
Abstract
DNA methylation is an epigenetic modification essential for gene regulations in plants, but understanding on how it is involved in fruit development, especially in non-climacteric fleshy fruit, is limited. The diploid woodland strawberry (Fragaria vesca) is an important model for non-climacteric fruit crops. In this study, we identified DNA methyltransferase genes and demethylase genes in Fragaria vesca and other angiosperm species. In accordance with previous studies, our phylogenetic analyses of those DNA methylation modifiers support the clustering of those genes into several classes. Our data indicate that whole-genome duplications and tandem duplications contributed to the expansion of those DNA methylation modifiers in angiosperms. We have further demonstrated that some DNA methylase and demethylase genes reach their highest expression levels in strawberry fleshy fruits when turning from white to red, suggesting that DNA methylation might undergo a dramatic change at the onset of fleshy fruit-ripening process. In addition, we have observed that expression of some DNA demethylase genes increases in response to various abiotic stresses including heat, cold, drought and salinity. Collectively, our study indicates a regulatory role of DNA methylation in the turning stage of non-climacteric fleshy fruit and responses to environment stimuli, and would facilitate functional studies of DNA methylation in the growth and development of non-climacteric fruits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Strawberry is an important model for fruit crop plant species. Strawberry has several distinct characteristics such as an ethylene-independent fruit-ripening process (non-climacteric) and fruit flesh tissues derived from receptacle not typically from ovary walls. The cultivated strawberry (F. × ananassa) has an extremely complex octoploid genome harboring 56 chromosomes (2n = 8x = 56) derived from four diploid ancestors, which makes genetic studies extremely complicated. Also, because of its small genome size (240Mb, 2n = 2x = 14) and the availability of the genome sequence (Shulaev et al. 2011), the diploid woodland strawberry Fragaria vesca is emerging as a model for fruit crop plant species. Recently, DNA methylation has been shown to be involved in the fleshy fruit development in tomato that is an ethylene-dependent, climacteric fruit species. However, studies on DNA methylation in non-climacteric fruits are limited. Therefore, identification and characterization of DNA methylation modifiers in strawberry would be of great significance for understanding of epigenetic regulatory mechanisms underlying non-climacteric fruit development.
In eukaryotes, genomic DNA is packaged into chromatin, which consists of DNAs, histones, associated chromosomal proteins and small RNAs. Modifications on DNA itself, specifically DNA methylations, play important roles in chromatin structure and gene expressions in eukaryotes. The regulatory mechanisms of DNA methylation in plants and animals are quite similar in some aspects, such as heavy methylation on transposable elements (TEs) in heterochromatin, depletion of methylation at promoters of active genes and some methylation over gene bodies (He et al. 2011).
In both plants and animals, methylation on the fifth carbon of cytosine residues (5mC) is most common and has been extensively studied. In animals, the majority of 5mC happens in the CG context (Ehrlich et al. 1982), while in plants, 5mC happens on both dinucleotide (CG) and trinucleotide sequence contexts (CHG and CHH, H = A, C or T) (Henderson and Jacobsen 2007; Law and Jacobsen 2010). In total, four functional DNA methyltransferases (DNMTs), DNA METHYLTRANSFERASE 1 (MET1), DOMAINS REARRANGED METHYLTRANSFERASE 1 and 2 (DRM1/2) and CHROMOMETHYLASE 3 (CMT3), were identified in the Arabidopsis thaliana genome (Chan et al. 2005). All DNMTs use S-adenosyl-l-methionine (SAM) as the methyl donor, and the enzymatic activity is mediated by the DNA methylase domain. MET1 is essential for the maintenance of 5mC in the CG context during meiosis and mitosis, similar to its mammalian ortholog Dnmt1. DRM1/2 catalyzes de novo methylation at all sequence contexts0, and CMT3 is a plant-specific DNA methyltransferase responsible for the maintenance of 5mC in the CHG context (Law and Jacobsen 2010). Dnmt2 enzymes were identified as a class of DNA methylase domain-containing proteins as well, but they were proved to be methyltransferases targeting tRNAs instead of DNAs (Goll et al. 2006).
How DNA methylation is controlled is of great interest. It has been revealed that the establishment and maintenance of DNA methylation are induced by a RNA-dependent DNA methylation (RdDM) mechanism and that there is an association between DNA methylation and histone modification (Saze et al. 2012). Once established, DNA methylation is not static: the local DNA methylation is regulated by different pathways involving both methylation and demethylation. Loss of methylated cytosine could result passively from the improper operation of DNA methylation maintenance pathways through rounds of DNA replication, or actively from the activities of DNA demethylases independent of replication (Furner and Matzke 2011). Four functional DNA demethylases were identified in Arabidopsis thaliana, ROS1, DEMETER (DME), DEMETER LIKE 2 and 3 (DML2/3). All of the four demethylases have a glycosylase domain responsible for the active removal of 5-methylcytosine from DNA by cleavage of the phosphodiester backbone and two additional domains A and B (DME domain) with no known homology to other proteins (Penterman et al. 2007; Zhang and Zhu 2012). In Arabidopsis, DME is essential for the genome-wide demethylation concurrent with transposon reactivation during gametogenesis. ROS1 and DML2/3 are expressed in vegetative cells and are able to demethylate unwanted methylations at specific loci to permit transcription from various genes, TEs and intergenic regions (Zhang and Zhu 2012). These different DNA demethylases share similar enzyme activities and are partially redundant. How DNA demethylases achieve any specificities in their target is largely unknown. It is suggested that some small RNA-binding proteins might be involved in the recruitment of DNA demethylases to their specific targets (Zheng et al. 2008).
