 19 in Bjun_A07-2 (gene density of 1 gene/5.26 kb) to as high as 29 in B. stricta and (C) sativa Chromosome 16 (1 gene/3.44 kb; Supplementary Fig. 4).
Comparative genomic analysis spanning the 100 kb region across all the 29 genomic segments revealed none of the protein-coding genes to be conserved in all the genomes. Alpha-dioxygenase (AT1g73680) was found to be present in all the genomes, except Bjun_B03-1 (27 out of 28 genomes analyzed; Fig. 3a); ETHYLENE INSENSITIVE 3-like 3 (AT1g73730) was present in all the genomes except Bnap_C06 and Bjun_B05 (Fig. 2b; encircled; 93%; Fig. 3a); homologs of AT1G73820 were detected in only 3 out of 28 genomes (10.7%). Homolog of AT1G73770 was deleted from all the genomes and sub-genomes in a Brassica-lineage-specific manner. Two genes in the 100 kb segment surrounding MIR159A in A. thaliana were predicted by FGENESH that were found to be unannotated in the A. thaliana genome release version 10 (TAIR10), and were identified as unknown and Cation transporter (Fig. 2a, b; marked X and Y, respectively). The Cation transporter locus was found to be present in A. lyrata, Csa_chr9, and Csa_chr7.
Analysis of gene content and order revealed gene duplications of either dispersed or tandem class, and specific to a particular genome (such as Prephanate dehydratase family gene in (A) lyrata genome; marked as square box with letter A), Phosphoethanolamine N-methyltransferase 3 (AT1G73600; 99 bp and 506 bp) in (B) rapa_A07-2.; or shared across multiple genomes such as Cysteine-rich receptor kinase 10 across T. halophila and T. salsuginea; Retrovirus-related Pol Poly from transposon TNT 1–94 in T. halophila, and B. stricta (Fig. 2a; square boxes-B and C, respectively). MIR159A itself was found to be duplicated (dispersed type) in B. juncea on chromosome A02 (named as A02-1 and A02-2). In several cases, the orientation of transcription of the duplicated genes was found to be different from each other (e.g., At1g73680 in Fig. 2a, b; red arrow and line). In Csa_Chr16, homologs of genes present upstream of At1g73670 in A. thaliana are completely missing implying a large segmental deletion or rearrangement specific to (C) sativa chromosome 16. 100 kb segment of Bjun_A02 also exhibits the duplication of uncharacterized protein LOC103853400, and triplication of F-box kelch-repeat At4g39560-like. Some more examples of duplication and deletion have been summarized in Table 2.
Synteny across genomic segments harboring MIR159B
BLASTN analysis identified at least one homolog each of MIR59B in all the genomes, except in allopolyploids B. juncea and B. napus, where two copies of MIR159B were identified; no homologs of MIR159B were identified in T. halophila and T. salsuginea. Global sequence alignment revealed that the 100 kb genomic segment of B. napus chromosome C05 (Bnap_C05) does not contain any additional genes homologous to the genomic segment of A. thaliana and was thus not included for synteny analysis. The number of genes predicted ranged from 32 in (A) thaliana (density of one gene/3.125 kb), 33 in (B) stricta (one gene/3.03 kb) to as low as 19 in (C) sativa MF sub-genome (gene density of one gene/5.26 kb; Supplementary Fig. 4B; Supplementary Table 3). Among the 15 genomes and sub-genomes that were analyzed for synteny, we found only MOTHER of FT and TFL1 (AT1G18100), and MIR159B that were shared across all (Supplementary Fig. 2A). Synteny was disrupted on the account of several duplication events including one involving MIR159B in B-genome-specific manner on Chromosome B04 in B. juncea and scaffold 86 in B. nigra (Supplementary Fig. 2A; oval). UNC93-1 and FMN-linked oxidoreductases superfamily genes are deleted in Brassica lineage-specific manner (Supplementary Fig. 2A). Some more examples of duplication and deletion are summarized in Table 2.
Synteny across genomic segments harboring MIR159C
MIR159C is annotated as AT2G46255 in (A) thaliana. At least one homolog of MIR159C was identified in majority of the genomes analyzed; two copies of MIR159C were identified each in (B) juncea genome (Bjun_Contig157 and Bjun_B01), B. napus (Bna_A05 and Bna_C04), and B. nigra (Bnig_scff570 and Bnig_Scff734); and three copies in (C) sativa. In the 100 kb genomic segment that was analyzed across the 18 genomes, the gene density ranged from one gene per 2.77 kb in (A) thaliana (36 genes in 100 kb) to as low as one gene per 5.26 kb in (B) nigra scaffold 570 (19 genes in 100 kb) (Supplementary Fig. 4). The conservation of A. thaliana homologs ranged from genes present in only 5.9% of the genomes (At4g46290, present only in A. thaliana and (C) rubella), to all the genomes surveyed (100%; At4g46230 and At4g46255-MIR159C; Supplementary Fig. 4). Examples of duplication and deletion in the segments harboring MIR159C are summarized in Table 2.
Genome fractionation
A consequence of polyploidization is the creation of multiple copies of the genome that over the course of evolution experiences gene loss creating distinct sub-genomes and homologous copies. The genomes of Brassica and C. sativa within Brassicaceae are known to have experienced triplication and composed of three distinct sub-genomes annotated as least fractionated (LF), moderately fractionated (MF1), and most fractionated (MF2). Given the triplicated status of the Brassica and C. sativa genomes, at least three copies of the MIRNA genomic segments are expected in B. rapa, B. nigra, and B. oleracea, and C. sativa; and six copies in B. napus and B. juncea. We analyzed the impact of genome fractionation across the genomic segments containing MIR159A, MIR159B, and MIR159C in Brassica and C. sativa (Supplementary Tables 5, 6).
