
Abstract
Members of basic helix-loop-helix (bHLH) gene family found in all eukaryotes play crucial roles in response to stress. Though, most eukaryotes carry the proteins of this family, biological functions of the most bHLH family members are not deeply evaluated in plants. In this study, we conducted a comprehensive genome-wide analysis of bHLH transcription factors in salt tolerant common bean. We identified 155 bHLH protein-encoding genes (PvbHLH) by using in silico comparative genomics tools. Based on the phylogenetic tree, PvbHLH genes were classified into 8 main groups with 21 subfamilies. Exon–intron analysis indicated that proteins belonging to same main groups exhibited a closely related gene structure. While, the PvbHLH gene family has been mainly expanded through segmental duplications, a total of 11 tandem duplication were detected. Genome-wide expression analysis of bHLH genes showed that 63 PvbHLH genes were differentially expressed in at least one tissue. Three of them displayed higher expression values in both leaf and root tissues. The in silico micro-RNA target transcript analyses revealed that totally 100 PvHLH genes targeted by 86 plant miRNAs. The most abundant transcripts, which were targeted by all 18 plant miRNA, were belonging to PvHLH-22 and PvHLH-44 genes. The expression of 16 PvbHLH genes in the root and leaf tissues of salt-stressed common bean was evaluated using qRT-PCR. Among them, two of PvbHLHs, PvbHLH-54, PvbHLH-148, were found to be up-regulated in both tissues in correlation with RNA-seq measurements. The results of this study could help improve understanding of biological functions of common bean bHLH family under salt stress. Additionally, it may provide basic resources for analyzing bHLH protein function for improving economic, agronomic and ecological benefit in common bean and other species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Among various transcription factor families, dehydration responsive element (DRE) binding factor, ERF (ethylene responsive factors), WRKY, MYB, bZIP and basic helix-loop-helix (bHLH) families play crucial roles in coordination of regulatory networks related with stress responses (Zhou et al. 2009; Cui et al. 2013; Lata et al. 2014; Yan et al. 2014). The latter is a universal transcription factor found in all eukaryotes. Although, most eukaryotes carry the proteins of this family, the biological functions of the most bHLH family members are not deeply evaluated in plants (Liu et al. 2014). Up to now, studies related with function of plant bHLH proteins were relatively limited when compared with their animal orthologs (Song et al. 2014). There are a few reports showing the role of bHLH genes under salt stress in plants such as Arabidopsis, rice, and tomato (Zhou et al. 2009; Feng et al. 2013; Liu et al. 2014). The bHLH proteins are characterized with specific signature domain consisting of about 60 amino acids. Each domain has two distinct regions having different function. The first segment, which consist of about 15 amino acids with typical six basic residues that they have role in binding to DNA, is called as basic region, which settled in the N-terminus (Atchley and Fitch 1997). With the increasing number of completed and drafted genomes, a numbers of identified bHLH gene families have been also increased. For instance, a total of 225, 480 and 206 bHLH-encoding genes from Arabidopsis, soybean, and potato have been reported in plant transcription factor databases, respectively (Jin et al. 2014). It has been also reported that plants includes 26 subfamilies of bHLH proteins according to the phylogenetic analyses (Pires and Dolan 2010).
Soil salinity is one of the most important environmental stress factors affecting crop yield worldwide (Munns and Tester 2008). High salinity effects wide range of developmental process and cause to retardation of growth, decrease in biomass, and leaf senescence (Munne-Bosch and Alegre 2004). Due to their sessile nature, plants are exposed to any of abiotic stresses use different physiological, biochemical and molecular mechanisms including reprogrammed transcriptome pattern to cope with the detrimental effects of those conditions (Yamaguchi-Shinozaki and Shinozaki 2006). Molecular and physiological responses against diverse stress factors are controlled by the genetic regulation at transcriptional level, which are mainly regulated by transcription factors (TF) (Płażek et al. 2013; Dou et al. 2014; Thamilarasan et al. 2014). TF is defined as a group of proteins involved in regulation of specific gene expression either positively or negatively by binding to promoter or enhancer region of DNA (Latchman 1997).
Pulse crops have a crucial role in sustainable agriculture because of theirs nitrogen fixing ability with rhizobia. Common bean or Phaseolus vulgaris L., one of the most consumed pulse crops, is an important source for human diet by providing an important part of proteins, micronutrients and calories of daily needs (Petry et al. 2015). Common bean is available as dry, canned, and frozen forms in market. The dried seeds have more economical value than others. The annual global bean production is about 23 million metric tons. With an annual production of 3.3 million tons, India is in the first place (Food and Agriculture Organization of the United 2014). The complete genome sequence of common bean, a true diploid with genome size estimated to be 587 Mbp, was recently published (Schmutz et al. 2014) and publicly accessible (http://www.phytozome.net/commonbean.php). It will be easier to do genomic and comparative analyses, such as genome-wide identification of transcription factors, by using this genome data (Schmutz et al. 2014). However, analysis of bHLH in common bean has not been carried out at genome-wide level against different stress conditions.
In this study, we conducted a comprehensive genome-wide analysis of bHLH TFs in common bean. We also characterized genomic structures, chromosomal locations and sequence homologies of all common bean bHLH TF genes. The expression analysis of selected bHLH genes was conducted using qRT-PCR and previously uploaded RNAseq data in root and leaf tissues under salt stress conditions.
Materials and methods
Sequence retrieval, analysis and phylogenetic tree construction
The bHLH amino acid sequences belong to P. vulgaris were identified by the combination of three approaches. Firstly, bHLH encoding amino acid sequences pertaining to 13 different plants (Arabidopsis thaliana, Carica papaya, Brachypodium distachyon, Cucumis sativus, Physcomitrella patens, Oryza sativa subsp. japonica, Populus trichocarpa, Sorghum bicolor, Glycine max, Medicago truncatula, Vitis vinifera, Lotus japonicus and Zea mays) were downloaded from plant transcription factor database 3.0 (http://planttfdb.cbi.pku.edu.cn) (Jin et al. 2014). Totally, 2736 amino acid sequences encoding bHLH transcription factors were retrieved. In order to determine homologous peptides from common bean, a BLASTP search at PHYTOZOME v10.1 database (http://www.phytozome.net) were carried out using default parameters (Goodstein et al. 2012). Additionally, the keyword search was performed with bHLH in PHYTOZOME v10.1 database (http://www.phytozome.net). Moreover, The Hidden Markov Model (HMM) profiles of the bHLH domains in the Pfam database (http://pfam.sanger.ac.uk) were searched against the PHYTOZOME v10.1 database (http://www.phytozome.net) of P. vulgaris. All hits with expected values less than 1.0 were downloaded as fastq files and redundant sequences were removed using the decrease redundancy tool (http://web.expasy.org/decrease_redundancy) and elimdupes (http://hcv.lanl.gov/content/sequence/elimdupes/elimdupes.html). Every non-redundant sequence identified was subsequently checked for the presence of the conserved bHLH domain by SMART (Letunic et al. 2012) and Pfam (http://pfam.sanger.ac.uk) searches to confirm that it belong to bHLH transcription factors family.
