
Abstract
The wild species in the Triticeae tribe are tremendous resources for crop breeding due to their abundant natural variation. However, their huge and highly repetitive genomes have hindered the establishment of physical maps and the completeness of their genome sequences. To develop molecular markers for the efficient utilization of their valuable traits while avoiding their genome complexity, we assembled RNA sequences of ten representative accessions of Aegilops tauschii, the progenitor of the wheat D genome, and estimated single nucleotide polymorphisms (SNPs) and insertions/deletions (indels). The deduced unigenes were anchored to the chromosomes of Ae. tauschii and barley. The SNPs and indels in the anchored unigenes, covering entire chromosomes, were sufficient for linkage map construction, even in combinations between the genetically closest accessions. Interestingly, the resolution of SNP and indel distribution on barley chromosomes was slightly higher than on Ae. tauschii chromosomes. Since barley chromosomes are regarded as virtual chromosomes of Triticeae species, our strategy allows capture of genetic markers arranged on the chromosomes in order based on the conserved synteny. The resolution of these genetic markers will be comparable to that of the Ae. tauschii whose draft genome sequence is available. Our procedure should be applicable to marker development for Triticeae species, which have no draft sequences available.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The tribe Triticeae includes many economically valuable cereals (e.g., barley, rye, and wheat) and their wild relatives. Aegilops tauschii Coss., the D-genome donor of bread wheat (Triticum aestivum L.), has been utilized for breeding wheat as a donor of important agricultural traits such as abiotic and biotic stress resistance (Matsuoka et al. 2013, 2015; Yang et al. 2014; Sehgal et al. 2015). The wild related species provide morphologically and genetically high diversity, acting as resources for potentially useful variability for cereal breeding. To accelerate introduction of these traits to crop species, it is necessary to develop efficient methods to find genetic markers linking the traits. However, the genome size of the Triticeae species is too large to develop high-quality physical maps, and whole genome sequencing of these species is still challenging.
Barley (Hordeum vulgare L.) provides one of the highest-quality genome sequences in the Triticeae. A high-resolution physical map and over 20,000 full-length cDNAs have also been released (Matsumoto et al. 2011; The International Barley Genome Sequencing Consortium, IBSC 2012). Barley is evolutionarily closer to wheat and its wild species than are other monocot species with high-quality genome sequences. Barley and wheat ancestors diverged 13 million years ago (Gaut 2002). Chromosomal synteny of gene location is highly conserved between barley and wheat (Mayer et al. 2011; Wicker et al. 2011). Using this conserved synteny, genetic markers linked to phenotypes have been isolated. For example, Nishijima et al. (2014) found tightly linked markers of the Iw2 locus of Ae. tauschii, which encodes a cuticular wax inhibitor and provides an evolutionary clue for the establishment of bread wheat. The D-genome donor of bread wheat is assumed to have been Ae. tauschii accessions with a recessive iw2 allele, the habitat of which is limited to the area roughly from Transcaucasia to the southern coastal region of the Caspian Sea (Tsunewaki et al. 1966; Nishijima et al. 2014). Although the genome sequence of Ae. tauschii is available (Jia et al. 2013), many contigs have failed to be connected to chromosomal positions. A physical map of Ae. tauschii was constructed as well using more than 400,000 bacterial artificial chromosomes (Luo et al. 2013), but not all the sequences are accessible, preventing identification of tightly linked markers. To solve these problems, several SNP markers derived from RNA sequencing analyses (Iehisa et al. 2012, 2014) were assigned to the Iw2 region, and the Ae. tauschii genome contigs that carry these markers were selected. Based on the synteny between barley and Ae. tauschii, additional contigs were assigned near the marker-carrying contigs, and the sequences of these additional contigs were searched for simple sequence repeat (SSR) motifs (Nishijima et al. 2014).
Designing useful SSR markers is laborious. Commonly, SSR markers are built based on repeats in the genome sequence and tested to determine whether these markers can detect variations in recombinant populations. To avoid this arduous process, a systematic way of designing genetic markers is needed. Whole genome sequencing of multiple samples from wild Triticeae species is not necessarily a convenient approach due to their large genome and the costs of sequencing. RNA sequencing will be a powerful approach to solve these problems. Here are some of its advantages: (1) it is applicable to species that have large and complex genomes, as it excludes intergenic repetitive regions, (2) it allows mapping of detected polymorphisms to chromosomes based on conserved coding regions and synteny between other grass species that have high-quality genome sequences, such as barley, rice, and Brachypodium distachyon, and (3) it can be used to measure gene expression level as well.
Iehisa et al. (2014) performed RNA sequencing from leaf and young spike tissues of two Ae. tauschii accessions using the 454 Life Sciences pyrosequencer and identified 13,347 high-confidence SNPs. The short reads were aligned to de novo assembled transcripts to call SNPs, and the assembled transcripts were mapped to the barley genome. At that time, barley full-length cDNA information was unavailable. The comparisons between two accessions provided a limited number of genetic markers for the Ae. tauschii population (Iehisa et al. 2014). The Triticeae can be treated as a single genetic system, sharing homoeologous chromosomes (Mayer et al. 2011; Wicker et al. 2011), and the highly conserved synteny among the Triticeae species enables conducting genetic analyses of wild Triticeae species whose genome sequences are unavailable.