The proper regulation of DNA methylation has a profound impact on genome stability, transcription and development, particularly, DNA methylation is involved in fleshy fruit-ripening of tomato (Zhong et al. 2013; Liu et al. 2015b). In addition, extensive studies have suggested that DNA methylation is of great importance for abiotic stress response in plants by directly or indirectly regulating various stress-responsive genes (Cavrak et al. 2014; Le et al. 2014; Bharti et al. 2015; Liu et al. 2015a; Rutowicz et al. 2015; Xu et al. 2015).
Strawberry is emerging as an important fruit model species because of its many unique characteristics such as ethylene-independent regulation of the fruit-ripening process, fruit flesh tissues derived from receptacle not typically from ovary wall, and a sequenced small diploid genome. Despite its profound impact on all kinds of biological processes in plants, detailed investigation of DNA methyltransferases and demethylases is still lacking in strawberry. In this study, we identified the methylase domain-containing methyltransferase genes and glycosylase domain-containing demethylase genes in various angiosperm species, including five species in Roseasae, three species in Cruciferae, three species in Cucurbitaceae, one species in Solanaceae, and three reference genomes. We performed an extensive phylogenetic analysis to investigate their evolutionary characteristics. Particularly, we profiled their dynamic expression patterns during fruit development, and responses to various stresses in diploid strawberry Fragaria vesca, which provides clues in their possible roles in various cellular processes in strawberry. Our study should facilitate the functional characterization of those epigenetic regulators in strawberry and other plant species.
Materials and methods
Proteins sequences
In total, 16 whole-genome sequenced species, including three from Cruciferae, five from Rosaceae, three from Cucurbitaceae, one from Solanaceae, two monocots, the basal angiosperm Amborella trichopoda, and one fern genome Selaginella moellendorffii were sampled. Except for Cucumis melo, the proteome of the other 15 species were downloaded from PGDD (Plant Genome Duplication Database (http://chibba.agtec.uga.edu/duplication/index/files). The complete protein sequence of Cucumis melo was downloaded from http://melenomics.net/genome. If two or more proteins are annotated for the same gene from alternative splicing, the longest one was selected for further analyses. The versions and the website are listed in Table S1.
Acquisition of DNA methyltransferase and DNA demethylase protein sequences
We use DNA methylase domain (PF00145) as query by hmmsearch to find DNA methyltransferase proteins of these species, HhH-GPD domain (PF00730) and RRM_DME domain (PF15628) to find DNA demethylase proteins. The full domain alignments were downloaded from pfam database V27.0 (Finn et al. 2010). The presence of the target domains was verified by Pfam database (http://pfam.xfam.org/search), the Simple Modular Architecture Research Tool database (SMART; http://smart.embl-heidelberg.de/), or the Conserved Domain Database (CDD; http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) available from NCBI, with the e value threshold of 10−4. All identified putative methyltransferase and demethylase genes are listed in Table S2 and S3.
Sequence alignment and phylogenetic analyses
The DNA methyltransferase or demethylase protein sequences were aligned by MUSCLE (Edgar 2004). The phylogenetic tree was constructed by the maximum likelihood method, using MEGA 6.0 (Tamura et al. 2013), with JTT+G+F model for methyltransferase tree, and JTT+I+G+F model for demethylase trees with 1000 bootstrapping replicates. The model was determined by ModelGenerator (Keane et al. 2006). The major clustering of the trees was verified by MrBayes v3.2 (Ronquist et al. 2012). For the Bayes tree construction, the model Jones was used with a gamma-shaped rate of variation and a proportion of invariable sites, printfreq = 100, samplefreq = 1000, nchains = 4, ngen = 2,000,000 and sump burnin = 250, and other model parameters were kept at default values.
Analyses of domain structure and motif structure
The presence of other domains was identified by Pfam database, the SMART, or the Conserved Domain Database with the e value threshold of 10−4. Motif analysis was performed by MEME (MEME, Version 4.10.2, http://meme-suite.org/tools/meme) (Bailey and Elkan 1994). The number of motifs was set to no more than 15 with the length from 6 to 150 amino acids for each search.
Expression analysis
The transcriptome data for different development stages or abiotic stresses were downloaded from (http://bioinformatics.towson.edu/strawberry/) for Fragaria vesca (Kang et al. 2013) and from (http://jsp.weigelworld.org/expviz/expviz.jsp) for Arabidopsis. The expression data (RPKM) were directly retrieved from the website, and the expression levels for the genes of interest were plotted by log2 scale.
Plant growth conditions, stress treatments and material collection
All plant materials were collected from a seventh generation inbred line of Fragaria vesca, Ruegen (kindly provided by Janet Slovin). For strawberry fruit collection, strawberry plants were grown in 10 cm × 10 cm pots in a growth chamber, set at 16 h light/8 h dark cycles, 22 °C and 65 % relative humidity. Strawberry fruits at 12-day-old big-green stage, white, pre-turning stage, pink stage (slight pink flesh and red seeds) and red stage (2–3 after the pink stage) were collected and immediately put into liquid nitrogen.