MIR159A
MIR159A was found to be present in all the three sub-genomic fractions of B. rapa, B. napus C and C. sativa; in B. oleracea (CC) and B. napus A, it is presented only in MF1 and MF2 fractions, indicating LF fraction-specific deletion in B. oleracea (CC). In B. napus C (AACC) genome, the MIR159A was lost from LF of A genome after natural hybridization between B. rapa (AA) and B. oleracea (CC).
In B. rapa, gene prediction and annotation revealed the presence of 30, 17, and 20 genes on A07-1 (LF), A02 (MF1), and A07-2 (MF2), respectively. Only two genes, i.e., Alpha-dioxygenase 2-like (Supplementary Fig. 5I; marked as A) and Ethylene insensitive 3-like 3 (Fig. 8I; marked B), are shared among all three genome fractions; four genes are shared among LF and MF1, whereas six genes are shared among LF and MF2 genome fractions, of which probable inactive serine–threonine-kinase fnkc (Supplementary Fig. 5I; marked C) gene is present in three copies in MF2 fraction.
In B. oleracea, genomic segments on LF (C06-1), MF1 (C02), and MF2 (C06-2) contain 26, 68, and 23 genes, respectively, with most of the genes be unique to each sub-genomes. Only two genes alpha-dioxygenase 2 (Fig. 5II; marked A) and ethylene insensitive 3-like 3 (Supplementary Fig. 5II; marked B) are shared among all three sub-genomes; and Detoxification 17 (Supplementary Fig. 5II; marked as D) and Suppressor of mec-8 and unc-52 homolog 1 (Supplementary Fig. 5II; marked E) were shared among LF and MF2 sub-genome.
B. napus, an allotetraploid (AACC) of B. rapa (AA) and B. oleracea, (CC) shows least retention of genes between sub-genomes. Analysis of genomic segment encompassing MIR159A revealed 14, 21, and 24 genes on A07-1 (LF), A02 (MF1), and A07-2 (MF2), respectively, with no gene shared among all three sub-genomes. LF and MF2 share three genes of which Probable inactive serine–threonine-kinase fnkc gene (Supplementary Fig. 5III; marked as C) is presented in three copies in MF2 fraction. The three sub-genomic segments corresponding to C genome of B. napus have 43, 32, and 26 genes on C06-1 (LF), C02 (MF1), and C06-2 (MF2), respectively. LF and MF1 fractions do not share any gene, while LF and MF2 fractions share nine genes of which Detoxification 17 has two copies in LF fraction (Supplementary Fig. 5IV; marked I).
In C. sativa, three genomic segments LF (Chr16), MF1 (Chr07), and MF2 (Chr09) contain 67, 42, and 41 genes of which as many as 28 genes are shared among all the three sub-genomes (Supplementary Fig. 5V; Table 3). Several genes on MF1 such as Bifunctional inhibitor lipid-transfer seed storage 2S albumin superfamily (marked J), ERAD-associated E3 ubiquitin- ligase component HRD3A-like (marked K), Detoxification 16-like isoform X1 (marked L), Ssu72-like family (marked M), Receptor kinase At4g00960 (marked N), and DNA ligase (marked O) are duplicated on LF and/or MF2 segments; Thaumatin isoform X1 (marked P) present as two copies on MF1 fraction is shared among the rest two genomic segments. A total of four genes—Coiled-coil domain-containing 1-like (marked S), Non-specific lipid-transfer 2-like (marked T), SAR-Deficient 1 (marked U), Core-2 I-branching beta-1,6-N-acetylglucosaminyltransferase family (marked V), are shared between LF and MF2 fractions of which Core-2 I-branching beta-1,6-N-acetylglucosaminyltransferase family has three copies in LF fraction. Some more examples of duplication and deletion are summarized in Table 3.
MIR159B
Analysis of homologous fragments corresponding to sub-genomes revealed loss of MIR159B in several genomes as a result of genome fractionation as it was detected in only LF fractions of B. rapa, B. oleracea, and B. napusA, and completely deleted from all three sub-genomes of C genome in B. napus. MIR159B was found to be present on all the sub-genomic fractions of C. sativa.
In B. rapa, the three sub-genomic fragments have variable number of genes with LF-A06 (29 genes) fraction genomic segment having the most number of genes (MF1-A08-21 genes and MF2-A09-17 genes). Most of the genes are unique to respective genomic fragments, and only 5, 2, and 3 genes are shared among LF-MF1, LF-MF2, and MF1-MF2 genomic fragments, respectively (Table 3).
In B. oleracea, sub-genomic segments on C06-1 (LF), C02 (MF1), and on C06-2 (MF2) contain 60, 31, and 17 genes, respectively, with most of the genes unique to sub-genomes. A single gene—Nuclear poly (A) polymerase 1 (Supplementary Fig. 6II; marked A)—was found to be shared by all three sub-genomes; only four genes shared between LF and MF1 sub-genomes; three genes between LF and MF2; and only a single gene, Cyclin-dependent kinase D-3 (Supplementary Fig. 6II; marked I) shared among MF1 and MF2 (Table 3).
The sub-genomes of the A genome of B. napus contain highly variable number of genes, 43 in LF (A06), 17 in MF1 (A08), and 15 in MF2 (A09). Most of the genes from LF have no homologs on MF1 and MF2. Eukaryotic peptide chain release factor G5TP- binding subunit ERF3A gene (Supplementary Fig. 6IV; marked as P) was found to be shared across all the sub-genomic fractions of B. napus C, indicating that deletion of MIR159B is not because of the deletion of the whole segment (Table 3; Fig. 4; Supplementary Fig. 6IV).
In C. sativa, LF and MF1 contain 38 genes each, and MF2 contains 34 genes. Four genes are shared only between LF and MF1, and five between only LF and MF2. A single gene Peptidyl-prolyl cis-trans isomerase FKBP17-chloroplastic-like (Supplementary Fig. 6V; marked as O) is shared between only MF1 and MF2. Most of the other genes are shared among all the three segments (Supplementary Fig. 6V; Laccase 1-marked L; 2 copies in LF).