Here, we performed RNA sequencing of ten accessions of Ae. tauschii on the Illumina MiSeq platform to develop molecular markers. Through the RNA sequencing assembly, a large number of SNPs was found between accessions, even within the same lineages. The SNPs and indels were anchored to the chromosomes of Ae. tauschii and barley, and were distributed over the entire chromosomes. Using this approach, a large number of genetic markers can be efficiently obtained in Triticeae species even if the reference genome is incomplete.
Materials and methods
Plant materials, library construction and RNA sequencing
Based on population structure analyses, Ae. tauschii can be divided into two major lineages L1 and L2 (Table 1) (Mizuno et al. 2010; Matsuoka et al. 2013). Ae. tauschii that has given rise to the establishment of common wheat is presumed to belong to L2 (Mizuno et al. 2010; Matsuoka et al. 2013; Nishijima et al. 2014). The large part of the gene pool of Ae. tauschii remains unexplored for wheat breeding, especially in L1. The ten accessions of Ae. tauschii used were chosen as representative of the two major lineages, L1 and L2 (Table 1). Total RNA was extracted from leaves at the seedling stage using a Plant Total RNA Extraction Miniprep System (Viogene, Taipei Hsien, Taiwan, ROC). A total of 4 µg of RNA was used for constructing paired-end libraries following the manufacturer’s instructions. The libraries were sequenced for 300 cycles × 2 on an Illumina MiSeq sequencer with 300-bp paired-end reads. Sequencing data have been deposited to DDBJ BioProject PRJDB4683.
De novo assembly of short reads from RNA sequencing
Quality control of short reads was performed using the FastQC tool of the Galaxy portal (Giardine et al. 2005; Blankenberg et al. 2010; Goecks et al. 2010). Low-quality bases (average Phred quality score per 4 bp < 30), adapter sequences and reads <100 bp were removed using the Trimmomatic version 0.32 tool (Bolger et al. 2014). After filtering, paired short reads were merged into single reads based on overlapping regions using PEAR version 0.9.7 software (Zhang et al. 2014). The merged reads were assembled de novo using Trinity version 2.1.1 software (Grabherr et al. 2011; Haas et al. 2013) to reconstruct transcripts for each accession of Ae. tauschii. Trinity generates a fasta file, in which each nucleotide sequence harbors a header with an isoform number (e.g., i1 = isoform #1, i2 = isoform #2, etc.). If a gene had multiple isoforms, the transcript sequence of isoform #1 was picked up from the fasta file to construct a set of unigenes. The assembled transcripts have been deposited to Transcriptome Shotgun Assembly (TSA) division of DDBJ BioProject PRJDB4683.
Mapping the assembled transcripts to Ae. tauschii and barley genome sequences
The representative transcripts were mapped to the Ae. tauschii reference genome “v1.25 masked” (Jia et al. 2013) and barley reference genome “v1.26 masked” (IBSC 2012) in Ensembl Plants (Kersey et al. 2015) using Gmap version 2013-03-31 software (Wu and Watanabe 2005; Quinlan and Hall 2010). For mapping to scaffolds of the Ae. tauschii genome, if the identity in the aligned region was >95 % and the coverage of the aligned region per total length of a transcript was >90 %, the transcript was regarded as mapping to a scaffold of Ae. tauschii. For mapping to the barley genome, if the identity was >78 % and the coverage was >77 %, the transcript was regarded as mapping to a barley chromosome.
Quantification of gene expression and identification of SNPs and indels between tested accessions
The merged reads of each Ae. tauschii accession were aligned pairwise to their corresponding unigenes using the Bowtie2 tool (Langmead and Salzberg 2012) with local alignment, generating 10 × 10 alignment outputs. We estimated depth of coverage over a transcript using SAMtools software with the command “samtools idxstats” (Li et al. 2009) and calculated counts per million (CPM) as a measure of gene expression level. Correlation coefficients for the CPM of each transcript between accessions were calculated using R software.
SNPs and indels were called from the alignment outputs using SAMtools and Coval software with the option “-freq 0.95 -m 1000000 -n 10” (Kosugi et al. 2013). SNP calling with Coval is successful in identification of causal SNPs and genetic markers linked to phenotypes (Abe et al. 2012; Takagi et al. 2013). To acquire novel SNP markers between the Ae. tauschii accessions, we chose only homozygous SNPs and indels, because Ae. tauschii is a self-fertilizing species. Anchoring of SNPs and indels to the scaffolds of Ae. tauschii and barley chromosomes was performed based on the outputs of Gmap for mapping unigenes to these scaffolds and chromosomes. To link the scaffolds of Ae. tauschii to the genetic map of Ae. tauschii (Luo et al. 2013), the positional information from Supplementary Material 4 in Iehisa et al. (2014) was used. All SNP information is available from our laboratory web site (http://www.lab.kobe-u.ac.jp/ans-plantgenetics/index.html).