For abiotic stresses, the strawberry seedlings were grown in MS media in growth chamber set at 16 h light/8 h dark cycles and 22 °C. Four-week-old seedlings were transferred to the growth chamber set at either 4 °C (at dark) or 38 °C (at dark) for cold/heat shocks. Cold-shocked seedlings were collected at 1, 3 and 8 h. Heat-shocked seedlings were collected at 1, 3, 4 (3 h heat shock and 1 h recovery at 22 °C) and 8 h (3 h heat shock and 5 h recovery at 22 °C). Seedlings kept in dark for 12 h were used as controls for cold/heat treatments.
In addition, 4-week-old seedlings were transferred to 1/2 MS liquid media with gentle agitation (100 rpm) for salinity and drought stress treatments. After setting in 1/2 MS media for 12 h, seedlings were transferred to liquid 1/2 MS media supplemented by 150 mM sodium chloride or 20 % PEG for salinity and drought stresses. Drought-stressed seedlings were collected at 1, 3 and 8 h. Seedlings put in liquid 1/2 MS media for 12 h were used as controls for salinity and drought-stressed materials. The collected materials were immediately put into liquid nitrogen for RNA processing.
RNA extraction and quantitative PCR analyses
Both de-seeded strawberry flesh and abiotic stress-treated seedlings were processed to RNA isolation by a modified CTAB method. The isolated RNAs were treated with DNase I and proceeded to cDNA synthesis using the Primerscript RT reagent Kit with gDNA Eraser (Takara). Quantitative PCR was performed using SYBR Premix Ex Tag (Takara) using the cDNA as the template. Primers used are listed in Table S4. Results were analyzed using the ΔΔCT method (Livak and Schmittgen 2001) using GAPDH (Amil-Ruiz et al. 2013) as the control locus. Three biological and three technical replicates were performed and analyzed.
Results
Identification of genes containing DNA methylase domains
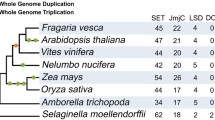
A sequence-based searching was performed to identify the methyltransferase catalytic domain-containing genes in F. vesca and other dicot species from Cruciferae, Rosaceae, Cucurbitaceae, Solanaceae, two monocot species and the basal angiosperm Amborella trichopoda. In the investigated 15 species, the number of identified DNA methylase domain-containing genes ranges from 5 (in Cucumis melo) to 15 (in Pyrus bretschneideri) (Fig. 1). The Arabidopsis genes with known functions were named as published; while genes in other species were named as methylase 1–15 following the species abbreviation.

Numbers of genes encoding DNA methyltransferase and demethylase in the 15 species investigated. The Taxonomy Common Tree constructed online by Taxonomy Browser in NCBI (http://www.ncbi.nlm.nih.gov/Taxonomy/CommonTree/wwwcmt.cgi) is shown on the left. The divergence time labeled on the phylogenetic tree was from listed references (Arabidopsis Genome 2000; Paterson et al. 2004; Huang et al. 2009; Guo et al. 2013; Wu et al. 2013)
To investigate the classification of putative DNA methyltransferase genes, phylogenetic trees (constructed by the maximum likelihood method, see Methods for details) based on the amino acid sequence conservation of whole proteins were constructed (Fig. 2). According to the tree topology and gene annotations in Arabidopsis, the methyltransferase genes could be grouped into four classes, MET1, CMT3, DRM1/2, and DNMT2 (Fig. 2). To validate the major clustering of the ML trees, the phylogenetic tree was constructed by a different method as well (Fig. S1 and S2, by MrBayes). Although the topologies of trees constructed by different methods have some differences (some inner branching within each class might be slightly different), the main four clustering with high bootstrap values are consistent between different methods (Fig. 1, Fig. S1 and S2). The F. vesca genome consists of 1, 3, 4, 1 genes in each class, respectively. For DNMT2 class enzymes conferring tRNA methyltransferase activity, 13 out of the 15 species (except for Pyrus bretschneideri and Cucumis sativus) consist only one gene in each genome, while the number of genes in the other three classes conferring DNA methyltransferase activity varies. For example, the CMT3 class consists of two to three members in each species, except in Pyrus bretschneideri and Malus × domestica. The phylogenetic trees indicate that both the duplications happened in the common ancestor of Pyrus bretschneideri and Malus × domestica, and lineage-specific duplications that happened in the two species contributed to this expansion (Figs. 1, 2). Overall, the four classes of DNA methylase domain-containing genes have different evolutionary characteristics.