MIR159C
Genome fractionation analysis in B. rapa revealed that the three sub-genomic segments harbor gene numbers ranging from only 3 genes (MF1), 11 (MF2), and 38 in LF (Supplementary Fig. 7I), with no shared genes among all the three sub-genomes. Only five genes were found to be shared between LF and MF2 fraction segments.
The MF1 sub-genomic segment of A-genome in B. napus was found to contain several incomplete sequence stretches, and was thus excluded from analysis. Seven genes are shared between LF (total 33 genes) and MF2 (total 13 genes) sub-genomic segment. The sub-genomic segments—LF, MF1, and MF2 of C genome in B. napus—contain 44, 8, and 16 genes, respectively. Two genes (Supplementary Fig. 7IV; marked A and B) are shared among all three fractions; LF and MF1 share four genes, while LF and MF2 share five (Fig. 4a; Table 3).
In C. sativa, three sub-genomic segments LF (Chr04), MF1 (Chr06), MF2 (Chr05) contain 35, 39, and 34 genes of which 27 genes are common to all. Two genes—Myosin heavy chain and Eukaryotic translation initiation factor 3 subunit I isoform X1—from LF sub-genome are duplicated on MF1 and/or MF2 segments (Supplementary Fig. 7V; marked C and D).
Comparison of synteny across the sub-genomic fractions for MIR159A (Supplementary Fig. 5), MIR159B (Supplementary Fig. 6), and MIR159C (Supplementary Fig. 7) revealed that among all the genomic segments, C. sativa displayed most number of genes to be conserved in all the fractions (Fig. 4). Gene density was found to be as high as one gene/3.37 kb (BnapusCMF2) to as low as one gene/7.53 kb (Bol_LF) in MIR159A genomic segments; from one gene/2.87 kb (Brapa MF2) to one gene/7.0125 kb (BnapusC LF) in MIR159B segments; and from one gene/3.39 kb (BnapusA MF2) to as low as one gene/8.82 kb (Brapa MF1) in MIR159C segments (Fig. 4b).
Segmental duplication
Phylogenetic reconstruction revealed that the paralogous relationship between MIR159A and MIR159B is specific to Brassicaceae. Whether this paralogy arose as a result of local duplication or as a part of segmental duplication was analyzed by performing pairwise synteny analysis between paralogs, i.e., between genomic segments harboring MIR159A and MIR159B, across genomes of Brassicaceae. Eight genes apart from MIR159A–MIR159B were paralogous and syntenic in S. irio, six genes in A. thaliana (Fig. 5a) and C. rubella, five in (A) lyrata, four in (B) stricta, between one and three genes in (C) sativa, and a single gene in B.rapa—MF2 and B. oleracea—LF (Fig. 5). The Serine–threonine-kinase EdR-like was found to be retained across paralogous blocks in majority of the genomes (Fig. 5; brown box).

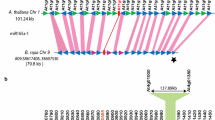
Analysis of synteny between genomic segments harboring MIR159B (At1g18075) and MIR159A (At1g73687) to establish segmental duplication as a cause of paralogy. a Diagrammatic representation of genomic segment containing MIR159B and MIR159A present on the p and q arms, respectively, on chromosome 1 of A. thaliana, shows the presence of six common genes. b Diagrammatic representation of comparison of genes present in genomic segment containing MIR159B and MIR159A for 11 genomes (Ath—A. thaliana, Alyr—(A) lyrata, Crub—C. rubella, Siri—S. irio, and genomic fractions of Csa—C. sativa, Bol—(B) oleracea, and Bra—B. rapa). Colored blocks represent the genes in the each genomic segment. (Color figure online)
Phylogeny of MYB33, MYB65, and MYB101
We estimated the phylogenetic relationship between 67 homologs of selected members of MYB family—MYB33, MYB65, and MYB101—from Brassicaceae using the CDS data set through GTR and doublet models implemented under the Bayesian method. These are reported to be PTGS targets of miR159 in A. thaliana. We also compared the conservation and divergence in the microRNA-binding site (MBS), along with the conservation of the mature 21-nt miRNA sequence along the phylogenetic cluster.
Homologs of MYB33 were not detected in any of the Brassica species. We observed three distinct clusters for the three MYBs with MYB33 and MYB65 sharing a recent common ancestor (Fig. 6). Within each of the clades, the tree topology reflected the clustering of homologs of base genome, and homoeologs from sub-genomes together. For instance, homologs from Arabidopsis species, Capsella species, Thellungiella species grouped with each other; similarly, homoeologs from LF1 of A genome, B genome, and C genome grouped together (and so on for MF1 and MF2). We further located the MBS within the sequences, and analyzed sequence and length polymorphisms in Brassicaceae (Supplementary Fig. 8A, B; Fig. 7). The mature miR159 sequence was found to be conserved across the Brassicaceae except in MIR159B of Brassica species, and in the MIR159C derived from B genome (B. nigra and B. juncea). Analysis of the MBS showed that the miRNA-binding region was entirely missing in MYB65 from A genome in B. napus (Fig. 7; Supplementary Fig. 8A, B). The MBS, especially the target cleavage site (at 9/10 position, 5′-TTCA-3′), was found to be conserved within the homologs of MYB65 and MYB33 across members of Brassicaceae (Fig. 7a). The MBS in MYB101, however, showed variation. In A. thaliana, the MBS was similar/identical to that found in MYB65/33, especially with reference to the nucleotide composition at the putative cleavage site at 9/10 position (TTCA/TTCT) (Fig. 7b). In all the other members of Brassicaceae, the putative MBS along with the cleavage site revealed sequence polymorphisms—ACCG in Arabidopsis lyrata and TGCG in the rest of the species (Fig. 7; Supplementary Fig. 8).