Marker construction and genotyping
To assess the usefulness of the SNP library, cleaved amplified polymorphic sequence (CAPS) or derived CAPS markers were developed to the chromosomal region around Iw2, a dominant non-glaucous allele, on the short arm of chromosome 2D. In our previous study, some Ae. tauschii scaffolds were anchored to this region (Nishijima et al. 2014). SNPs mapping to these scaffolds in silico were used to develop molecular markers. The primer sequences for each SNP marker and relevant restriction enzymes are summarized in Supplementary Table S1. PCR and subsequent analyses were performed as described previously (Iehisa et al. 2012, 2014).
SNP assignment and association analysis
In Nishijima et al. (2014), we developed a genetic map of Iw2 using F2 mapping population derived from a cross between KU-2003 (L1 lineage and non-glaucous phenotype) and KU-2124 (L2 lineage and glaucous phenotype). Newly generated SNP markers through the RNA sequencing analysis were assigned to this framework. Another 210 Ae. tauschii accessions (Takumi et al. 2011; Nishijima et al. 2014) were also genotyped using these markers to assess the genetic associations among the markers and glaucous phenotype. Association was analyzed using a mixed linear model function in TASSEL ver. 5.0 software (Bradbury et al. 2007). The Q matrix of population membership probabilities was derived from population structure analysis of Ae. tauschii accessions and kinship was calculated in TASSEL based on the genotyping information of the 169 DArT markers for the 206 Ae. tauschii accessions (Matsuoka et al. 2013).
Results
Reconstruction of transcript sequences of the ten accessions of Aegilops tauschii
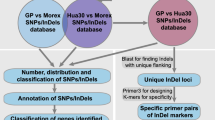
To obtain novel genetic markers for the D genome, we performed RNA sequencing of the ten accessions of Ae. tauschii (Table 1). To cover the entire genetic variation of Ae. tauschii, we chose representative accessions from both lineages I (L1) and II (L2) including sublineages (Mizuno et al. 2010; Matsuoka et al. 2013, 2015). Altogether, 4.8–5.8 million paired-end short reads were obtained for each accession (Supplementary Table S2) and analyzed according to the workflow shown in Fig. 1. After filtering and merging of these short reads, 2.6–3.6 million high-quality reads were acquired. The average length of the merged reads was about 327 bp. After de novo assembly of the merged reads, 33,680–65,827 transcripts were reconstructed for each accession, with N50 values of 1369–1519 bp. If a transcript had more than one isoform, one of the isoforms from the transcript was selected as a unigene. Finally, a set of unigenes for each accession, containing 29,386–55,268 representative isoforms, was obtained (Supplementary Table S3).

The RNA sequencing analysis workflow for constructing genetic markers of Ae. tauschii
To evaluate how many unigenes link to the published genome sequences of Ae. tauschii (Jia et al. 2013) and barley (IBSC 2012), the unigene sequences for each accession were aligned to these genome sequences. Of the total unigenes, 65.7–79.7 % were mapped to Ae. tauschii and 29.8–39.1 % to barley genome sequences (Supplementary Table S3). As expected, the number of unigenes that mapped to Ae. tauschii sequences was higher than to barley sequences. Only a few percent of the unigenes specifically mapped to the barley sequences. The unigenes unmapped to both genome sequences occupied 15.6–29.8 % of the total and could have been derived from accession-specific genomic regions or could be due to the incompleteness of the reference genome.
In order to compare nucleotide sequences of transcripts between Ae. tauschii accessions, plants were grown under the same conditions and their leaves were sampled at the same stage. To assess the pattern of gene expression in these samples, we estimated CPM, a commonly used parameter of gene expression, and calculated the correlation coefficient of CPM between all the possible pairs of the ten accessions (Supplementary Table S4). All the pairwise comparisons showed significant positive correlations, indicating that there was no drastic difference in gene expression between the tested samples.
Detection of nucleotide polymorphisms in Ae. tauschii
To estimate how many potential genetic markers between accessions could be constructed, we counted SNPs and indels from the pairwise comparisons according to the thresholds with read depth >3 (Supplementary Table S5) or >10 (Table 2). More SNPs and indels were found under the thresholds with read depth >3. Although the set of SNPs and indels with read depth >3 may include some false positives, it may be useful in searching for genetic markers to use for genetic linkage analysis when no marker candidates are available in the set with read depth >10. Here, we conducted downstream analyses using the set of high-confidence SNPs and indels with read depth >10.
The number of SNPs and indels between accessions is dependent on comparisons between and within lineages (Table 2). In pairwise comparisons between the same accessions, 98–1492 SNPs and 4–20 indels were detected. These SNPs and indels could be false positives generated by nucleotide differences between paralogous genes in the same genome. The average number of SNPs between lineages L1 and L2 was 37,408 and the average number of indels was 2041. Within L1, the average number of SNPs and indels was 16,778 and 919, respectively. Within L2, the average number of SNPs and indels was 22,875 and 951, respectively. Even within the lineages, a large number of potential genetic markers was obtained.