A most likelihood phylogenetic tree of predicted DNA methylase domain-containing genes in the 15 species. The phylogenetic tree was constructed based on the whole protein sequences from the 15 species with 1000 bootstrapping replicates, and the results of the bootstrapping analysis larger than 50 % are shown. The domain composition is shown on the right
Within a protein family, domains and motifs are conserved sequences most relevant to its function. To investigate the putative functional conservation during the evolution of methyltransferase genes, domain composition was identified by pfam and SMART, and the 15 most common motifs in the whole proteins were identified by MEME. The results suggest that domain and motif compositions are conserved within each class, but divergent among different classes (Fig. 2, Fig. S3 and S4). CMT3 class has a BAH domain, a specific chromo domain adjacent to the methylase domain, and motif 1, 2, 3, 4, 6, 7, 9, 10 and 13. MET1 class is characterized by two DNMT1-RFD domains, two BAH domains and motifs 1, 2, 3, 4, 6, 7, 10, 11 and 13. DRM1/2 class has a shorter DNA methylase domain and motifs 3, 5, 9 and 15. All the DRM genes have no other domain identified except the YTH domain in Md-methylase7, and the Pkinase domain in Fv-methylase6, Pb-methylase10 and Pm-methylase3 (Fig. 2). The majority of genes in the DNMT2 class have only one single methylase domain identified and is characterized by motifs 3, 5, 8, 12 and 14. It is noted that Fv-methylase3 in the DNMT2 class is extremely long and has an extra DYW_deaminase domain. DYW proteins are implicated in chloroplast and mitochondrial RNA transcript maturation by numerous C to U editing events (Salone et al. 2007). Whether Fv-DNMT2 functions in this pathway is of great interest. Collectively, each methyltransferase class is characterized by specific combinations of domains/motifs, and variations within a particular class are observed as well.
Identification of genes encoding putative DNA demethylases
Previous studies on Arabidopsis have suggested that active DNA demethylases have three conserved domains, a helix-hairpin-helix-Gly-Pro-Asp (HhH-GPD) domain conferring glucosylase activity, domain B (DME domain) at the C-terminal and domain A at the N-terminal mediating DNA binding (Mok et al. 2010). Domain A and domain B are DNA demethylase-specific domains, and all the three domains are required for the proper 5mC glucosylase activity (Mok et al. 2010). Thus, in our analysis, genes encoding the proteins consisting of both hHh-GPD and DME domains were identified as a putative DNA demethylase genes, and the domain A was further confirmed by sequence homology.
This sequence-based searching identified two (in A. trichopoda) to nine (in Pyrus bretschneideri) putative DNA demethylase genes in each species, with F. vesca having four (Fig. 1). Based on the phylogenetic trees, DNA demethylase genes were grouped into two classes, the DME and the DML (Fig. 3). DME class genes are present in all eudicot species, but absent in monocots (Figs. 1, 3). All the species in Solanaceae, Cruciferae and Cucurbitaceae have a single DME gene, while the number of DME genes varies in Rosaceae, with Pyrus bretschneideri having the most of 4. It is noted that A. trichopoda does not have any DME genes either, thus it is more likely that DME was a new class originated in the common ancestor of eudicots and specifically expanded in Rosaceae. While DML genes are present in all the plant species investigated, the number of DML demethyalse genes ranges from 2 to 5, with Pyrus bretschneideri having the most (5).

A most likelihood phylogenetic trees and a schematic diagram for domain composition of DNA demethylase genes in the 15 species investigated. The results of the bootstrapping analysis larger than 50 % are shown. The phylogenetic tree was constructed based on the amino acids of the whole protein
The domain compositions identified by SMART show that the majority of the DNA demethylase genes have four common domains, ENCO3C (the DNA glycosylase domain), FES, Perm-CXXC (Permuted single zf-CXXC unit) and RRM-DME domain (Fig. 3). ENCO3C and FES are two adjacent domains, while Perm-CXXC and RRM-DME are next to each other as well. The conserved domain A has not been identified by Pfam or SMART, but was recognized by motif analyses as motifs 2 and 13 (Fig. S5).
WGD contributed to the duplication of DNA methyltransferase/demethylase genes in angiosperms
Some of the 14 angiosperm species had several rounds of whole-genome duplication/triplications (WGD) after their split from A. trichopoda (Fig. 1 data from the PGDD website, http://chibba.agtec.uga.edu/duplication/). The analyses above show that the number of DNA methyltransferase and demethylase genes varies in angiosperms. It is obvious that there is no clear correlation between the number of DNA methyltransferase or demethylase genes and the number of WGD events, though some species with more identified WGDs do have more genes identified (Table 1). Thus, other factors may have played a role in the duplication of DNA methyltransferase and demethylase genes as well. The expansion of gene families could result from WGDs, segmental, tandem or dispersed duplications. To disentangle the relative contributions of each mechanisms, the chromosomal locations of those genes were mapped back to the chromosomes and the possible mechanisms responsible for the duplication events were analyzed by MCscan (Wang et al. 2012).
The MCscan results revealed that out of the total 127 methyltransferase genes (including the DNMT2 class), 20 were related to WGDs, 9 were from tandem duplications and the rest of the 98 genes resulted from either proximal, dispersed duplications or identified as singletons (Table 1). Six out of the 15 species have methyltransferase genes originated from WGDs. Pyrus bretschneideri is the species having the most WGD-related duplicated genes (8 out of the total 15 genes). It is noted that among the duplicated gene pairs identified by MCscan resulting from WGDs, some of the respective genes degenerated quickly, with four genes losing the DNA methylase domain already (Table S5). Furthermore, 5 out of the 15 species had tandem duplication events contributed to the expansion of methyltransferase genes, and Brassica rapa is the species having the most tandem duplication events detected (Table 1). In addition, 16 out of the 56 DNA demethylase genes were identified as having originated from WGDs, 3 from tandem duplications and the rest of the 37 from other events (Table 1). Thus, WGDs played an important role in the expansion of DNA demethylase genes in angiosperms as well. Among the duplicated demethylase gene pairs identified as resulting from WGDs by MCscan, two genes had already lost the glycosylase domain (Table S5). Collectively, WGD and tandem duplications contributed to the expansion of both DNA methyltransferase and demethylase genes in angiosperms.