The final amino acid sequences of bHLH proteins were aligned in ClustalW by using CLC Genomics Workbench 7.0 software (CLC bio, Aarhus, Denmark) with default parameters. Then, a phylogenetic trees of aligned common bean bHLH proteins was constructed using MEGA 6.0.1 and CLC Genomics Workbench via the Neighbor-Joining (NJ) method with the following parameters: WAG protein substitution model, gamma distribution, and bootstrap (1000). The phylogenetic tree was visualized with ITOL (Letunic and Bork 2007). The density of bHLH was determined as the ratio of number of bHLH to genome size in Mb (Song et al. 2014). The conserved motifs in full-length bHLH proteins were identified CLC Genomics Workbench 7.0.
Gene ontology (GO) and functional annotation
The functional characterization and annotation of bHLH sequences were performed using CLC Genomics Workbench Blast2GO plugin with default parameters. First, the common bean amino acids sequences were used as queries in a BLASTP search launched from CLC Genomics Workbench. The BLAST search is run against the non-redundant (NR) database at NCBI with an e-value of 1xE-10 and we used the top 50 alignments for each sequence. The resulting Multi Blast data collection is then converted into a Blast2GO Project. GO annotation is carried out by applying the Blast2GO annotation rule, which computes an annotation score for each candidate GO term.
Chromosomal location, gene-structure prediction and genomic distribution of bHLH genes
The exact positions of bHLH genes on common bean chromosomes were determined by BLASTN search of the P. vulgaris sequences against the PHYTOZOME v10.1 database using default settings (http://www.phytozome.net). The genes were aligned separately onto eleven bean chromosomes based on their ascending order of physical position (bp), from the short-arm telomere to the long-arm telomere and finally displayed using MapChart v2.2 (http://www.wageningenur.nl/en/show/Mapchart.htm) (Voorrips 2002). The exon–intron structure of each bHLH gene was determined by aligning the full-length cDNA sequence or predicted coding sequence (CDS) with the genomic sequence in the bean genome database (http://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Pvulgaris). The gene schematic structure was drawn by the Gene Structure Display Server (http://gsds.cbi.pku.edu.cn/index.php) (Hu et al. 2014). Tandem and segmental duplications were also identified according to the method of Plant Genome Duplication Database (Tang et al. 2008). In detail, adjoining genes of same sub-family located within 30 kbp of each other were characterized as tandem duplication (Du et al. 2013; Shiu and Bleecker 2003). Firstly, BLASTP search was performed against all predicted peptide sequences of common bean for segmental duplications and top five matches with ≤1e−05 was identified as potential anchors. Then, MCScan was used for evaluation of collinear blocks and finally alignments with ≤1e−10 were considered as significant matches (Tang et al. 2008; Du et al. 2013).
Estimation of synonymous and non-synonymous substitution rates
To understand the expansion of this important gene family, the ratios of non-synonymous (Ka) versus synonymous (Ks) substitution rates (Ka/Ks) were estimated. The amino-acid sequences tandem and segmental duplicated protein-encoding bHLH genes were aligned using CLUSTALW according to multiple sequence alignment tool. For estimation of the synonymous (Ks) and non-synonymous (Ka) substitution rates, alignments of the amino-acid sequences and their respective original cDNA sequences of bHLH genes were performed in the CODEML program of PAML interface tool of PAL2NAL (http://www.bork.embl.de/pal2nal) (Suyama et al. 2006). Time of duplication (million years ago, Mya) and divergence of each bHLH genes were calculated using a synonymous mutation rate of λ substitutions per synonymous site per year, as T = Ks/2λ (λ = 6.5 × 10e−9) (Lynch and Conery 2000; Yang et al. 2008).
3D protein homology modeling and protein properties
First of all, BLASTP search with the default parameters was performed in the Protein Data Bank (PDB) (Berman et al. 2000) with all bHLH proteins for identification of the best template having similar sequence and known three-dimensional structure. Using ‘intensive’ mode in Protein Homology/Analogy Recognition Engine (Phyre2) (Kelley and Sternberg 2009), the data was analyzed for prediction of protein structure of bean bHLHs. The theoretical isoelectric point (pI) and protein statistics were analyzed using ExPASy (Gasteiger et al. 2003) and Sequence Manipulation Suite (http://www.bio-soft.net/sms/), respectively. The PROSOII program (http://mips.helmholtz-muenchen.de/prosoII/prosoII.seam) was used to predict the sequence based solubility of proteins (Smialowski et al. 2012).
In silico identification of miRNAs targeting the bHLH genes
The perfect or closely perfect complementarity between miRNAs and their target genes can be used for identification of miRNAs specifically targeted a gene (Rhoades et al. 2002). To predict potential miRNAs targeting the bHLH genes, the web-based psRNA Target Server (http://plantgrn.noble.org/psRNATarget) was used with default parameters. Additionally, identified miRNA targets were further analyzed using BLASTX searches with ≤1e−10 against P. vulgaris EST sequences at NCBI database. The interaction between identified miRNAs and their bHLH target transcripts were visualized by using CYTOSCAPE v3.2.1 (http://www.cytoscape.org/).
Expression analysis of PvbHLH genes using transcriptome data
To evaluate the common bean bHLH gene expression patterns, we used the Illumina RNA-seq data downloaded from Sequence Read Archive (SRA), following accession number SRR957668 (salt-treated leaf), SRR957667 (control leaf), SRR958472 (salt-treated root and SRR958469 (control root) that were reported previously (Hiz et al. 2014). Original RNA-seq data includes 52,858, 60,590, 51,564 and 59,510 unigenes in control leaf, control root, salt-treated leaf and salt-treated root, respectively. All raw reads, downloaded as single SRA files, was spitted into two paired-end files and converted to “fastq” format by the NCBI SRA Toolkit’s fastq-dump command. The quality of fastq files were evaluated with FASTQC and low-quality reads [Phred quality (Q) score <20] were trimmed with CLC Genomics Workbench 7.0. After final quality check, the reads were aligned to Phaseolus vulgaris genome (v1.0), downloaded from PHYTOZOME V10.1 database (http://www.phytozome.net), by using CLC Genomics Workbench with default parameters.