Nucleotide differences of 3 bp between PCR amplicons can be detected by 6 % polyacrylamide gel electrophoresis. The estimated indels >3 bp can be directly used as co-dominant markers. To evaluate how many indels are available for development of co-dominant markers, the distribution of the length of indels was examined (Supplementary Fig. S1). The average number of indels >3 bp was 863, 378, and 411, respectively, between the lineages, within L1, and within L2. These indels can be used for initial rough mapping.
Transcript expression levels may affect SNP and indel frequencies per transcript. To evaluate the impact of gene expression on the SNP and indel frequencies, the correlation between CPM and the number of polymorphisms per transcript was calculated (Supplementary Fig. S2). No clear correlation was found between CPM and the number of polymorphisms, indicating that the gene expression levels did not influence the SNP and indel frequencies under our criteria for SNP and indel calling.
Anchoring SNPs and indels to scaffolds and chromosomes of Ae. tauschii
To develop genetic markers anchored to the chromosomes of Ae. tauschii, we evaluated how many SNPs and indels were anchored to scaffolds of the draft genome or the chromosomes of Ae. tauschii (Table 3, Supplementary Table S6). Of the total SNPs in the pairwise comparisons, an average of 68.5 and 17.8 % were, respectively, anchored to the scaffolds and chromosomes. There was no large difference in the percentage of SNPs mapping to the scaffolds (61.0–79.8 %) or the chromosomes (10.8–21.9 %) for any pairwise comparisons. The number of these anchored SNPs was directly proportional to the total number of SNPs (Tables 2, 3). Unfortunately, 78.1–89.2 % of SNPs remained unmapped to the chromosomes, even when the SNPs and indels were anchored to the scaffolds. Indels showed the same tendency as the SNPs (Table 2, Supplementary Table S6). Although the scaffolds cover 84.3 % of the Ae. tauschii genome (4.23-Gb), only 1.72-Gb is anchored to the chromosomes (Jia et al. 2013). Thus, the number of SNPs and indels mapped to the chromosomes were limited as well.
The average number of SNPs per chromosome within L1, within L2 and between L1 and L2 was 312.2–678.0, 209.3–413.0 and 816.2–1356.8, respectively (Supplementary Table S7). Except for chromosome 7, a large coefficient of variation in the number of SNPs within L1 (0.66–0.83) was detected (Supplementary Fig. S3; Table S7). The number of SNPs for some pairwise comparisons within L1 was greater than between L1 and L2 on six chromosomes. The average number of SNPs on chromosome 6D was smaller than on the other chromosomes, but no clear differences were found among the chromosomes when the average number of SNPs per transcript was estimated. The same tendency was detected for the indels (Supplementary Fig. S3; Table S8).
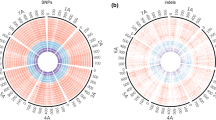
To develop molecular markers for genetic linkage analysis, uniform distribution of SNPs and indels over the chromosomes is desirable. The distributions of transcripts, SNPs and indels over the seven chromosomes were visualized in representative pairs within L1 (Fig. 2a), within L2 (Fig. 2b), and between L1 and L2 (Fig. 2c). These representative pairs represent the most and least SNP- and indel-rich pairs in each combination of lineages. Although the SNPs were not evenly distributed, they covered all chromosomes moderately well in intra- and interlineage comparisons. Compared with the SNPs, indels were more unevenly distributed. At least a small number of indels was located on each chromosome. A high density of SNPs and indels was detected around the centromeric regions. This observation could be explained by the suppression of recombination in these regions, generating gaps between physical and genetic distance.

Distribution of SNPs and indels detected in pairwise comparison of Ae. tauschii accessions on the genetic map of Ae. tauschii (a–c, scale in cM) and the physical map of barley (d–f scale in Mb). Three circles of the same color show the number of transcripts, SNPs, and indels from the outer to inner circles. The red circles represent the richest SNP and indel pairs, and the green circles, the least rich pairs in each combination of lineages: a L1 vs L1, KU-2087 read-mapped to PI499262 transcript and AT76 to KU-2003, b L2 vs L2, KU-2093 to KU-2124 and KU-2078 to KU-2075, c L1 vs L2, KU-2087 to KU-2078 and KU-2025 to KU-2093, d L1 vs L1, KU-2087 to PI499262 and AT76 to KU-2627, e L2 vs L2, KU-2075 to KU-2124 and KU-2078 to KU-2075, and f L1 vs L2, KU-2075 to AT76 and PI499262 to KU-2078. Arrowheads indicate centromeric positions of each chromosome, except for chromosome 1H, which only had whole-chromosome sequence (IBSC 2012)
Anchoring SNPs and indels to the chromosomal pseudomolecules of barley
To evaluate whether the barley physical map is useful for assigning SNPs and indels between Ae. tauschii accessions in the same order on their chromosomes, the estimated SNPs and indels were mapped to the barley chromosomes (Table 4, Supplementary Table S9). An average of 29.1 % of the total SNPs and 25.4 % of the indels were anchored to the barley chromosomes. The percentages of anchored SNPs and indels were almost the same in any pairwise comparison. Interestingly, the number of SNPs and indels anchored to the barley chromosomes was larger than the number anchored to the chromosomes of Ae. tauschii in all pairwise comparisons (Tables 3, 4).