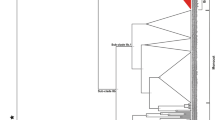
Phylogenetic reconstruction of homologs of MYB33, MYB65, and MYB101 from Brassicaceae using GTR and doublet model of Bayesian method. MYB33 (Magenta) and MYB65 (Blue) form sister clade. MYB33 was found to be completely absent from Brassica. (Color figure online)

Sequence comparison of mature 21-nt miR159A, miR159B, and miR159C to the miRNA-binding site (MBS) in the target transcription factors—MYB33, MYB65, and MYB101—across Brassicaceae to reveal polymorphism and conservation. Sequence alignment revealed sequence divergence and the genomes can be categorized into various groups (details in Supplementary Fig. 8). Group 1 refers to A. thaliana homolog in each category. Identical nucleotides are marked by dot; any polymorphism is marked by respective nucleotide; sequence complementarity is marked by vertical line. Boxed region and arrow indicates seed region (9/10) and cleavage site. Brassica specific deletion of “T” can be observed in miR159B group 2, 3, and 4 (boxed). Maximum conservation in miRNA-binding site (MBS) and mature miRNA was detected in MYB33 and MYB65 (a). MYB101 shows extensive sequence polymorphism at MBS across Brassicaceae (b). (Color figure online)
Discussion
Understanding the genome organization, evolutionary history, and genomics of regulatory elements, especially of those in polyploid genomes, and in crop species remains a major challenge and is an important area of research. To the best of our knowledge, this is the first detailed analysis of the evolutionary history of MIR159 family and co-evolution of miRNA-binding sites (MBS) in PTGS targets. Phylogenetic reconstruction across green plants revealed paralogy between MIR159A–MIR159B only in Brassicaceae. Analysis and comparison of phylogeny and synteny both across orthologous and paralogous segments across Brassicaceae implicate a segmental duplication in ancestral Brassicaceae being responsible for origin of MIR159A–MIR159B paralogy. The impact of polyploidy including genome fractionation was evident when homoeologous segments were analyzed with respect to gene content and conservation status. Homology search indicated that MYB33 is completely lost in Brassica species, but retained in rest of the members of Brassicaceae. A comparison of the mature miRNA from miR159a, miR159b, and miR159c, and the miRNA-binding site (MBS) of the PTGS targets showed that the mature miRNA isoforms are capable of targeting MYB65 across Brassicaceae, MYB33 in all species except Brassica, and MYB101 only in A. thaliana. Results from the present study, thus, reveal novel insights into the genomics and evolution of MIR159 family, and comparative analysis of the regulatory pair of miR159—MYB33/MYB65/MYB101 unraveled intricacies between components of regulatory module that are involved in sequence-dependent interactions, such as in post-transcriptional gene silencing (PTGS). The full impact of polyploidization, genome fractionation, and sequence polymorphism in the mature miRNA and the cognate targets on functional diversification including neo- and sub-functionalization can only be quantified when functional analysis of the regulatory modules vis-à-vis their role in various developmental and adaptive processes are undertaken in future.
A combinatorial analysis that investigates genome organization and structure in a phylogenetic context is useful to gain insights into evolutionary and functional aspects of modules of regulatory elements that act in pairs. An example is miRNA–PTGS target as module pairs in plants that largely function through post-transcriptional gene silencing (PTGS) based on strict criteria of perfect or near-perfect sequence complementarity (Voinnet 2009; Axtell and Bowman 2008). Polyploid genomes with paralogous copies can accumulate polymorphisms and exhibit sequence diversity in either component of the module. Such polymorphisms can lead to disruption of function and possibility of perturbation of existing networks, and may allow the formation of novel regulatory interactions and networks. The present investigation was designed to understand the evolutionary history of MIR159, a three member gene family in A. thaliana with mature 21-nt isoforms that differ by one nucleotide (Allen et al. 2010); they regulate several developmental processes such as transition to flowering time (Achard et al. 2004; Millar and Gubler 2005), anther development (Allen et al. 2007), and biotic (Du et al. 2014) and abiotic (Li et al. 2016) stress responses. The regulation of selected member of MYBs-MYB33, MYB65, and MYB101 by miR159a, miR159b, and miR159c through strict complimentary base pairing via PTGS is known (Palatnik et al. 2007; Li et al. 2011). However, evolutionary history, impact of polyploidy on MIR159 gene family, sequence variation, if any, encountered in the mature 21-nt sequence in MIR159 family in the other species outside A. thaliana, spectrum of potential PTGS targets, and on miRNA-target pairing that is critical for sequence-dependent PTGS remains unexplored.
Our mining of plant databases revealed that, from a single copy in Physcomitrella patens and Selaginella moellendorffii, MIR159 has undergone several events of genome and family-specific expansion, and we could detect as many as 11 copies in B. juncea and Zea mays and 10 copies in B. napus and O. sativa. The single copy in Selaginella moellendorffii coincides with the lack of evidence of whole-genome duplication (Baniaga et al. 2016; Banks et al. 2011; Jiao et al. 2011). Phylogenetic reconstruction revealed the clustering of homologs in a family-/lineage-specific manner, implying that the expansion in gene family is an outcome of family-/lineage-specific expansion events. Such expansion of gene families along the plant phylogenetic tree has also been demonstrated in KCS gene family (Singh et al. 2018). A clear demarcation and paralogy between MIR159A and MIR159B was detected only in Brassicaceae. We, thus, analyzed data from members of Brassicaceae, exploiting the availability of a number of complete genome sequences including those after various stages polyploidization such as paleo-polyploidy (A. thaliana; Blanc and Wolfe 2004), meso-polyploidy (B. rapa/B.oleracea; Wang et al. 2011a, b), and neo-polyploidy (B. napus; C. sativa; Chalhoub et al. 2014; Kagale et al. 2014). Brassicaceae-specific whole-genome duplication events such as mesoploidy (triplication in B. rapa and B. oleracea, allo-tetraploidization in B. napus and B. juncea) and neopolyploidy (hexaploidy in C. sativa) resulting in duplication, retention, and losses of several genes, and has been proposed to be responsible for evolution of a group of plants diverse in form and function (Kellogg 2016; Tank et al. 2015). In the present study, synteny across Brassicaceae revealed origin and expansion of genes in genome- or lineage-specific manner (e.g., homolog Brassica—lineage-specific deletion of AT1G73770; Thellungiella—lineage-specific duplication of Cysteine-rich receptor kinase 10), and disruption of synteny.