The maintenance of both genes from a duplication event could lead to fast diversification or functional compromise of the two redundant copies (Kaltenegger and Ober 2015). The domain and motif analyses suggest that functional redundancy did lead to faster evolution of those recently duplicated genes. Among the duplicated events that happened in the last 50 million years (Myr) which could still be detected, some had obvious domain diversifications (Figs. 2, 3). For example, in the CTM3 class, Md-methylase4/8 and Pb-methylase1/2 resulted from a duplication event that happened in their common ancestor. Among those four genes, three lost the BAH domain quickly (Fig. 2). In the DME class, the Pb-demethylase4 resulted from lineage-specific duplication and had already lost part of the DME domain (Fig. 3). Considering that within a class, genes from different species are highly conserved in the domain and motif composition, those recent duplicated gene pairs did evolve much faster than singletons. The fast gain and loss of domains in the duplicated gene pairs indicate that there was a wave of relaxed natural selection constraints since the duplication events happened.
Expression profiles of DNA methyltransferase and demethylase genes during fruit development in F. vesca
DNA methyltransferases and demethylases were revealed to be actively involved in fruit ripening in tomato (Zhong et al. 2013; Liu et al. 2015b). Strawberry is special as the fruits originated from the receptacle and are regarded as a non-climactic model crop species. To investigate whether and how DNA methyltransferases and demethylases might be involved in strawberry fruit development and ripening, their expressions were profiled in early-stage fruit development (data from Kang et al. 2013) and ripening (quantitative PCR data produced in this study). The genomic transcriptome data suggested that DNA methylation modifiers have quite diversified expression patterns. Fv-methylase6 (DRM class) is the only DNA methyltransferase gene showing relatively ubiquitous expression in fruits and also in vegetative tissues, while other DNA methyltransferase genes show specificity in different tissues/stages during early-stage fruit development. For the four DNA demethylase genes, Fv-demethylase1 and Fv-demethylase2 exhibit higher expression levels and more universal expression patterns in different tissues, while the mRNA levels of Fv-demethylase3 and Fv-demethylase4 are relatively lower (Fig. 4a). In summary, DNA methylation modifier genes have different tissue specificity, and each tissues has unique combinatorial patterns which might be consistent with their functions.

Expression profiles of the identified DNA methyltransferase and demethylase genes in F. vesca. a The mRNA levels of the identified DNA methylation modifiers in the early-stage fruit development. Data from Kang et al. (2013). Expression levels were calculated in the log2 scale. b Expression profiles of DNA methyltransferase and demethylase genes during fruit ripening. The expression levels relative to GAPDH were measured by quantitative RT-PCR and displayed in the log2 scale. Three biological replicates and three technical replicates were obtained for each data point. More details are shown in Fig. S4. c The schematic diagram for the five stages of fleshy fruit investigated in b
To study the expression patterns of DNA methylation modifier genes during fruit ripening, the expression levels of DNA methyltransferase and demethylase genes were investigated in strawberry fleshy fruits (stripped with seeds, including pith and cortex only) at big green (12 days after anthesis), white (white flesh with green seed), pre-turning (white flesh with red seeds), pink (light pink flesh with red seeds) and red (2–3 day after pink stage) stage by quantitative PCR reaction (Fig. 4b, c). For the expression levels of the eight DNA methyltransferase genes, four of them peaked at the pre-turning stage and three peaked at the pink stage (Fig. 4b, Fig. S6 and Fig. S7). Furthermore, all the four DNA demethylase genes reached their peaks at the pre-turning stage. Overall, the qPCR data suggest that the expression levels of DNA methylation modifier genes are dynamic during fruit ripening, and the majority of them peaked at either pre-turning or pink stage, indicating that DNA methylation patterns might undergo a dynamic change at the onset of fruit ripening.
Expression profiles of DNA demethylase genes upon abiotic stresses in F. vesca
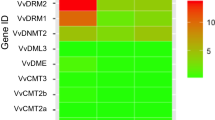
DNA methylations are suggested to play an important role in the regulation of gene expression in response to abiotic stresses (Sanchez and Paszkowski 2014; Xu et al. 2015). Salinity, drought, cold and heat are recurrent stresses land plants will encounter in the natural environment. The Arabidopsis transcriptome data shown that under those stresses, the expression levels of DNA demethylase genes underwent substantial changes (Fig. S8). To study how strawberry demethylase genes respond to abiotic stresses, the expression profiles of DNA demethylase genes were investigated in strawberry seedlings. The qPCR results demonstrated that DNA demethylase genes responded differentially to a particular abiotic stress (Fig. 5, Fig. S9 and S10). Fv-demethylase1 belonging to the DML class showed higher expression after cold, drought and salinity stresses, but it did not increase during heat shock. The mRNA level of Fv-demethylase2 decreased after heat or cold treatment, but increased after drought or salinity stresses. It is noted that Fv-demethylase2 mRNA level was lower after 3 h of heat shock, but recovered after 8 h (3 h heat shock + 5 h recovery), indicating that the effect of heat shock did not last long. Fv-demethylase4 expression was higher in all four treatments. As the only DME class gene, Fv-demethylase3 did not have dramatic response to any of the four treatments. In summary, some demethylase genes show dynamic expression patterns upon cold, heat, salinity and drought treatments, which indicates that active demethylation may be involved in F. vesca’s responses to abiotic stresses.