Uniquely mapped reads were used in the expression level determination. Normalization of the gene expression values were carried out by the reads per kilobase of exon model per million mapped reads (RPKM) algorithm (Mortazavi et al. 2008). To identify differentially expressed bHLH genes, a FDR-value ≤0.001, fold change (RPKM-tr/RPKM-cont) ≥2 and the absolute ratio of log2 (RPKM-tr/RPKM-cont) ≥1 were used as threshold values. Finally, the heat maps of hierarchical clustering were visualized with PermutMatrix (Caraux and Pinloche 2005).
Plant materials, growth conditions, and salt stress applications
The salt tolerant “Ispir” variety was used for gene expression analysis (Dasgan and Koc 2009). Surface sterilization of seeds was carried out by applying 10 % (v/v) hypochlorite solution for 15 min and rinsing three times with distilled water. The seeds were germinated in vermiculite containing plug trays at 24/20 °C cycle under a 16 h/8 h photoperiod with 350 μmol m−2 s−1 light intensity, and 50–60 % relative humidity. They watered daily with Hoagland’s solution (Hoagland and Arnon 1950) in a growth chamber up to trifoliate leaf stage. During salt treatment, seedlings were kept at same conditions and watered with same solution including 150 mM NaCl. After 9 days, root and leaf tissues were collected from salt treated and control plants in order to conduct physiological and gene expression analyzes.
RNA extraction and quantitative real-time PCR analysis
Total RNAs of leaf and root tissues were isolated by RNeasy Plant Mini Kit (Qiagen, Valencia, California, USA) according to the manufacturer’s protocol and quantified using NanoDrop 2000D UV–VIS spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and quality-checked using Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The first strand of cDNA was synthesized by using 1 µg of DNAse I treated total RNA in a 20 µl reaction volume with RevertAid™ First Strand cDNA Synthesis Kit (Thermo Scientific, USA).
qRT-PCR experiments were conducted in order to measure tissue-specific expression levels of the salt responsive PvbHLH TF family genes that showed differentially upregulated expression pattern in RNA-seq analysis upon salt stress. qRT PCR analysis was performed by following the previously reported method (Turktas et al. 2013) using SYBR Green I Master Kit (Roche Germany) on LightCycler 480 Instrument II (Roche, Germany). Primers were designed individually for root and leaf tissues in P. vulgaris cv. İspir. Leaf specific primers were PvbHLH-54, PvbHLH-148, PvbHLH-69, PvbHLH-53, PvbHLH-98, PvbHLH-28, PvbHLH-124, PvbHLH-82, PvbHLH-128 and root specific primers were PvbHLH-54, PvbHLH-148, PvbHLH-69, PvbHLH-81, PvbHLH-104, PvbHLH-04, PvbHLH-133, PvbHLH-116, PvbHLH-119 and PvbHLH-97. Besides PvbHLH-54, PvbHLH-148 and PvbHLH-69 were designed for both leaf and root tissues since they showed higher expression level in RNA-seq analysis. The common bean 18S rRNA gene was used as an internal control (Budak et al. 2013; Wang et al. 2013). A list of the primers used in qRT-PCR is given in Table S1. The qRT-PCR was carried out in 96-well optical plates. PCR reactions were performed in a total volume of 20 μl containing 0.1 μl reverse and forward primers (100 pmol), 2 μl of cDNA, 10 μl FastStart SYBR Green I Master Mix and nuclease-free water was added up to 20 μl. The qRT-PCR conditions were set up as follows: preheating at 95 °C for 5 min; followed by 55 cycles of 95 °C for 10 s; 53 °C or 55 °C for 20 s; and 72 °C for 10 s. The melting curves were adjusted to 95 °C for 5 s and 55 °C for 1 min and then cooled to 40 °C for 30 s. All reactions were repeated three times with triple biological replicates. The expression levels were calculated as the mean-signal intensity across the three replicates.
MDA and proline analysis
To assess membrane damage because of salinity stress treatments, lipid peroxidation in terms of malondialdehyde (MDA) content was measured with spectrophotometer by using the method of Ohkawa et al. (1979). For measurement of membrane damage, about 0.2 g fresh shoot tissues were homogenized with liquid nitrogen by the addition of 1 ml of 5 % trichloroacetic acid (TCA). The homogenates were transferred to fresh tubes and centrifuged at 11,200 g for 15 min at room temperature. Equal volumes of supernatant and 0.5 % thiobarbituric acid (TBA) in 20 % TCA (freshly prepared) were put into eppendorf tubes and incubated for 25 min at 96 °C. The tubes were placed in ice bath after incubation period and then centrifuged at 7800g for 5 min. Absorbance of the supernatant was determined at 532 nm and the correction for non-specific turbidity was performed by subtracting the absorbance at 600 nm. MDA contents were calculated using an extinction coefficient of 155 mM−1 cm−1.
Proline contents of salt treated and control samples were estimated according to the method of Bates et al. (1973). 0.3 g of each collected sample was grinded with liquid nitrogen and the homogenized extracts were suspended with 1 ml of 3 % sulphosalicilic acid. Supernatants were obtained with centrifugation at 15,000g for 5 min at 4 °C and 0.1 ml of supernatants were mixed with 0.2 ml acid ninhydrin, 0.2 ml 96 % acetic acid and 0.1 ml 3 % sulphosalicilic acid. Toluene was added to this mixture after the incubation at 96 °C for 1 h. In order to achieve phase separation, another centrifugation was performed at 15,000g for 5 min at 4 °C. Absorbance measurements were carried out with upper phases at 520 nm. In order to determine the proline amounts in samples, proline standard curve was used. The curve was formed from the measurement of absorbance of proline standard (1, 10, 50, 100, 150, 200 and 300 µM) suspended in 3 % sulphosalicilic acid.