The average number of SNPs per chromosome within L1, within L2, and between L1 and L2 was 413.4–929.4, 481.6–851.5, and 1306.4–1849.0, respectively (Supplementary Table S10). The average number of indels per chromosome within L1, within L2, and between L1 and L2 was 18.9–46.0, 22.9–43.7, and 67.4–89.9, respectively (Supplementary Table S11). These numbers were larger than for those anchored to the Ae. tauschii chromosomes. A similar observation was detected in the average number of SNPs and indels per transcript on each chromosome. As observed for the SNPs anchored to the Ae. tauschii chromosomes, a large coefficient of variation in the number of SNPs (0.62–0.83) and indels (0.55–0.76) within L1 was detected, except for chromosome 7H (Supplementary Fig. S4; Tables S10, S11).
To determine whether the SNPs and indels that were anchored to the barley chromosomes were evenly distributed over the entire chromosomes, the distribution of these SNPs and indels was visualized. We chose pairs having the most or least numbers of SNPs and indels within L1 (Fig. 2d), within L2 (Fig. 2e), and within L1 and L2 (Fig. 2f). In all combinations excluding the pairs with the least number of SNPs between L1 and L2 (between PI499262 and KU-2078), SNPs covered the entire region of the chromosomes except for the unigene-sparse regions, one of which corresponded to the centromeric region of chromosome 2H. In the pairs with the least number of SNPs between L1 and L2 (between PI499262 and KU-2078), a high-density localization of SNPs was observed only for chromosomes 1H and 7H. Although the distribution of indels was not as uniform as that of SNPs, every chromosome carried a small number of indels in any comparison.
Integration of SNPs and indels that were anchored to the chromosomes of Ae. tauschii and barley
If the SNPs and indels anchored to Ae. tauschii chromosomes are different from those anchored to barley chromosomes, utilization of these SNPs and indels increases the number of potential genetic markers. We tested overlaps between the SNPs and indels that were anchored to Ae. tauschii and barley chromosomes (Fig. 3). Venn diagrams of mean values of SNPs and indels showed that most SNPs and indels were specifically anchored to Ae. tauschii or barley chromosomes. Only 15.6 and 14.9 % of the total SNPs and indels overlapped. These results indicated that the integration of SNPs and indels anchored to the chromosomes of Ae. tauschii and barley increased the available genetic markers for linkage analysis.

Venn diagrams of SNPs mapped to the Ae. tauschii and barley genomes. The numbers indicate mean values of SNPs and indels
Application of CAPS markers to linkage and association analyses
To evaluate the usefulness of the SNP dataset, Iw2 region specific markers were designed based on in silico mapping of transcript models to Ae. tauschii scaffolds. We developed 14 markers on 4 scaffolds (Supplementary Fig. S5). For ten markers, the genotyping results were consistent with the allelic type detected in RNA-seq. For the other four markers, however, genotypes of some accessions (scaf43829_1, 2, and 7 for PI499262 and KU-2025 and scaf10812_2 for KU-2124) contradicted the results of RNA-seq. This observation suggests that the transcript models were constructed by reads derived from paralogues on other chromosomes. No specific PCR products were detected for a few SNP markers in several accessions, for which reads were absent at the SNP sites, e.g., scaf10812_6 for KU-2093, KU-2075, and KU-2078 and scaf10812_8 for six L1 accessions. This result indicated that some chromosomal rearrangements had occurred during divergence among the Ae. tauschii accessions, and that the genomic segments from which these markers were derived did not exist in the accessions that had no reads on the sites.
To confirm whether these markers were precisely linked to the Iw2 locus on chromosome 2DS, we genotyped F2 individuals of the KU-2003/KU-2124 population using ten markers that were polymorphic between KU-2003 and KU-2124. Of the selected markers, seven mapped to the Iw2 region and the remaining three markers were assigned to another chromosome (Fig. 4). The Iw2 locus was mapped within a 0.9 cM interval between the most closely linked markers and co-localized with two markers. The chromosomal order of selected scaffolds at the Iw2 region was consistent with that predicted in a previous study (Nishijima et al. 2014). Association analysis showed that five out of the seven markers that mapped to the Iw2 region were significantly (P < 1E−3) associated with the glaucous/non-glaucous trait in Ae. tauschii (Table 5). All 20 glaucous accessions had the KU-2124-type allele at scaf10812_2 and scaf10812_6, and most non-glaucous accessions had the KU-2003-type allele at scaf43829_9. These results demonstrated that a fine map could be constructed efficiently using SNPs derived from RNA-seq. Thus, the SNP dataset might be useful for molecular marker development in the specific chromosomal region of the D genome or across the entire genome.