Genomes of Brassica and C. sativa have undergone triplication in past, and thus, the genomes of B. rapa, B. nigra, B. oleracea, and C. sativa are composed of three distinct sub-genomes annotated as LF, MF1, and MF2, with the pattern of gene retention being LF > MF1 > MF2 (Wang et al. 2011a, b). We expected at least three copies of the MIRNA genomic segments in B. rapa, B. nigra, B. oleracea, and C. sativa; and six copies in B. napus and B. juncea. The present study did not show a clear pattern of LF > MF1 > MF2 among the MIR159 genomic segments. In contrast, the previous comparative genomic analyses have revealed that miRNA-encoding genes are subjected to similar rules of genome and sub-genome fractionation and diversification as protein encoding genes do (Jain and Das 2016).This discordance implies that rules of genome fractionation are not uniform across the entire genome landscape.
When synteny analysis was correlated with results obtained from genome fractionation analysis using homologous segments, the extent and the complexity of gain and loss of genes and genetic elements become evident. For instance, MIR159B is retained in only LF fractions of B. rapa (A genome), B. oleracea (C-genome), and B. napus A genome but not in any of the sub-genomes of C genome of B. napus (including LF) implying B. napus C-genome-specific loss of LF counterpart. In contrast, Nuclear poly (A) polymerase and EF1A were found to be retained across all the sub-genomes of A and C genomes in MIR159B segment. EF1A is involved in translation termination in response to termination codons and stimulates the activity of ERF1 (Valouev et al. 2002). Nuclear Poly (A) Polymerase generates 3′-poly (A) tail of mRNAs and also required for endo-ribonucleocytic cleavage reaction at some polyadenylation sites (Proudfoot 2011). Conservation of such genes across all fractions probably reflects their indispensable role in transcriptional and translational processes. A detailed investigation on the selection pressure operative on the genetic elements flanking MIR159 family will throw an additional light on the evolutionary trajectory. Loss/retention and copy-number expansion of genes in multiple genomes and sub-genomes are best addressed through character and phylogenetic state reconstruction as was shown recently (Singh et al. 2018). Synteny and genome fractionation analysis revealed extensive gene loss and gain in Brassica species as compared to C. sativa given the older age of polyploidization in Brassica lineage than Camelina (Kagale et al. 2014).
Phylogenetic analysis among green plants clearly revealed that MIR159A and MIR159B share a result of paralogous relationship limited to Brassicaceae. A combination of synteny among the paralogous segments of MIR159A and MIR159B in a pairwise manner across each genome showed the presence of several other genes other than MIR159A–MIR159B raising the probability that the genomic segments harboring MIR159A and MIR159B have arisen as an outcome of segmental duplication. MIR159B and MIR159A are present on the top arm and bottom arm of chromosome 1 of A. thaliana, which has been shown to be related by a large segmental duplication and also responsible for expansion of MIR395A–B–C and MIR395D–E–F family and KCS5–KCS6 paralogy (Rathore et al. 2016; Singh et al. 2018). Taken together data that have been previously published (Rathore et al. 2016; Singh et al. 2018) and data obtained in the present investigation, we can conclude that MIR159A–MIR159B paralogy arose due to a segmental duplication that is shared across Brassicaceae.
Small RNAs such as miRNAs involved in the regulation of their targets through PTGS require precise base pairing. This is in contrast to translational repression where a relaxed pairing criterion is applicable (Carthew and Sontheimer 2009). It is evident that the 21-nt mature miRNA sequence, and the MBS in the target are under a strict selection pressure which does not permit any mutation to accumulate (Zhang et al. 2006). Any mutation in mature miRNA or in the MBS of the target, thus, has the potential to disrupt the RNA:RNA interaction necessary for PTGS, and can lead to formation of novel regulatory interaction (Chen and Rajewsky 2007; Wang and Adams 2015). The probability of such disruptions in interaction is higher in polyploid genomes.
The potential targets of MIR159—MYB33, MYB65, and MYB101—in A. thaliana, are post-transcriptionally silenced through target cleavage (Li et al. 2011; Zheng et al. 2017). Mere presence of miRNA-binding site (MBS) in the target transcript is not sufficient to ensure PTGS through target cleavage; accessibility of the MBS in the target is governed through the formation of secondary structure and polymorphism in the sequences flanking MBS that influences secondary structure of the target has been shown to be responsible for efficiency of cleavage of miR159 target across plants (Zheng et al. 2017). The MBS for miR159 was found to be conserved among eight MYB-TFs including MYB33, MYB65, MYB81, MYB97, MYB101, MYB104, MYB120, and DUO1. However, the flanking sequences—100 bp each on 5′- and 3′- of MBS of only MYB33 and MYB65—was predicted to permit the formation of a RNA stem-loop structure which correlated with highly efficient cleavage of the transcripts by miR159 (Zheng et al. 2017). We did not detect any homologs of MYB33 in Brassica species leaving only MYB65 and MYB101 as being potential targets in Brassica. A comparison of sequence complementarity between mi159a/b/c isoforms and MYB33/MYB65/MYB101 revealed that MYB65 is a universal potential target across Brassicaceae, MYB33 remains a target in all species examined except Brassica species, and MYB101 acts as a target only in A. thaliana. Existence of such sequence polymorphism in MBS of MYB101 can lead to change in spectrum of target leading to specialization.