Results
Identification of PvbHLH genes and their phylogeny
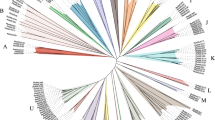
A total of 155 bHLH genes encoding bHLH domain was identified in common bean genome (Table S2). The protein coding sequences in the representative genomes of 13 plant species was searched for comparative genomic analyses (Table S3). Finally, 2060 genes encoding bHLH domain were identified in these selected plant species. The density of PvbHLH is about 0.2640 which is lower than most of the analyzed plants. Zea mays (0.098), Physcomitrella patens (0.2304) and Lotus japonicus (0.2038) has lower density when compared to ratio of the number of PvbHLH to genome size (Mb). Unrooted phylogenetic trees, constructed by CLC Genomics Workbench by ClustalW (Thompson et al. 1997) alignment by the Neighbor-Joining method (Saitou and Nei 1987) and bootstrap analysis (1000 replicates), was used in order to elucidate the phylogenetic relationships between PvbHLH domain sequences. The conserved motifs were predicted by the CLC Genomics Workbench 7.0 (Sup. Figure 1). The alignment indicated that the residues Arg-20, Arg-21, Leu-31 and Pro-37 were completely conserved in bHLH domains of all identified PvbHLH proteins. In general, bHLH proteins clustered in together shared similar motif composition. Based on the conserved motifs and phylogenetic tree, we identified 8 major groups of PvbHLH consisting of 21 subfamily (Fig. 1).

Relationship of neighbor-joining phylogenetic tree of the PvbHLH domains
The identified members of PvbHLH genes have significantly different size, fundamental chemical and physical characters. Although, the bHLH domains generally located close to N-terminal, location of the domains within the protein also differs. Protein length of PvbHLHs is changed from 90 to 725 amino acids. According to an instability index (II), most of the PvbHLH proteins were estimated as unstable in a test tube. EXPASY analysis revealed that the PvbHLH protein sequences had large variations in isoelectric point (pI) values (ranging from 4.67 to 10.18) and molecular weight (ranging from 10.365 to 79.679 kDa). Sequence based solubility prediction was performed and almost half of the PvbHLH proteins were found as soluble in Escherichia coli. The details of PvbHLH protein sequences were summarized in Table S2.
Chromosomal distribution of bHLH genes
PvbHLH genes were placed on 11 chromosomes of common bean based on BLASTN analysis. The exact position (in kp) of each bHLHs on bean chromosome is shown in Fig. 2. Among all, chromosome 3 contains the highest number of bHLH genes, 24/155 (15.5 %) and chromosome 4 contains the least, 5/155 (3.2 %) (Sup. Figure 2). Distribution arrangement of bHLH genes on individual chromosomes also indicated certain physical regions with a relatively higher accumulation of gene clusters. Common bean bHLH genes are mainly located on both ends of the chromosomes. For example, bHLH genes found on chromosomes 1, 2, 3, 7 and chromosomes 10 appear to be congregate at the lower end and upper ends of the arms. In addition, some bHLH genes are located near the telomere regions of the chromosomes, especially on chromosome 5, 6, 8 and 9.

Distribution of 155 PvbHLH genes onto eleven bean chromosomes. The chromosomes are numbered between 1 and 11 and shown at the top of each chromosome (Chr represented as bars). Chromosomal distances are given in Mbp. Black regions showed telomere regions of the chromosomes
Duplication and divergence rate of the bHLH genes
Tandem and segmental duplication of PvbHLH gene members in common bean were determined. A total of 11 tandem duplicate PvbHLH genes were detected (Table S4), which means that a small portion of tandem duplication events have caused gene expansion of bHLH family members in bean. PvbHLH genes located on chromosome 10 showed the highest numbers of tandem duplications. Several direct tandem repeats were also found on chromosome 5 (PvbHLH-65, PvbHLH-66, PvbHLH-67 and PvbHLH-68). Totally, 61 segmental duplicated bean bHLH genes have been detected, accounting for around 39 % (61/155) of total PvbHLH genes (Table S5). The most surprising aspect of the data is high segmental duplicated ratio which is firstly shown for PvbHLH genes in common bean.
The relationships between Darwin’s positive selection in divergence and duplication of PvbHLH genes were also examined to better understand family expansion of these important family members. For this, the ratios of non-synonymous (Ka) versus synonymous (Ks) substitution rates (Ka/Ks) were estimated for 11 tandem (Table S4) and 61 segmentally (Table S5) duplicated gene-pairs. The ratios of Ka/Ks for tandem duplication ranged from 0.02 to 1.11 with an average of 0.38, whereas Ka/Ks for segmentally duplicated gene-pairs varied from 0.05 to 0.54 with an average of 0.22. If Ka/Ks ratio is below 1, it is generally suggested that genes are found intense selection pressure. Therefore, average Ka/Ks ratios indicated that tandem and segmental duplicated genes of bean bHLHs are under strong purifying selection pressure. In addition, tandem and segmental duplication events of these genes may be estimated to have occurred around 4–14 and 6–20 Mya, respectively (Sup. Figure 3).
Exon–intron organization of PvbHLH genes in bean
Gene structure analysis of the 155 PvbHLHs was performed to gain some information about exon–intron organization (Sup. Figure 4). A total of 11 bHLH genes without intron were found, which accounts for 7 % of total PvbHLH genes. Most of these intronless genes were clustered into the Cluster 6 and Cluster 7c. Intron organization and numbers of bHLH genes in common bean showed different variation and distribution into different clusters. According to exon–intron organization of bHLH genes in bean, phylogenetically related proteins exhibited a closely related gene structure, in terms of intron number or exon length.
3D homology modeling of PvbHLH proteins
To construct 3D homology model for bHLH proteins, BLASTP search was performed against the PDB. Twelve PvbHLH proteins (PvbHLH-23-50-59-62-74-78-79-80-86-103-105-123) with high homology rate were selected. To predict homology modeling in Phyre2, which utilizes the alignment of hidden Markov models via HMM–HMM search (Soding 2005), the detection rate method was used. The intensive mode was selected in Phyre 2 to increase accuracy of alignment. In addition, it integrates a new ab initio folding simulation termed as Poing (Jefferys et al. 2010) to model areas of proteins without any significant homology for known structures. The 3D protein modelling of selected 12 bHLH proteins are predicted at >90 % confidence and the percentage residue varied from 80 to 100 (Fig. 3). The secondary structures were predominantly constituted of α helices and having rare incurrence of β sheets. Thus, all suggested protein structures are assessed to be highly reliable which offers a preliminary basis for understanding the molecular function of PvbHLH proteins.

Predicted 3-D structures of bHLH proteins in bean. The structure of 12 bean bHLH proteins with >90 % confidence level are shown
GO annotation terms
The identified common bean bHLH genes were classified into the three Gene ontology (GO) categories, which were Cellular Component (5 %), Biological Process (28 %), and Molecular Function (67 %) (Fig. 4, Table S6). Within each three main categories, genes that annotated for “binding” (155), “developmental process” (20), “single-organism process” (21), “response to stimulus” (17) and “multicellular organismal process” were the most abundant (Fig. 4, Table S6).