Expression profiles of DNA demethylase genes in response to abiotic stresses including heat, cold, drought and salinity. The expression levels relative to GAPDH were measured by quantitative RT-PCR and displayed in log2 scale. Three biological replicates and three technical replicates were obtained for each data point. More details are shown in Fig. S5
Discussion
In this study, we performed a sequence-based searching and phylogenetic analysis to identify the putative DNA methyltransferase and demethylase genes in F. vesca and other higher species. Nine methyltransferase genes and four demethylase genes were identified in F. vesca. Some of those F. vesca genes have very special domain/motif compositions and are of particular interest. For the 15 species investigated, the number of particular classes of methyltransferase and demethylase genes varies, and each class has different evolutionary characteristics. All four classes of DNA methyltransferase gene are present in all angiosperms, but one class of the demethylase genes, the DME, is absent in monocots (Fig. 1). A search for DNA demethylase genes in a reference species Selaginella moellendorffii demonstrates that there is only one gene encoding both the glycosylase domain and the DME domain identified in the genome, which belongs to the DML class (Fig. S11). In line with previously reported (Zemach et al. 2010), the lack of DME class DNA demethylase genes in A. trichopoda, S. moellendorffii and all monocots indicates that the DME is a newly evolved class of DNA demethylase gene that originated in the common ancestor of eudicots.
The phylogenetic studies suggested that WGDs and tandem duplication events contributed to the expansions of DNA methylation modifiers in angiosperms. But in F. vesca and the other three species, all identified genes resulted from dispersed duplications (Table 1). Pyrus bretschneideri has the largest number of both DNA methyltransferase and demethylase genes, for which lineage-specific duplications contributed substantially. Those recently duplicated gene pairs had a faster gain and loss of domains/motifs and experienced relaxed selective forces after the duplication events happened.
Strawberry flesh is of great interest, as it is a model fruit species for non-climacteric ripening. Both genomic transcriptome and qPCR data show diversified expression patterns of those methyltransferase and demethylase genes in fruit development and ripening. Particularly, some of the DNA methyltransferase genes and all of the DNA demethylase genes reach their expression peaks at either pre-turning or turning stage (Fig. 4b). This indicates that DNA methylation patterns might undergo a dramatic change at this critical stage of fruit ripening. Interestingly, an independent study on histone lysine methylation modifiers revealed that the expression levels of histone lysine methyltransferase and demethylase genes peaked at the pink stage as well (Gu et al. unpublished data). This concordance suggests a possible overall change of both DNA methylation and histone methylation, and a regulatory role of epigenetic factors at the onset of fruit ripening.
Plants are sessile and thus their fast response to abiotic stresses is of great importance for their adaptation to the ever-changing environment. Chromatin modifications play an essential role in regulating gene expression during abiotic stresses. Our data showed that DNA demethylase genes were found to respond to abiotic stresses including salinity, drought, cold and heat in F. vesca (Fig. 5). In accordance with the studies in Arabidopsis, the expression level of the DML class demethylase gene Fv-demethylase4 was found to increase substantially during heat shock. DNA methylation is important for suppressing the transcription of transposons and DNA repeats. This suppression can be transiently released under prolonged heat stress (Sanchez and Paszkowski 2014). The dramatically increased expression of demethylase genes in both Arabidopsis and strawberry under heat shock indicates that DML class demethylase genes might be involved in this ectopic TE activation in plants. In addition, during various abiotic stresses, the expression levels of DNA demethylase genes change substantially, suggesting that demethylation of some critical genes might be crucial for abiotic responses in plants. On the other hand, each DNA demethylase has its special target (Stroud et al. 2013). This specificity ensures that active DNA methylation by demethylases could regulate a set of related genes and that cells could respond more accurately. Considered that passive loss of DNA methylation is replication dependent while active demethylation is not, active DNA methylation by demethylases could respond more quickly and is more advantageous for plants suffering through severe stresses.
DNA methylation is essential in regulating developmental processes and responses to environment in plants. How DNA methylases and demethylases are involved in various cellular processes is of great interest. In this study, genes encoding DNA methyltransferases and demethylases are identified in F. vesca and other plant species. The phylogenetic studies suggest that whole-genome duplications and tandem duplication contributed to the expansion of those genes in plants. The expression data show that some DNA methylation modifier genes reach their highest expression levels in strawberry fleshy fruits when turning from white to red, demonstrating a possible regulatory role of DNA methylation in the ripening process of strawberry fruits. Also, our data show that expression of some DNA methylation modifier genes responds to various abiotic stresses including heat, cold, drought and salinity, which suggests that DNA methylation is most likely involved in some of the environmental influences on fruit growth and development. We therefore believe that the work presented here sheds some light on the possible roles of DNA methylation in fruit development under normal and abiotic stress environments, and also provides a basis for further characterization of epigenetic regulatory mechanisms on fruit development in F. vesca.