References
Achard P, Herr A, Baulcombe DC, Harberd NP (2004) Modulation of floral development by a gibberellin-regulated microRNA. Development 131:3357–3365
Allen RS, Li J, Stahle MI, Dubroue A, Gubler F, Millar AA (2007) Genetic analysis reveals functional redundancy and the major target genes of the Arabidopsis miR159 family. Proc Natl Acad Sci 104:16371–16376
Allen RS, Li J, Alonso-Peral MM, White RG, Gubler F, Millar AA (2010) MicroR159 regulation of most conserved targets in Arabidopsis has negligible phenotypic effects. Silence 1:18–36
Alonso-Peral MM, Li J, Li Y, Allen RS, Schnippenkoetter W, Ohms S, White RG, Millar AA (2010) The microRNA159-regulated GAMYB-like genes inhibit growth and promote programmed cell death in Arabidopsis. Plant Physiol 154(2):757–771
Alonso-Peral MM, Sun C, Millar AA (2012) MicroRNA159 can act as a switch or tuning microRNA independently of its abundance in Arabidopsis. PLoS One 7:e34751
Axtell MJ, Bartel DP (2005) Antiquity of microRNAs and their targets in land plants. Plant Cell 17:1658–1673
Axtell MJ, Bowman JL (2008) Evolution of plant microRNAs and their targets. Trends Plant Sci 13:343–349
Axtell MJ, Snyder JA, Bartel DP (2007) Common functions for diverse small RNAs of land plants. Plant Cell 19:1750–1769
Aya K, Ueguchi-Tanaka M, Kondo M, Hamada K, Yano K, Nishimura M, Matsuoka M (2009) Gibberellin modulates anther development in rice via the transcriptional regulation of GAMYB. Plant Cell 21:1453–1472
Baniaga AE, Arrigo N, Barker MS (2016) The small nuclear genomes of Selaginella are associated with a low rate of genome size evolution. Genome Biol Evol 8:1516–1525
Banks JA, Nishiyama T, Hasebe M, Bowman JL, Gribskov M, dePamphilis C (2011) The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 332:960–963
Blanc G, Wolfe KH (2004) Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16:1679–1691
Blazquez MA, Weigel D (2000) Integration of floral inductive signals in Arabidopsis. Nature 404:889–892
Bouckaert R, Heled J, Kuhnert D, Vaughan T, Wu CH, Xie D, Drummond AJ (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537
Bray N, Dubchak I, Pachter L (2003) AVID: a global alignment program. Genome Res 13:97–102
Carthew RW, Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655
Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, Wang X, Chiquet J, Belcram H, Tong C, Samans B, Corréa M (2014) Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345:950–953
Chaney L, Sharp AR, Evans CR, Udall JA (2016) Genome mapping in plant comparative genomics. Trends Plant Sci 21:770–780
Chen K, Rajewsky N (2007) The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8:93–103
Cheng F, Liu S, Wu J, Fang L, Sun S, Liu B, Wang X (2011) BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol 11:136–141
Cheng F, Wu J, Fang L, Sun S, Liu B, Lin K, Bonnema G, Wang X (2012) Biased gene fractionation and dominant gene expression among the subgenomes of Brassica rapa. PloS one 7(5):e36442
Cheng F, Mandakova T, Wu J, Xie Q, Lysak MA, Wang X (2013) Deciphering the diploid ancestral genome of the mesohexaploid Brassica rapa. Plant Cell 25:1541–1554
Cheng F, Wu J, Cai X, Liang J, Freeling M, Wang X (2018) Gene retention, fractionation and subgenome differences in polyploid plants. Nat Plants 4:258–268
Comai L, Tyagi AP, Winter K, Holmes-Davis R, Reynolds SH, Stevens Y, Byers B (2010) Phenotypic instability and rapid gene silencing in newly formed Arabidopsis allotetraploids. Plant Cell 12(9):1551–1567
Csukasi F, Donaire L, Casanal A, Martinez-Priego L, Botella MA, Medina-Escobar N, Valpuesta V (2012) Two strawberry miR159 family members display developmental-specific expression patterns in the fruit receptacle and cooperatively regulate Fa-GAMYB. New Phytol 195:47–57
Dodsworth S, Chase MW, Leitch AR (2015) Is post-polyploidization diploidization the key to the evolutionary success of angiosperms? Bot J Linn Soc 180:1–5
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973
Du Z, Chen A, Chen W, Westwood JH, Baulcombe DC, Carr JP (2014) Using a viral vector to reveal the role of microRNA159 in disease symptom induction by a severe strain of Cucumber mosaic virus. Plant Physiol 164:1378–1388
Franzke A, Lysak MA, Al-Shehbaz IA, Koch MA, Mummenhoff K (2011) Cabbage family affairs: the evolutionary history of Brassicaceae. Trends Plant Sci 16:108–116
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32:273–279
Ghani MA, Li J, Rao L, Raza MA, Cao L, Yu N, Zou X, Chen L (2014) The role of small RNAs in wide hybridisation and allopolyploidization between Brassica rapa and Brassica nigra. BMC Plant Biol 14:272–284
Ghiurcuta CG, Moret BM (2014) Evaluating synteny for improved comparative studies. Bioinformatics 30:9–18
Gocal GF, Sheldon CC, Gubler F, Moritz T, Bagnall DJ, MacMillan CP, Li SF, Parish RW, Dennis ES, Weigel D, King RW (2001) GAMYB-like genes, flowering, and gibberellin signaling in Arabidopsis. Plant Physiol 127:1682–1693
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Rokhsar DS (2011) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40:1178–1186
Griffiths-Jones S (2004) The microRNA registry. Nucleic Acids Res 32:109–111
Griffiths-Jones S, Grocock RJ, Van Dongen S, Bateman A, Enright AJ (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 34:140–144
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2007) miRBase: tools for microRNA genomics. Nucleic Acids Res 36:154–158