Gene ontology (GO) patterns for the bHLH proteins. The CLC Genomics Workbench-BLAST2GO-ANNEX categorized the gene ontology as cellular component, biological process, and molecular function
In silico identification of bHLH gene targeting miRNAs
The miRNAs targeting the PvbHLH genes were predicted. Totally, 100 PvbHLH genes targeted by 86 plant miRNAs were identified in this study. Most of the PvbHLH genes were targeted by 3 or more plant miRNAs. The most abundant transcripts, which were targeted by all 18 plant miRNAs, were belonging to PvbHLH-22 and PvbHLH-44 genes (Sup. Figure 5). miR396, miR530 and miR165 are one of the most important miRNAs found by targeting and BLASTn analysis.
bHLH gene expression profiling
We analyzed expression levels of bHLH genes in two different tissue samples of common bean. According to normalized RPKM values, expression of 20 bHLH genes (PvbHLH-01, PvbHLH-02, PvbHLH-04, PvbHLH-150, PvbHLH-143, PvbHLH-120, PvbHLH-105, PvbHLH-108, PvbHLH-90, PvbHLH-66, PvbHLH-67, PvbHLH-68, PvbHLH-39, PvbHLH-40, PvbHLH-46, PvbHLH-17, PvbHLH-80, PvbHLH-27, PvbHLH-30 and PvbHLH-141) were not detected in reads belong to either root and leaf tissues. A total of 63 PvbHLH genes were differentially expressed in at least one tissue. Among the 155 PvbHLHs, 16 PvbHLHs were differentially upregulated in leaf tissue, including PvbHLH-28, PvbHLH-53, 82 and PvbHLH-69. In root tissue, a total of 11 PvbHLHs including PvbHLH-69, PvbHLH-104, PvbHLH-119 and PvbHLH-116 showed higher expression pattern. Interestingly, three of total PvbHLHs (54, 148 and 69) were highly expressed both in leaf and root tissues. Genome-wide expression analysis also showed that there were a total of 55 up-regulated PvbHLHs and 63 down-regulated PvbHLHs in leaf tissues after salt stress (Table S7). Likewise, there were 69 up-regulated PvbHLH genes and 54 down-regulated PvbHLH genes in root tissues. An expression profile of all identified PvbHLH genes was shown as heat map in Fig. 5.

Heat map of bHLH genes expressed in both tissues
Gene expression levels of salt responsive bHLH transcription factor family genes
Expression levels of 9 bHLH family members (PvbHLH-54, PvbHLH-148, PvbHLH-69, PvbHLH-53, PvbHLH-98, PvbHLH-28, PvbHLH-124, PvbHLH-82, PvbHLH-128) and 10 bHLH family members (PvbHLH-54, PvbHLH-148, PvbHLH-69, PvbHLH-81, PvbHLH-104, PvbHLH-04, PvbHLH-133, PvbHLH-116, PvbHLH-119 and PvbHLH-97) in salt-stressed leaf and root tissues were measured, respectively. qRT-PCR results revealed that all nine selected genes were up-regulated in common bean leaf tissues after salt treatment. Among them, PvbHLH-53 and PvbHLH-148 showed relatively higher expression level when compared to control sample. Therefore, we validated all the 9 PvbHLHs in leaf tissue by using qRT-PCR. But we found that the genes PvbHLH-69, PvbHLH-81, PvbHLH-104, PvbHLH-133, and PvbHLH-116, were down-regulated and PvbHLH-54, PvbHLH-148, PvbHLH-04, PvbHLH-119 and PvbHLH-97 were up-regulated after salt treatment in root tissue. Among the 3 PvbHLHs (PvbHLH-54, PvbHLH-148, PvbHLH-69), which was differentially up-regulated in transcriptome data of both root and leaf tissues, PvbHLH-54 and PvbHLH-148 were up-regulated in both tissues according to the qRT-PCR results (Fig. 6).

Relative expression levels of bHLH TF family genes for root and leaf tissues under salt stress conditions (salt stress treatment) and untreated (control) in Phaseolus vulgaris (P. vulgaris cv. İspir)
MDA and proline analysis
In this study, we measured MDA content of both root and leaf tissues in control and salt-treated plants. We observed that there was no significantly difference between control root and salt-treated root tissues. Likewise, there was no significant change in spite of an increase in MDA content in control and salt-treated leaf tissues (Table S8). Additionally, we measured proline, an osmoprotectant, in order to compare control and salt-treated common bean plants. Both root and leaf tissues showed significantly increased proline content when compared to control groups under salinity stress (Table S8). Therefore, we confirmed that accumulation of proline in stress tolerant plant species under salt stress has been correlated with stress tolerance.
Discussion
Common bean is an important crop for human diet being a rich source of proteins, micronutrients and calories of daily needs. In spite of its importance, there is scarce information on genome-wide characterization of transcription factors. Likewise, there is a few reports related with genome-wide identification of bHLH transcription factors in plants compared to metazoans. Up to date a total of 150 and 45 bHLH gene family members were identified in Arabidopsis and soybean by Toledo-Ortiz et al. (2003), Pires and Dolan (2010) and Osorio et al. (2012), respectively. Among the plant bHLH transcription factors, the highest number of bHLH transcription factor genes was discovered in Chinese cabbage which contains 230 BrabHLH genes in Chinese cabbage genome (Song et al. 2014). In a recent study, Sun et al. (2015) detected 159 bHLH protein-encoding genes (SlbHLH) in tomato. We have also identified 155 bHLH genes located on 11 chromosomes of common bean. The phylogenetic analysis showed that PvbHLHs were separated into the eight groups consist of 21 subfamily consistent with previous finding (Sun et al. 2015). Although, number of common bean PvbHLH genes is similar to Arabidopsis and tomato, they are divergent in different plant species. So, it can be concluded that a similar gene evolution of bHLH transcription factor genes might occur in bean, Arabidopsis and tomato.