References
Amil-Ruiz F, Garrido-Gala J, Blanco-Portales R, Folta KM, Munoz-Blanco J, Caballero JL (2013) Identification and validation of reference genes for transcript normalization in strawberry (Fragaria × ananassa) defense responses. PLoS ONE 8:e70603
Arabidopsis Genome I (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815
Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2:28–36
Bharti P, Mahajan M, Vishwakarma AK, Bhardwaj J, Yadav SK (2015) AtROS1 overexpression provides evidence for epigenetic regulation of genes encoding enzymes of flavonoid biosynthesis and antioxidant pathways during salt stress in transgenic tobacco. J Exp Bot 66:5959–5969
Cavrak VV, Lettner N, Jamge S, Kosarewicz A, Bayer LM, Mittelsten Scheid O (2014) How a retrotransposon exploits the plant’s heat stress response for its activation. PLoS Genet 10:e1004115
Chan SW, Henderson IR, Jacobsen SE (2005) Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat Rev Genet 6:351–360
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10:2709–2721
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A (2010) The Pfam protein families database. Nucleic Acids Res 38:D211–D222
Furner IJ, Matzke M (2011) Methylation and demethylation of the Arabidopsis genome. Curr Opin Plant Biol 14:137–141
Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, Golic KG, Jacobsen SE, Bestor TH (2006) Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311:395–398
Guo S, Zhang J, Sun H, Salse J, Lucas WJ, Zhang H, Zheng Y, Mao L, Ren Y, Wang Z, Min J, Guo X, Murat F, Ham BK, Zhang Z, Gao S, Huang M, Xu Y, Zhong S, Bombarely A, Mueller LA, Zhao H, He H, Zhang Y, Zhang Z, Huang S, Tan T, Pang E, Lin K, Hu Q, Kuang H, Ni P, Wang B, Liu J, Kou Q, Hou W, Zou X, Jiang J, Gong G, Klee K, Schoof H, Huang Y, Hu X, Dong S, Liang D, Wang J, Wu K, Xia Y, Zhao X, Zheng Z, Xing M, Liang X, Huang B, Lv T, Wang J, Yin Y, Yi H, Li R, Wu M, Levi A, Zhang X, Giovannoni JJ, Wang J, Li Y, Fei Z, Xu Y (2013) The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 45:51–58
He XJ, Chen T, Zhu JK (2011) Regulation and function of DNA methylation in plants and animals. Cell Res 21:442–465
Henderson IR, Jacobsen SE (2007) Epigenetic inheritance in plants. Nature 447:418–424
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas WJ, Wang X, Xie B, Ni P, Ren Y, Zhu H, Li J, Lin K, Jin W, Fei Z, Li G, Staub J, Kilian A, van der Vossen EA, Wu Y, Guo J, He J, Jia Z, Ren Y, Tian G, Lu Y, Ruan J, Qian W, Wang M, Huang Q, Li B, Xuan Z, Cao J, Asan WuZ, Zhang J, Cai Q, Bai Y, Zhao B, Han Y, Li Y, Li X, Wang S, Shi Q, Liu S, Cho WK, Kim JY, Xu Y, Heller-Uszynska K, Miao H, Cheng Z, Zhang S, Wu J, Yang Y, Kang H, Li M, Liang H, Ren X, Shi Z, Wen M, Jian M, Yang H, Zhang G, Yang Z, Chen R, Liu S, Li J, Ma L, Liu H, Zhou Y, Zhao J, Fang X, Li G, Fang L, Li Y, Liu D, Zheng H, Zhang Y, Qin N, Li Z, Yang G, Yang S, Bolund L, Kristiansen K, Zheng H, Li S, Zhang X, Yang H, Wang J, Sun R, Zhang B, Jiang S, Wang J, Du Y, Li S (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41:1275–1281
Kaltenegger E, Ober D (2015) Paralogue interference affects the dynamics after gene duplication. Trends Plant Sci 20:814–821
Kang C, Darwish O, Geretz A, Shahan R, Alkharouf N, Liu Z (2013) Genome-scale transcriptomic insights into early-stage fruit development in woodland strawberry Fragaria vesca. Plant Cell 25:1960–1978
Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McLnerney JO (2006) Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol 6:29
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11:204–220
Le TN, Schumann U, Smith NA, Tiwari S, Au PC, Zhu QH, Taylor JM, Kazan K, Llewellyn DJ, Zhang R, Dennis ES, Wang MB (2014) DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol 15:458
Liu J, Feng L, Li J, He Z (2015a) Genetic and epigenetic control of plant heat responses. Front Plant Sci 6:267
Liu R, How-Kit A, Stammitti L, Teyssier E, Rolin D, Mortain-Bertrand A, Halle S, Liu M, Kong J, Wu C, Degraeve-Guibault C, Chapman NH, Maucourt M, Hodgman TC, Tost J, Bouzayen M, Hong Y, Seymour GB, Giovannoni JJ, Gallusci P (2015b) A DEMETER-like DNA demethylase governs tomato fruit ripening. Proc Natl Acad Sci USA 112:10804–10809
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25:402–408
Mok YG, Uzawa R, Lee J, Weiner GM, Eichman BF, Fischer RL, Huh JH (2010) Domain structure of the DEMETER 5-methylcytosine DNA glycosylase. Proc Natl Acad Sci USA 107:19225–19230
Paterson AH, Bowers JE, Chapman BA (2004) Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc Natl Acad Sci USA 101:9903–9908