Ha M, Lu J, Tian L, Ramachandran V, Kasschau KD, Chapman EJ, Carrington JC, Chen X, Wang XJ, Chen ZJ (2009) Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc Natl Acad Sci 106:17835–17840
Jain A, Das S (2016) Synteny and comparative analysis of miRNA retention, conservation, and structure across Brassicaceae reveals lineage-and sub-genome-specific changes. Funct Integr Genom 16:253–268
Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE (2011) Ancestral polyploidy in seed plants and angiosperms. Nature 473:97–100
Jiao Y, Li J, Tang H, Paterson AH (2014) Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. Plant Cell 26:2792–2802
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Kagale S, Koh C, Nixon J, Bollina V, Clarke WE, Tuteja R, Spillane C, Robinson SJ, Links MG, Clarke C, Higgins EE (2014) The emerging biofuel crop Camelina sativa retains a highly undifferentiated hexaploid genome structure. Nat Commun 5:3706–3716
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780
Katoh K, Rozewicki J, Yamada KD (2017) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 1–7
Kellogg EA (2016) Has the connection between polyploidy and diversification actually been tested? Curr Opin Plant Biol 30:25–32
Kozomara A, Griffiths-Jones S (2010) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39:152–157
Kozomara A, Griffiths-Jones S (2013) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:68–73
Li Y, Li C, Ding G, Jin Y (2011) Evolution of MIR159/319 microRNA genes and their post-transcriptional regulatory link to siRNA pathways. BMC Evol Biol 11:122–140
Li Y, Alonso-Peral M, Wong G, Wang MB, Millar AA (2016) Ubiquitous miR159 repression of MYB33/65 in Arabidopsis rosettes is robust and is not perturbed by a wide range of stresses. BMC Plant Biol 16:179–191
Liu S, Liu Y, Yang X, Tong C, Edwards D, Parkin IA, Wang X (2014) The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun 5:3930–3941
Liu B, De Storme N, Geelen D (2017) Gibberellin induces diploid pollen formation by interfering with meiotic cytokinesis. Plant Physiol 173:480–512
Luo Y, Guo Z, Li L (2013) Evolutionary conservation of microRNA regulatory programs in plant flower development. Dev Biol 380:133–144
Lysak MA, Koch MA, Pecinka A, Schubert I (2005) Chromosome triplication found across the tribe Brassiceae. Genome Res 15:516–525
Lysak MA, Cheung K, Kitschke M, Bure SP (2007) Ancestral chromosomal blocks are triplicated in Brassiceae species with varying chromosome number and genome size. Plant Physiol 145:402–410
Mallory AC, Vaucheret H (2006) Functions of microRNAs and related small RNAs in plants. Nat Genet 38:31–36
Millar AA, Gubler F (2005) The Arabidopsis GAMYB-like genes, MYB33 and MYB65, are microRNA-regulated genes that redundantly facilitate anther development. Plant Cell 17:705–721
Murray F, Kalla R, Jacobsen J, Gubler F (2003) A role for HvGAMYB in anther development. Plant J 33:481–491
Nagaharu U (1935) Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. J Jpn Bot 7:389–452
Nozawa M, Miura S, Nei M (2012) Origins and evolution of microRNA genes in plant species. Genome Biol Evol 4:230–239
Palatnik JF, Wollmann H, Schommer C, Schwab R, Boisbouvier J, Rodriguez R, Warthmann N, Allen E, Dezulian T, Huson D, Carrington JC (2007) Sequence and expression differences underlie functional specialization of Arabidopsis microRNAs miR159 and miR319. Dev Cell 13:115–125
Panchy N, Lehti-Shiu M, Shiu SH (2016) Evolution of gene duplication in plants. Plant Physiol 171:2294–2316
Proudfoot NJ (2011) Ending the message: poly(A) signals then and now. Genes Dev 25:1770–1782
Rakow G (2004) Species origin and economic importance of Brassica. Springer, Berlin, pp 3–11
Rathore P, Geeta R, Das S (2016) Microsynteny and phylogenetic analysis of tandemly organised miRNA families across five members of Brassicaceae reveals complex retention and loss history. Plant Sci 247:35–48
Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP (2002) MicroRNAs in plants. Genes Dev 16:1616–1626
Singh NK, Anand S, Jain A, Das S (2017) Comparative genomics and synteny analysis of KCS17–KCS18 cluster across different genomes and sub-genomes of Brassicaceae for analysis of its evolutionary history. Plant Mol Biol Report 35:237–251
Singh S, Das S, Geeta R (2018) A segmental duplication in the common ancestor of Brassicaceae is responsible for the origin of the paralogs KCS6-KCS5, which are not shared with other angiosperms. Mol Phylogenet Evol 126:331–345
Song Q, Chen ZJ (2015) Epigenetic and developmental regulation in plant polyploids. Curr Opin Plant Biol 24:101–109
Tank DC, Eastman JM, Pennell MW, Soltis PS, Soltis DE, Hinchliff CE, Brown JW, Sessa EB, Harmon LJ (2015) Nested radiations and the pulse of angiosperm diversification: increased diversification rates often follow whole genome duplications. New Phytol 207:454–467
Valouev IA, Kushnirov VV, Ter-Avanesyan MD (2002) Yeast polypeptide chain release factors eRF1 and eRF3 are involved in cytoskeleton organization and cell cycle regulation. Cytoskeleton 52:161–173
Van de Peer Y, Mizrachi E, Marchal K (2017) The evolutionary significance of polyploidy. Nat Rev Genet 18:411–424
Vanneste K, Baele G, Maere S, Van de Peer Y (2014) Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous–Paleogene boundary. Genome Res 24:1334–1347