While there is no report related with genome-wide identification of bHLH under salt stress conditions, OrbHLH2 and AtbHLH122 genes were identified and functionally characterized in terms of resistance to high concentration of salt (Zhou et al. 2009; Liu et al. 2014). Here, we identified salt responsive bHLH genes at transcriptome level in common bean. Gene expression patterns can provide important clues for gene function. Therefore, a total of 19 genes (9 for leaf and 10 for root tissues) were selected for further confirmation via qRT PCR. PvHLH-148, an orthologs of AtbHLH122, showed higher expression in both tissues after salinity stress in consistent with previous report (Liu et al. 2014). Likewise, we observed higher expression of PvHLH-154, another bHLH122 like gene, only in salt stressed leaf tissues As a result, expression of all selected PvbHLH genes were validated in leaf tissues and a strong consistency with RNA-seq data was obtained. On the other hand, half of the measured PvbHLH genes were consistent with transcriptome analysis of root tissues. The inconsistency might be resulted due to the quality of RNA-seq data or growing conditions of plants (Fang and Cui 2011).
Although expression of selected PvbHLH genes was mostly validated in leaf and root tissues and they showed strong consistency with RNA-seq data, we also evaluated the physiological responses of common bean by measuring MDA and Proline content under salt stress. Increased proline concentration was generally evaluated as a good indicator of stress tolerance in many plants. Stress-tolerant plants accumulate more proline compared to sensitive species upon stress conditions (Ashraf and Foolad 2007; Kavas et al. 2013). As a results of MDA and Proline assays, we observed a meaningful relationship between these two substances in salt-stress-tolerant common bean cultivar. Finally, we concluded that stress tolerant common bean cultivar (Ispir) might have well established physiological and molecular stress tolerance mechanism including higher proline accumulation and regulated expression pattern of PvbHLHs, respectively.
Segmental and tandem duplications play a crucial roles in the evolution and expansion of gene families in plants (Cannon et al. 2004). These results match those observed in earlier studies which indicate that segmental duplication has a major impact for the expansion of gene families (Baloglu 2014). Gene duplication occur frequently and leads to evolution of related genes in organisms (Mehan et al. 2004). Studies in rice and Arabidopsis show that tandem and segmental duplication events of bHLH genes were observed. So, this supported a prominent role for expansion of this important gene family (Heim et al. 2003; Toledo-Ortiz et al. 2003; Li et al. 2006). A total of 14 tandem duplication were determined in tomato bHLH genes (Sun et al. 2015). The findings observed in this study are consistent with these studies that have examined prominent role for genome segments and tandem duplication in the expansion of bHLH gene family (Heim et al. 2003; Toledo-Ortiz et al. 2003; Li et al. 2006). This observation may support the idea that evolution of those genes might arise from two ways including specific gene duplications or integration into genomic region following a reverse transcription (Lecharny et al. 2003). Our findings provide further support for another idea that duplicated genes have different functions, which provides evolutionary novelty for organisms (Nam et al. 2004; Nei and Rooney 2005).
Whole-genome tandem and segmental duplications cause formation of multiple copies of genes in many plant TF gene-families such as bZIP, NAC, MBF as well as in HSPs (Cannon et al. 2004; Jain et al. 2007; Nijhawan et al. 2008; Wang et al. 2011; Puranik et al. 2012; Baloglu et al. 2014). The ratios of Ka/Ks were below 1 for tandem and segmentally duplicated gene-pairs for bHLH transcription factor members in bean. All duplicated bean PvbHLHs genes were found under strong purifying selection pressure. These results are consistent with those of other studies and suggest that the segmental and tandem duplication events have an important role in evolution, for shaping and expansion of gene families in different organisms (Nijhawan et al. 2008; Wang et al. 2011; Baloglu et al. 2014).
In order to obtain information about gene structures, detection of exon–intron organization have been widely used method (Baloglu 2014). We have found that phylogenetically related proteins have an identical gene structure. Similar cases have also been observed in Arabidopsis, poplar, rice, moss and algae (Li et al. 2006; Carretero-Paulet et al. 2010). Intron distribution pattern and its correlation with phylogenetic classification were also detected in Chinese cabbage BrabHLH (Song et al. 2014) and tomato SlbHLH genes (Sun et al. 2015). These findings may help us to understand evolutionary conservation of bHLH genes in different organisms. Amino acid motifs of PvbHLHs also show strong correlation with other plant`s bHLHs such as tomato and chinese cabbage.
MicroRNAs (miRNAs) are short (aprox. 21 nucleotides), single-stranded, noncoding RNAs characterized in different organisms including plants and animals and have functions in regulation of expression of their target (Bartel 2004). They play critical roles in plant development as well as response to environmental stress. Up to now, several salt responsive miRNAs such as miR159, miR394, miR396 and miR1866 have been identified (Liu et al. 2008; Ding et al. 2009). In the context of in silico identification of miRNAs target genes, we found that 100 PvbHLH genes targeted by 86 plant miRNAs. One of the identified miRNA was miR396, which was previously reported for salt stress response (Liu et al. 2008; Eren et al. 2014). In addition to the targeting of salt responsive genes, this family of miRNAs also target growth regulating factor (GRF) transcription factors, rhodenase-like proteins, and kinesin-like protein B.
In conclusion, a comprehensive genome-wide characterization of the common bean bHLH family genes was performed and their expression levels were analyzed in response to excess salt concentration for the first time. A total of 155 PvbHLH genes were identified by using in silico analysis. This analysis of the bHLH family in common bean with regard to other plant species reveals some common features, such as family size and global structure. Expression patterns of 19 PvbHLH genes have been analyzed under salt stress with qRT-PCR. Analysis of expression patterns shows that bHLH subfamily genes were expressed in both root and leaf tissues. Additionally, genomic structures, chromosomal locations, gene duplication and sequence similarity analyses of bHLH genes were reported. Our results suggest that chromosomal segment duplications may be the main factors for the expansion of the bHLH gene family in common bean. The results of this study revealed the importance of bHLH genes during salt stress response in roots and leaves. It may serve in a key role in elucidating bHLH family gene function in protein interactions, signaling pathway regulations and defense responses under salt stress conditions. Additionally, this results may become useful as tools to engineer stress-tolerant plants.