Comparison of linkage maps of the Iw2 region. a Genetic map of the KU-2154/KU-2126 F2 population (Nishijima et al. 2014). b The Ae. tauschii scaffolds anchored to the chromosomal region (Nishijima et al. 2014). The positions of genes and markers on the two maps are indicated. c The linkage map constructed using markers developed in this study
Discussion
RNA sequencing as an efficient tool for SNP discovery and its potential for evolutionary studies in Ae. tauschii
We performed 300-bp paired-end RNA sequencing of leaf tissues from ten representative Ae. tauschii accessions including both the L1 and L2 lineages with the purpose of SNP identification and molecular marker development using the Illumina MiSeq platform. RNA sequencing of samples obtained from tissues grown under the same conditions generated a similar pattern of gene expression between the accessions, facilitating the detection of a large number of SNPs. The average number of SNPs between L1 and L2 (78,519) was four times higher than discovered in previous studies by transcriptome sequencing of leaves and spikes of two Ae. tauschii accessions, one each from L1 and L2 (Iehisa et al. 2012, 2014). Iehisa et al. (2014) performed RNA sequencing of leaf and spike tissues from the two Ae. tauschii accessions using 454 sequencing and detected 16,148 high-confidence SNPs. However, 300-bp paired-end sequencing on the MiSeq platform can generate long reads of higher quality. The length of the merged reads is comparable to the length of expressed sequence tags. This distinguishing characteristic enables detection of more SNPs, even from RNA sequencing data of only leaf tissue.
The increased number of SNPs among various accessions will help to construct more detailed genetic maps of the D genome. Although most intralineage pairings had fewer SNPs than interlineage pairings (Table 2), the least polymorphic pair had as many SNPs as the previous report (Iehisa et al. 2014). This result suggests that even intralineage crosses could possess enough polymorphisms for construction of genetic linkage maps. Molecular markers derived from the SNP dataset were actually assigned around the Iw2 locus (Fig. 4), and were as densely distributed as our previous study (Nishijima et al. 2014), demonstrating that a fine map could be constructed efficiently using SNPs derived from RNA sequencing. This SNP dataset allows development of molecular markers in any specific chromosomal region of the D genome.
As expected, the number of indels was much smaller than that of SNPs. Since indels in exons often cause frame-shift mutations, purifying selection removes such deleterious mutations from the population. This is one of the disadvantages of marker development based on RNA sequencing. Indels, however, were detected across entire chromosomes as well (Fig. 2). The average number of indels that were anchored to the chromosomes was >10 on each chromosome in most of the pairwise comparisons of Ae. tauschii accessions. They should be useful for rough mappings, as PCR-based indel markers can be genotyped more easily than SNP markers.
Pairwise comparisons between the ten accessions of Ae. tauschii revealed a large variance in the number of SNPs and indels on chromosomes within L1 except for 7D, where some combinations between L1 and L2 accessions had a smaller number of SNPs and indels than between L1 accessions. These observations suggest the existence of gene flow between L1 and L2 lineages on these chromosomes, whereas the number of SNPs on chromosome 7D between the L1 and L2 lineages was clearly larger than within L1 or within L2. The intraspecific lineage structure of Ae. tauschii is associated with differences in seed production and flowering time (Matsuoka et al. 2015). In particular, the L1 lineage with its high seed production and early flowering time has spread across East Asia (Matsuoka et al. 2015). The L1 lineage may have loci related to adaption to different habitats, preventing gene flow on chromosome 7D and generating a clear divergence between the L1 and L2 lineages.
The present study used only ten accessions, which are not enough to address evolutionary questions such as the above hypothesis. To dissect the evolutionary dynamics of the Ae. tauschii population, it is necessary to study nucleotide polymorphisms in more Ae. tauschii accessions. We found no clear correlations between gene expression level and SNP frequency per transcript (Fig. S2). This allowed SNPs identified through RNA sequencing to capture the genome-wide genetic diversity in the Ae. tauschii population without any bias derived from differences in gene expression level. Since the costs of RNA sequencing are fairly low owing to the development of next-generation sequencing technology and improvements in library construction (Townsley et al. 2015), the approach of RNA sequencing will make revealing genome-wide nucleotide polymorphisms in over a hundred samples of Ae. tauschii accessions feasible in the near future.
Advantages of RNA sequencing in the development of novel genetic markers for Triticeae
To reduce the complexity of genome sequencing, many derivatives of next-generation sequencing have been developed for the development of genetic markers (Kumar et al. 2012). In genotyping-by-sequencing and restriction site-associated DNA sequencing, restriction enzymes are used to create reduced representation libraries for sequencing (Davey et al. 2011). Although this method has been useful for SNP identification, the choice of enzymes might be a critical factor, and a portion of the sequenced reads such as repetitive elements might be discarded from the following analyses, especially in the Triticeae species, which have a large and complex genome. On the other hand, RNA sequencing focuses on exons only and fails to detect abundant polymorphisms in intragenic regions. SNPs between tested samples can be detected only in expressed genes. However, our study demonstrated that RNA sequencing enables detection of a large number of SNPs, even between genetically close accessions of Ae. tauschii. In addition, our approach based on RNA sequencing assembly can make almost full use of the sequenced reads, and requires no prior information about the genome of interest. Based on the conserved gene order between barley and its crop relatives, SNPs can be anchored to specific chromosomal regions. From these points of view, RNA sequencing should be a reasonable approach for the development of molecular markers in the Triticeae.