Penterman J, Uzawa R, Fischer RL (2007) Genetic interactions between DNA demethylation and methylation in Arabidopsis. Plant Physiol 145:1549–1557
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Rutowicz K, Puzio M, Halibart-Puzio J, Lirski M, Kotlinski M, Kroten MA, Knizewski L, Lange B, Muszewska A, Sniegowska-Swierk K, Koscielniak J, Iwanicka-Nowicka R, Buza K, Janowiak F, Zmuda K, Joesaar I, Laskowska-Kaszub K, Fogtman A, Kollist H, Zielenkiewicz P, Tiuryn J, Siedlecki P, Swiezewski S, Ginalski K, Koblowska M, Archacki R, Wilczynski B, Rapacz M, Jerzmanowski A (2015) A specialized histone H1 variant is required for adaptive responses to complex abiotic stress and related DNA methylation in Arabidopsis. Plant Physiol 169:2080–2101
Salone V, Rudinger M, Polsakiewicz M, Hoffmann B, Groth-Malonek M, Szurek B, Small I, Knoop V, Lurin C (2007) A hypothesis on the identification of the editing enzyme in plant organelles. FEBS Lett 581:4132–4138
Sanchez DH, Paszkowski J (2014) Heat-induced release of epigenetic silencing reveals the concealed role of an imprinted plant gene. PLoS Genet 10:e1004806
Saze H, Tsugane K, Kanno T, Nishimura T (2012) DNA methylation in plants: relationship to small RNAs and histone modifications, and functions in transposon inactivation. Plant Cell Physiol 53:766–784
Shulaev V, Sargent DJ, Crowhurst RN, Mockler TC, Folkerts O, Delcher AL, Jaiswal P, Mockaitis K, Liston A, Mane SP, Burns P, Davis TM, Slovin JP, Bassil N, Hellens RP, Evans C, Harkins T, Kodira C, Desany B, Crasta OR, Jensen RV, Allan AC, Michael TP, Setubal JC, Celton JM, Rees DJ, Williams KP, Holt SH, Ruiz Rojas JJ, Chatterjee M, Liu B, Silva H, Meisel L, Adato A, Filichkin SA, Troggio M, Viola R, Ashman TL, Wang H, Dharmawardhana P, Elser J, Raja R, Priest HD, Bryant DW Jr, Fox SE, Givan SA, Wilhelm LJ, Naithani S, Christoffels A, Salama DY, Carter J, Lopez Girona E, Zdepski A, Wang W, Kerstetter RA, Schwab W, Korban SS, Davik J, Monfort A, Denoyes-Rothan B, Arus P, Mittler R, Flinn B, Aharoni A, Bennetzen JL, Salzberg SL, Dickerman AW, Velasco R, Borodovsky M, Veilleux RE, Folta KM (2011) The genome of woodland strawberry (Fragaria vesca). Nat Genet 43:109–116
Stroud H, Greenberg MV, Feng S, Bernatavichute YV, Jacobsen SE (2013) Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 152:352–364
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, Kissinger JC, Paterson AH (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40:e49
Wu J, Wang Z, Shi Z, Zhang S, Ming R, Zhu S, Khan MA, Tao S, Korban SS, Wang H, Chen NJ, Nishio T, Xu X, Cong L, Qi K, Huang X, Wang Y, Zhao X, Wu J, Deng C, Gou C, Zhou W, Yin H, Qin G, Sha Y, Tao Y, Chen H, Yang Y, Song Y, Zhan D, Wang J, Li L, Dai M, Gu C, Wang Y, Shi D, Wang X, Zhang H, Zeng L, Zheng D, Wang C, Chen M, Wang G, Xie L, Sovero V, Sha S, Huang W, Zhang S, Zhang M, Sun J, Xu L, Li Y, Liu X, Li Q, Shen J, Wang J, Paull RE, Bennetzen JL, Wang J, Zhang S (2013) The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res 23:396–408
Xu R, Wang Y, Zheng H, Lu W, Wu C, Huang J, Yan K, Yang G, Zheng C (2015) Salt-induced transcription factor MYB74 is regulated by the RNA-directed DNA methylation pathway in Arabidopsis. J Exp Bot 66:5997–6008
Zemach A, Kim MY, Silva P, Rodrigues JA, Dotson B, Brooks MD, Zilberman D (2010) Local DNA hypomethylation activates genes in rice endosperm. Proc Natl Acad Sci USA 107:18729–18734
Zhang H, Zhu JK (2012) Active DNA demethylation in plants and animals. Cold Spring Harb Symp Quant Biol 77:161–173
Zheng X, Pontes O, Zhu J, Miki D, Zhang F, Li WX, Iida K, Kapoor A, Pikaard CS, Zhu JK (2008) ROS3 is an RNA-binding protein required for DNA demethylation in Arabidopsis. Nature 455:1259–1262
Zhong S, Fei Z, Chen YR, Zheng Y, Huang M, Vrebalov J, McQuinn R, Gapper N, Liu B, Xiang J, Shao Y, Giovannoni JJ (2013) Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat Biotechnol 31:154–159
Acknowledgments
This study was funded by the National Natural Science Foundation of China (31400269) and the Fundamental Research Funds for the Central Universities (KJQN201537) to Tingting Gu.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by S. Hohmann.
Tingting Gu and Shuai Ren are considered to be co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gu, T., Ren, S., Wang, Y. et al. Characterization of DNA methyltransferase and demethylase genes in Fragaria vesca . Mol Genet Genomics 291, 1333–1345 (2016). https://doi.org/10.1007/s00438-016-1187-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-016-1187-y