References
Ashraf M, Foolad MR (2007) Roles of glycine betaine and proline in improving plant abiotic stress resistance. Environ Exp Bot 59(2):206–216
Atchley WR, Fitch WM (1997) A natural classification of the basic helix–loop–helix class of transcription factors. P Natl Acad Sci USA 94(10):5172–5176
Baloglu MC (2014) Genome-wide in silico identification and comparison of growth regulating factor (GRF) genes in Cucurbitaceae family. Plant Omics 7(4):260–270
Baloglu MC, Eldem V, Hajyzadeh M, Unver T (2014) Genome-wide analysis of the bZIP transcription factors in cucumber. PLoS One 9(4):e96014
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116(2):281–297
Bates LS, Waldren RP, Teare ID (1973) Rapid determination of free proline for water-stress studies. Plant Soil 39(1):205–207
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The Protein Data Bank. Nucleic Acids Res 28(1):235–242
Budak H, Akpinar B, Unver T, Turktas M (2013) Proteome changes in wild and modern wheat leaves upon drought stress by two-dimensional electrophoresis and nanoLC-ESI–MS/MS. Plant Mol Biol 83(1–2):89–103
Cannon S, Mitra A, Baumgarten A, Young N, May G (2004) The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol 4(1):1–21
Caraux G, Pinloche S (2005) PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 21(7):1280–1281
Carretero-Paulet L, Galstyan A, Roig-Villanova I, Martinez-Garcia JF, Bilbao-Castro JR, Robertson DL (2010) Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol 153(3):1398–1412
Cui MH, Yoo KS, Hyoung S, Nguyen HTK, Kim YY, Kim HJ, Ok SH, Yoo SD, Shin JS (2013) An Arabidopsis R2R3-MYB transcription factor, AtMYB20, negatively regulates type 2C serine/threonine protein phosphatases to enhance salt tolerance. FEBS Lett 587(12):1773–1778
Dasgan HY, Koc S (2009) Evaluation of salt tolerance in common bean genotypes by ion regulation and searching for screening parameters. J Food Agric Environ 7(2):363–372
Ding D, Zhang L, Wang H, Liu Z, Zhang Z, Zheng Y (2009) Differential expression of miRNAs in response to salt stress in maize roots. Ann Bot London 103(1):29–38
Dou L, Zhang X, Pang C, Song M, Wei H, Fan S, Yu S (2014) Genome-wide analysis of the WRKY gene family in cotton. Mol Genet Genomics 289(6):1103–1121
Du DL, Zhang QX, Cheng TR, Pan HT, Yang WR, Sun LD (2013) Genome-wide identification and analysis of late embryogenesis abundant (LEA) genes in Prunus mume. Mol Biol Rep 40(2):1937–1946
Eren H, Pekmezci MY, Okay S, Türktaş M, İnal B, İlhan E, Atak M, Eryaman M, Ünver T (2014) Hexaploid wheat (Triticum aestivum L.) root miRNome analysis in response to salt stress. Ann Appl Biol (in press)
Fang Z, Cui X (2011) Design and validation issues in RNA-seq experiments. Brief Bioinform 12(3):280–287
Feng H-L, Ma N-N, Meng X, Zhang S, Wang J-R, Chai S, Meng Q-W (2013) A novel tomato MYC-type ICE1-like transcription factor, SlICE1a, confers cold, osmotic and salt tolerance in transgenic tobacco. Plant Physiol Biochem 73:309–320
Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31 (13):3784–3788
Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, Rokhsar DS (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40:1178–1186
Heim MA, Jakoby M, Werber M, Martin C, Weisshaar B, Bailey PC (2003) The basic helix-loop-helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol Biol Evol 20(5):735–747
Hiz MC, Canher B, Niron H, Turet M (2014) Transcriptome analysis of salt tolerant common bean (Phaseolus vulgaris L.) under saline conditions. PLoS One 9(3):e92598
Hoagland DR, Arnon DI (1950) The water-culture method for growing plants without soil. Circ Calif Agric Exp Stn 347:1–32
Hu B, Jin J, Guo A-Y, Zhang H, Luo J, Gao G (2014) GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics
Jain M, Nijhawan A, Arora R, Agarwal P, Ray S, Sharma P, Kapoor S, Tyagi AK, Khurana JP (2007) F-box proteins in rice. Genome-wide analysis, classification, temporal and spatial gene expression during panicle and seed development, and regulation by light and abiotic stress. Plant Physiol 143(4):1467–1483
Jefferys BR, Kelley LA, Sternberg MJE (2010) Protein folding requires crowd control in a simulated cell. J Mol Biol 397(5):1329–1338
Jin J, Zhang H, Kong L, Gao G, Luo J (2014) PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res 42:1182–1187
Kavas M, Baloğlu MC, Akça O, Köse FS, Gökçay D (2013) Effect of drought stress on oxidative damage and antioxidant enzyme activity in melon seedlings. Turk J Biol 37:491–498
Kelley LA, Sternberg MJE (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat Protocols 4(3):363–371
Lata C, Mishra AK, Muthamilarasan M, Bonthala VS, Khan Y, Prasad M (2014) Genome-wide investigation and expression profiling of AP2/ERF transcription factor superfamily in foxtail millet (Setaria italica L.). PLoS One 9(11):e113092
Latchman DS (1997) Transcription factors: an overview. Int J Biochem Cell Biol 29(12):1305–1312
Lecharny A, Boudet N, Gy I, Aubourg S, Kreis M (2003) Introns in, introns out in plant gene families: a genomic approach of the dynamics of gene structure. J Struct Func Genom 3(1–4):111–116
Letunic I, Bork P (2007) Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23(1):127–128
Letunic I, Doerks T, Bork P (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res 40:302–305
Li N, Zhang DS, Liu HS, Yin CS, Li XX, Liang WQ, Yuan Z, Xu B, Chu HW, Wang J, Wen TQ, Huang H, Luo D, Ma H, Zhang DB (2006) The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell 18(11):2999–3014
Liu H-H, Tian X, Li Y-J, Wu C-A, Zheng C-C (2008) Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14(5):836–843
Liu W, Tai H, Li S, Gao W, Zhao M, Xie C, Li W-X (2014) bHLH122 is important for drought and osmotic stress resistance in Arabidopsis and in the repression of ABA catabolism. New Phytol 201(4):1192–1204
Lynch M, Conery JS (2000) The evolutionary fate and consequences of duplicate genes. Science 290(5494):1151–1155
Mehan MR, Freimer NB, Ophoff RA (2004) A genome-wide survey of segmental duplications that mediate common human genetic variation of chromosomal architecture. Hum Genomics 1(5):335–344
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth 5(7):621–628
Munne-Bosch S, Alegre L (2004) Die and let live: leaf senescence contributes to plant survival under drought stress. Funct Plant Biol 31(3):203–216
Munns R, Tester M (2008) Mechanisms of salinity tolerance. Annu Rev Plant Biol 59(1):651–681