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136:669–687
Wang S, Adams KL (2015) Duplicate gene divergence by changes in microRNA binding sites in Arabidopsis and Brassica. Genome Biol Evol 7:646–655
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Huang S (2011a) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039
Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, Bai Y, Mun JH, Bancroft I, Cheng F, Huang S (2011b) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43:1035–1039
Warwick SI, Francis A, Gugel RK (2009) Guide to wild germplasm of Brassica and allied crops (tribe Brassiceae, Brassicaceae). Agriculture and Agri-Food Canada, Canada
Woodger FJ, Millar A, Murray F, Jacobsen JV, Gubler F (2003) The role of GAMYB transcription factors in GA-regulated gene expression. J Plant Growth Regul 22:176–184
Zhang B, Pan X, Cobb GP, Anderson TA (2006) Plant microRNA: a small regulatory molecule with big impact. Dev Biol 289:3–16
Zheng Z, Reichel M, Deveson I, Wong G, Li J, Millar AA (2017) Target RNA secondary structure is a major determinant of miR159 efficacy. Plant Physiol 174:1764–1778
Funding
The study was supported by a DBT Grant number BT/PR628/AGR/36/674/2011 to SD. Financial assistance in the form of JRF/SRF to SA from DBT and non-NET fellowship to ML from DU/UGC is gratefully acknowledged. SD would also like to acknowledge Delhi University for the financial and infrastructural support through R&D Grants.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Akhilesh K. Tyagi.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2019_1540_MOESM1_ESM.ai
Reconstruction of phylogenetic relationship among homologs and homeologs of MIR159A, MIR159B and MIR159C across Brassicaceae using GTR and doublet model of Bayesian method. Homologs and homeologs of MIR159A-MIR159B can be observed forming sister clade. Within each branch, genome and sub-genome (LF/MF1/MF2) specific clustering can be observed reflecting that the MIR159A-MIR519B paralogy occurred in the ancestral Brassicaceae and precedes genome duplication. (AI 190 KB)
438_2019_1540_MOESM2_ESM.ai
Micro-synteny analysis across genomic regions (100 kb) flanking MIR159B(A) and MIR159C(B) from different genomes of Brassicaceae showing duplication, losses and rearrangement of Protein coding genes, and MIRNA in A. thaliana and their homologs are represented by different colours, and connected. Genes unique to a particular genomic segment are marked by black arrows. Details of genes in each genome and alphabetical letters marking specific duplications and insertion are discussed in detail in text, and in supplementary table 3 and 4. A: Black square blocks marked as A and B show Capsella grandiflora specific segmental duplication; black oval region show BB genome specific duplication in B. juncea and B. nigra. Rounded rectangle indicates A. thaliana specific gene duplication. (AI 1082 KB)
438_2019_1540_MOESM3_ESM.ai
Graphical representation of percentage conservation of genes predicted by AVID/VISTA and FGENESH analysis in homologous and homeologous genomic segments harbouring MIR159A (A), MIR159B (B) and MIR159C (C) across 14 genomes (A. lyrata, C. rubella, C. sativa, B. rapa, B. oleracea, B. napus, B. juncea, B. nigra, S.irio, T. halophila, T. salsuginea, B. sricta, C. grandiflora, and A. arabicum). (AI 2713 KB)
438_2019_1540_MOESM4_ESM.ai
Graphical representation of gene density (one gene per xx kb) in homologous and homeologous genomic segments harbouring miR159A (A), miR159B (B) and miR159C (C) across 14 genomes (A. lyrata, C. rubella, C. sativa, B.rapa, B. oleracea, B. napus, B. juncea, B. nigra, S. irio, T. halophila, T. salsuginea, B. sricta, C. grandiflora, and A. arabicum). (AI 5031 KB)
438_2019_1540_MOESM5_ESM.ai
Impact of genome fractionation on three sub-genomes (LF, MF1 and MF2) of B. rapa (I), B. oleracea (II), B. napus A (III), B. napus C (IV) and C. sativa (V) harbouring MIR159A. Alphabetical letters represent various genes exhibiting retention and the losses (explained in text, and supplementary tables 5, 6) (AI 1133 KB)
438_2019_1540_MOESM6_ESM.ai
Impact of genome fractionation on three sub-genomes (LF, MF1 and MF2) of B. rapa (I), B. oleracea (II), B. napus A (III), B. napus C (IV) and C. sativa (V) harbouring MIR159B. Alphabetical letters represent various genes exhibiting retention and the losses (explained in text, and supplementary tables 5, 6). (AI 1120 KB)
438_2019_1540_MOESM7_ESM.ai
Impact of genome fractionation on three sub-genomes (LF, MF1 and MF2) of B. rapa (I), B. oleracea (II), B. napus A (III), B. napus C (IV) and C. sativa (V) harbouring MIR159C. Alphabetical letters represent various genes exhibiting retention and the losses (explained in text, and supplementary tables 5, 6). (AI 1242 KB)
438_2019_1540_MOESM8_ESM.ppt
Sequence polymorphism in mature 21-nt miR159A, miR159B and miR159C (A) and miRNA binding site (MBS) in the target transcription factors- MYB33, MYB65 and MYB101 (B) across selected members of Brassicaceae. (PPT 516 KB)
Rights and permissions
About this article

Cite this article
Anand, S., Lal, M. & Das, S. Comparative genomics reveals origin of MIR159A–MIR159B paralogy, and complexities of PTGS interaction between miR159 and target GA-MYBs in Brassicaceae. Mol Genet Genomics 294, 693–714 (2019). https://doi.org/10.1007/s00438-019-01540-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-019-01540-4