The number of SNPs aligned to the Ae. tauschii genome was twice that of barley (Tables 3, 4). However, the number of SNPs mapping to chromosomes was slightly higher for barley (Tables 3, 4), consistent with a previous report (Iehisa et al. 2014), suggesting that molecular markers can be designed to specific target chromosomes using genomic information from barley as precisely as information from Ae. tauschii. These results also imply the versatility of the barley genome for the dissection of the genomes of other Triticeae species, since the synteny between barley and wheat relatives is widely conserved (Mayer et al. 2011; Wicker et al. 2011). In conclusion, our strategy, in which SNPs detected through RNA sequencing assembly are anchored to the seven chromosomes in silico based on the conserved synteny, can develop a large number of molecular markers and should be applicable to marker development for other wild species in the tribe Triticeae, at least diploids such as Ae. caudata (C genome), Ae. umbellulata (U genome) and Ae. comosa (M genome), which have no draft sequences available at all.
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M, Innan H, Cano L, Kamoun S, Terauchi R (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178
Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J (2010) Galaxy: a web-based genome analysis tool for experimentalists. In: Current protocols in molecular biology , Chap 19.10, vol 89, pp 19.10.1–19.10.21
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML (2011) Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet 12:499–510
Gaut BS (2002) Evolutionary dynamics of grass genomes. New Phytol 154:15–28
Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, Zhang Y, Blankenberg D, Albert I, Taylor J, Miller W, Kent WJ, Nekrutenko A (2005) Galaxy: a platform for interactive large-scale genome analysis. Genome Res 15:1451–1455
Goecks J, Nekrutenko A, Taylor J (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11:R86
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, Macmanes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, Leduc RD, Friedman N, Regev A (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512
Iehisa JCM, Shimizu A, Sato K, Nasuda S, Takumi S (2012) Discovery of high-confidence single nucleotide polymorphisms from large-scale de novo analysis of leaf transcripts of Aegilops tauschii, a wild wheat progenitor. DNA Res 19:487–497
Iehisa JCM, Shimizu A, Sato K, Nishijima R, Sakaguchi K, Matsuda R, Nasuda S, Takumi S (2014) Genome-wide marker development for the wheat D genome based on single nucleotide polymorphisms identified from transcripts in the wild wheat progenitor Aegilops tauschii. Theor Appl Genet 127:261–271
International Barley Genome Sequencing Consortium (2012) A physical, genetic and functional sequence assembly of the barley genome. Nature 491:711–716
Jia J, Zhao S, Kong X, Li Y, Zhao G, He W, Appels R, Pfeifer M, Tao Y, Zhang X, Jing R, Zhang C, Ma Y, Gao L, Gao C, Spannagl M, Mayer KFX, Li D, Pan S, Zheng F, Hu Q, Xia X, Li J, Liang Q, Chen J, Wicker T, Gou C, Kuang H, He G, Luo Y, Keller B, Xia Q, Lu P, Wang J, Zou H, Zhang R, Xu J, Gao J, Middleton C, Quan Z, Liu G, Wang J, International Wheat Genome Sequencing Consortium, Yang H, Liu X, He Z, Mao L, Wang J (2013) Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature 496:91–95
Kersey PJ, Allen JE, Armean I, Boddu S, Bolt BJ, Carvalho-Silva D, Christensen M, Davis P, Falin LJ, Grabmueller C, Humphrey J, Kerhornou A, Khobova J, Aranganathan NK, Langridge N, Lowy E, McDowall MD, Maheswari U, Nuhn M, Ong CK, Overduin B, Paulini M, Pedro H, Perry E, Spudich G, Tapanari E, Walts B, Williams G, Tello-Ruiz M, Stein J, Wei S, Ware D, Bolser DM, Howe KL, Kulesha E, Lawson D, Maslen G, Staines DM (2015) Ensembl genomes 2016: more genomes, more complexity. Nucleic Acids Res 44:574–580
Kosugi S, Natsume S, Yoshida K, MacLean D, Cano L, Kamoun S, Terauchi R (2013) Coval: improving alignment quality and variant calling accuracy for next-generation sequencing data. PLoS One 8:e75402
Kumar S, Banks TW, Cloutier S (2012) SNP discovery through next-generation sequencing and its applications. Int J Plant Genomics. doi:10.1155/2012/831460
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup 1000 Genome Project Data Processing (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
Luo M-C, Gu YQ, You FM, Deal KR, Ma Y, Hu Y, Huo N, Wang Y, Wang J, Chen S, Jorgensen CM, Zhang Y, McGuire PE, Pasternak S, Stein JC, Ware D, Kramer M, McCombie WR, Kianian SF, Martis MM, Mayer KFX, Sehgal SK, Li W, Gill BS, Bevan MW, Šimková H, Doležel J, Weining S, Lazo GR, Anderson OD, Dvorak J (2013) A 4-gigabase physical map unlocks the structure and evolution of the complex genome of Aegilops tauschii, the wheat D-genome progenitor. Proc Natl Acad Sci USA 110:7940–7945