Nam J, Kim J, Lee S, An GH, Ma H, Nei MS (2004) Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. P Natl Acad Sci USA 101(7):1910–1915
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152
Nijhawan A, Jain M, Tyagi AK, Khurana JP (2008) Genomic survey and gene expression analysis of the basic leucine zipper transcription factor family in rice. Plant Physiol 146(2):333–350
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95(2):351–358
Osorio MB, Bucker-Neto L, Castilhos G, Turchetto-Zolet AC, Wiebke-Strohm B, Bodanese-Zanettini MH, Margis-Pinheiro M (2012) Identification and in silico characterization of soybean trihelix-GT and bHLH transcription factors involved in stress responses. Genet Mol Biol 35(1):233–246
Petry N, Boy E, Wirth J, Hurrell R (2015) Review: the potential of the common bean (Phaseolus vulgaris) as a vehicle for iron biofortification. Nutrients 7(2):1144–1173
Pires N, Dolan L (2010) Origin and diversification of basic-helix-loop-helix proteins in plants. Mol Biol Evol 27(4):862–874
Płażek A, Tatrzańska M, Maciejewski M, Kościelniak J, Gondek K, Bojarczuk J, Dubert F (2013) Investigation of the salt tolerance of new Polish bread and durum wheat cultivars. Acta Physiol Plant 35(8):2513–2523
Puranik S, Sahu PP, Srivastava PS, Prasad M (2012) NAC proteins: regulation and role in stress tolerance. Trends Plant Sci 17(6):369–381
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP (2002) Prediction of plant microRNA targets. Cell 110(4):513–520
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Bio Evol 4(4):406–425
Schmutz J, McClean PE, Mamidi S, Wu GA, Cannon SB, Grimwood J, Jenkins J, Shu S, Song Q, Chavarro C, Torres-Torres M, Geffroy V, Moghaddam SM, Gao D, Abernathy B, Barry K, Blair M, Brick MA, Chovatia M, Gepts P, Goodstein DM, Gonzales M, Hellsten U, Hyten DL, Jia G, Kelly JD, Kudrna D, Lee R, Richard MMS, Miklas PN, Osorno JM, Rodrigues J, Thareau V, Urrea CA, Wang M, Yu Y, Zhang M, Wing RA, Cregan PB, Rokhsar DS, Jackson SA (2014) A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet 46(7):707–713
Shiu SH, Bleecker AB (2003) Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol 132(2):530–543
Smialowski P, Doose G, Torkler P, Kaufmann S, Frishman D (2012) PROSO II—a new method for protein solubility prediction. FEBS J 279(12):2192–2200
Soding J (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21(7):951–960
Song XM, Huang ZN, Duan WK, Ren J, Liu TK, Li Y, Hou XL (2014) Genome-wide analysis of the bHLH transcription factor family in Chinese cabbage (Brassica rapa ssp. pekinensis). Mol Genet Genomics 289(1):77–91
Sun H, Fan HJ, Ling HQ (2015) Genome-wide identification and characterization of the bHLH gene family in tomato. Bmc Genomics 16(1):9
Suyama M, Torrents D, Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34:W609–W612
Tang HB, Bowers JE, Wang XY, Ming R, Alam M, Paterson AH (2008) Perspective—synteny and collinearity in plant genomes. Science 320(5875):486–488
Thamilarasan SK, Park JI, Jung HJ, Nou IS (2014) Genome-wide analysis of the distribution of AP2/ERF transcription factors reveals duplication and CBFs genes elucidate their potential function in Brassica oleracea. Bmc Genomics 15:422
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25(24):4876–4882
Toledo-Ortiz G, Huq E, Quail PH (2003) The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell 15(8):1749–1770
Turktas M, Inal B, Okay S, Erkilic EG, Dundar E, Hernandez P, Dorado G, Unver T (2013) Nutrition Metabolism Plays an Important Role in the Alternate Bearing of the Olive Tree (Olea europaea L.). PloS one 8 (3):e59876
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93(1):77–78
Wang JZ, Zhou JX, Zhang BL, Vanitha J, Ramachandran S, Jiang SY (2011) Genome-wide expansion and expression divergence of the basic leucine zipper transcription factors in higher plants with an emphasis on Sorghum. J Integr Plant Biol 53(3):212–231
Wang C, Deng P, Chen L, Wang X, Ma H, Hu W, Yao N, Feng Y, Chai R, Yang G, He G (2013) A wheat WRKY transcription factor TaWRKY10 confers tolerance to multiple abiotic stresses in transgenic tobacco. PLoS One 8(6):e65120
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57:781–803
Yan H, Jia H, Chen X, Hao L, An H, Guo X (2014) The cotton WRKY transcription factor GhWRKY17 functions in drought and salt stress in transgenic Nicotiana benthamiana through ABA signaling and the modulation of reactive oxygen species production. Plant Cell Physiol 55(12):2060–2076
Yang ZF, Gu SL, Wang XF, Li WJ, Tang ZX, Xu CW (2008) Molecular evolution of the CPP-like gene family in plants: insights from comparative genomics of Arabidopsis and rice. J Mol Evol 67(3):266–277
Zhou J, Li F, Wang JL, Ma Y, Chong K, Xu YY (2009) Basic helix-loop-helix transcription factor from wild rice (OrbHLH2) improves tolerance to salt- and osmotic stress in Arabidopsis. J Plant Physiol 166(12):1296–1306
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by Research Fund of Ondokuz Mayıs University (PYO.ZRT.1901.13.017).
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by A. K. Tyagi.
Electronic supplementary material
Below is the link to the electronic supplementary material.
438_2015_1095_MOESM1_ESM.tif
Sup. Figure 1. Conserved amino acid motifs in the bHLH domains. The overall height of each letter and bar shows the conservation of the sequence at that position (TIFF 1473 kb)
438_2015_1095_MOESM3_ESM.jpg
Sup Figure 3. Time of duplication and divergence (MYA) of PvbHLH genes. Time of duplication and divergence (MYA) rates were calculated with synonymous substitution rate (Ks) and estimated using tandem and segmental duplicated PvbHLH genes (JPEG 111 kb)
438_2015_1095_MOESM4_ESM.jpg
Sup Figure 4. Exon–intron structures of the PvbHLH genes. The bHLH family members were classified according to Fig. 1 (Phylogenetic tree). The values in parentheses show the number of corresponding clusters of bHLH genes. Yellow boxes represent exons and black lines represent introns (JPEG 2608 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Kavas, M., Baloğlu, M.C., Atabay, E.S. et al. Genome-wide characterization and expression analysis of common bean bHLH transcription factors in response to excess salt concentration. Mol Genet Genomics 291, 129–143 (2016). https://doi.org/10.1007/s00438-015-1095-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-015-1095-6