Matsumoto T, Tanaka T, Sakai H, Amano N, Kanamori H, Kurita K, Kikuta A, Kamiya K, Yamamoto M, Ikawa H, Fujii N, Hori K, Itoh T, Sato K (2011) Comprehensive sequence analysis of 24,783 barley full-length cDNAs derived from 12 clone libraries. Plant Physiol 156:20–28
Matsuoka Y, Nasuda S, Ashida Y, Nitta M, Tsujimoto H, Takumi S, Kawahara T (2013) Genetic basis for spontaneous hybrid genome doubling during allopolyploid speciation of common wheat shown by natural variation analyses of the paternal species. PLoS One 8:e68310
Matsuoka Y, Takumi S, Kawahara T (2015) Intraspecific lineage divergence and its association with reproductive trait change during species range expansion in central Eurasian wild wheat Aegilops tauschii Coss. (Poaceae). BMC Evol Biol 15:213
Mayer KFX, Martis M, Hedley PE, Šimková H, Liu H, Morris JA, Steuernagel B, Taudien S, Roessner S, Gundlach H, Kubaláková M, Suchánková P, Murat F, Felder M, Nussbaumer T, Graner A, Salse J, Endo T, Sakai H, Tanaka T, Itoh T, Sato K, Platzer M, Matsumoto T, Scholz U, Doležel J, Waugh R, Stein N (2011) Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 23:1249–1263
Mizuno N, Yamasaki M, Matsuoka Y, Kawahara T, Takumi S (2010) Population structure of wild wheat D-genome progenitor Aegilops tauschii Coss.: implications for intraspecific lineage diversification and evolution of common wheat. Mol Ecol 19:999–1013
Nishijima R, Iehisa JCM, Matsuoka Y, Takumi S (2014) The cuticular wax inhibitor locus Iw2 in wild diploid wheat Aegilops tauschii: phenotypic survey, genetic analysis, and implications for the evolution of common wheat. BMC Plant Biol 14:246
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26:841–842
Sehgal D, Vikram P, Sansaloni CP, Ortiz C, Saint Pierre C, Payne T, Ellis M, Amri A, Petroli CD, Wenzl P, Singh S (2015) Exploring and mobilizing the gene bank biodiversity for wheat improvement. PLoS One 10:e0132112
Takagi H, Uemura A, Yaegashi H, Tamiru M, Abe A, Mitsuoka C, Utsushi H, Natsume S, Kanzaki H, Matsumura H, Saitoh H, Cano LM, Kamoun S, Terauchi R (2013) Methods MutMap-Gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol 200:276–283
Takumi S, Koyama K, Fujiwara K, Kobayashi F (2011) Identification of a large deletion in the first intron of the Vrn-D1 locus, associated with loss of vernalization requirement in wild wheat progenitor Aegilops tauschii Coss. Genes Genet Syst 86:183–195
Tsunewaki K (1966) Comparative gene analysis of common wheat and its ancestral species. II. Waxiness, growth habit and awnedness. Jpn J Bot 19:175–229
Townsley BT, Covington MF, Ichihashi Y, Zumstein K, Sinha NR (2015) BrAD-seq: Breath Adapter Directional sequencing: a streamlined, ultra-simple and fast library preparation protocol for strand specific mRNA library construction. Front Plant Sci 6:366
Wicker T, Mayer KFX, Gundlach H, Martis M, Steuernagel B, Scholz U, Šimková H, Kubaláková M, Choulet F, Taudien S, Platzer M, Feuillet C, Fahima T, Budak H, Dolezel J, Keller B, Stein N (2011) Frequent gene movement and pseudogene evolution is common to the large and complex genomes of wheat, barley, and their relatives. Plant Cell 23:1706–1718
Wu TD, Watanabe CK (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21:1859–1875
Yang C, Zhao L, Zhang H, Yang Z, Wang H, Wen S, Zhang C, Rustgi S, von Wettstein D, Liu B (2014) Evolution of physiological responses to salt stress in hexaploid wheat. Proc Natl Acad Sci 111:11882–11887
Zhang J, Kobert K, Flouri T, Stamatakis A (2014) PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30:614–620
Acknowledgments
Computations for the RNA sequencing assembly and alignments of short reads were performed on the NIG supercomputer at the ROIS National Institute of Genetics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by a Grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan [Grant-in-Aid for Scientific Research (B) Nos. 25292008 and 16H04862] to ST, and by MEXT as part of a Joint Research Program implemented at the Institute of Plant Science and Resources, Okayama University, Japan.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by S. Hohmann.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nishijima, R., Yoshida, K., Motoi, Y. et al. Genome-wide identification of novel genetic markers from RNA sequencing assembly of diverse Aegilops tauschii accessions. Mol Genet Genomics 291, 1681–1694 (2016). https://doi.org/10.1007/s00438-016-1211-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-016-1211-2