
Abstract
Equine piroplasmosis (EP) is a global worldwide infection, which can lead to the death of animals. Despite the causative agents of EP being well studied, there are no data on the distribution and genetic characteristics of EP agents in any region of Russia. In this study, blood samples from 750 horses from Novosibirsk province, Irkutsk province, and Altai region of Russian Siberia were examined for the presence of EP agents. Theileria equi and Babesia caballi were detected in all examined regions, with mean prevalence rates of 60.4% and 7.2%, respectively. The identified pathogens were genetically characterized by the 18S rRNA gene. The determined T. equi sequences were highly conserved and belonged to genotypes A and E, with genotype E being found in 88.6% of genotyped samples. In contrast to T. equi, B. caballi sequences were genetically diverse. Seven sequence variants of B. caballi were identified, and only two of them matched known sequences from the GenBank database. The determined B. caballi sequences belonged to four distinct branches within genotype A. Mixed infections with several variants of B. caballi or with T. equi and B. caballi were common. The conducted phylogenetic analysis based on all available B. caballi sequences of the 18S rRNA gene (> 900 bp) from GenBank and from this study first demonstrated the presence of five monophyletic clusters within genotype A and three clusters within genotype B. Thus, the genetic study of B. caballi from Siberia has significantly expanded the data on the genetic diversity of this pathogen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Theileria equi [Theileria, Theileriidae, Piroplasmida, Apicomplexa] and Babesia caballi [Babesia, Babesiidae, Piroplasmida, Apicomplexa] are the main causative agents of equine piroplasmosis (EP) worldwide (Tirosh-Levy et al. 2020a). Recently, a new species, T. haneyi, closely related to T. equi, has been discovered in equine blood based on complete genome sequencing (Knowles et al. 2018). EP is found mainly in Equidae—horses, donkeys, mules, zebras. Reported cases of infections in other animals are rare (de Sousa et al. 2018; Onyiche et al. 2019; Qablan et al. 2013).
The life cycles of both Theileria and Babesia include asexual reproduction (merogony) in the erythrocytes of equine hosts and sexual (gamogony) and asexual (sporogony) stages within the ticks; however, they have their own peculiarities. Thus, Theileria spp., but not B. caballi, undergoes asexual schizogony within equine lymphocytes before the penetration into red blood cells. Another difference is that only B. caballi can be effectively transmitted transovarially to the next generation of ticks (Scoles and Ueti 2015; Wise et al. 2013).
Equine piroplasmosis has emerged as a global worldwide infection in the beginning of twentieth century. The infection first emerged in Southern Africa and then spread to other continents, including Eurasia. The disease posed a serious threat to the economy due to high morbidity and mortality rates in horses. EP is a reportable infection by the World Organization for Animal Health (WOAH) (https://www.woah.org/). Today, only several countries report EP-free status, namely, Australia, Canada, Great Britain, Ireland, Japan, New Zealand, and, until a large outbreak in 2009, the United States (Tirosh-Levy et al. 2020a).
Equine piroplasmosis can occur in acute, subacute, and chronic forms. Acute infection is characterized by fever up to 40 °C, loss of appetite, weakness, anorexia, mucosal edema, splenomegaly, thrombocytopenia, and hemolytic anemia leading to hemoglobinuria and jaundice. In some cases, infection can lead to abortions (Dirks et al. 2021; Wise et al. 2013). Both parasites cause similar pathology, but clinical presentations are more severe in cases of T. equi infection (Tirosh-Levy et al. 2020a; Wise et al. 2013). Horses that recovered from an acute infection remain persistently infected by pathogens without any clinical manifestations; however, they can transmit protozoan pathogens to tick vectors. Typically, the persistence of T. equi is lifelong, while B. caballi is cleared within several years (Scoles and Ueti 2015; Wise et al. 2013). The level of long-termed parasitemia is usually low, so EP can be diagnosed mainly by serological or molecular methods, but not by pathogen finding in blood smears (Scoles and Ueti 2015). Expectedly, clinical cases of both T. equi and B. caballi are associated with higher mean parasite load compared to subclinically infected horses (Tirosh-Levy et al. 2020b). In endemic areas, the proportion of infected animals is often high, exceeding 60% (Bhoora et al. 2020; Dahmana et al. 2019; Díaz-Sánchez et al. 2018). Theileria equi is a more common agent than B. caballi. In 20-year period (2000–2019), the global estimated prevalence of T. equi and B. caballi infections was 34.6% and 7.4%, respectively, being the highest in South America (Tirosh-Levy et al. 2020a).
The natural foci of EP cannot be maintained in the absence of competent vectors. At least 33 ixodid species, mainly belonging to the genera Hyalomma, Dermacentor, and Rhipicephalus, have been implicated as competent vectors for both B. caballi and T. equi (Scoles and Ueti 2015; Onyiche et al. 2019).
Nowadays, the danger of EP is less significant because of a lower abundance of horses, the availability of modern acaracid and antiprotozoal drugs, and a wide distribution of pre-immune horse population. Cases of acute infection in endemic areas are rare; young animals are exposed to infected ticks when they are usually protected by maternal antibodies, and exposure to infection can lead to the development of strong immunity. Outbreaks primarily occur when naive horses are transported into endemic areas or when infected animals are imported into non-endemic areas where competent tick vectors are abundant (Scoles and Ueti 2015). Therefore, the outbreaks of EP periodically occur in both endemic and non-endemic regions from various countries (Fedulina et al. 2014; Ionita et al. 2018; Short et al. 2012; Tirosh-Levy et al. 2020b).
Significant genetic variability was shown for both B. caballi and T. equi. Based on the study of the 18S rRNA gene, three genotypes of B. caballi (A, B1, and B2) and five genotypes of T. equi (A–E) have been identified (Bhoora et al. 2009, 2020; Tirosh-Levy et al. 2020a); however, in 2018, some T. equi isolates of genotype C were assigned to a new species, T. haneyi (Knowles et al. 2018). Distribution of different T. equi genotypes and T. haneyi depends on geography (Tirosh-Levy et al. 2020a). Notably, T. equi and T. haneyi cannot be distinguished by the 18S rRNA gene phylogeny but only based on the presence of equi merozoite antigen 1 (ema-1) gene in T. equi and hypothetical protein gene of unknown function in T. haneyi isolates (Knowles et al. 2018).
Mixed infection with these three EP agents as well as simultaneous infection with several T. equi genotypes has been described in many studies (Bhoora et al. 2020; Coultous et al. 2020; Elsawy et al. 2021). Moreover, in some regions, infection of equines with multiple T. equi genotypes prevailed over single genotype infection (Coultous et al. 2020). There is no reliable evidence that a specific T. equi genotype correlates with increased virulence; however, two studies demonstrated a higher frequency of genotype A in clinically affected horses compared with genotype D (Tirosh-Levy et al. 2020b) and genotype B (Manna et al. 2018).
In Russia, EP was first recorded in 1906–1908 in the Ryazan province; subsequently, the infection was found in various provinces of Russia. In the 1930–1950s, the epizootic situation regarding EP significantly worsened due to the movement of a large number of horses to endemic areas; there was a massive loss of animals. This was the reason for conducting large-scale studies of this disease in the USSR. Both pathogens were identified in the blood of horses from endemic areas and the role of Hyalomma scupense, Rhipicephalus bursa, Dermacentor silvarum, Dermacentor nuttalli, and Dermacentor marginatus as carriers of these hemoparasites was proved (Abramov 1955; Budnik 1955; Ovchinnikov et al. 1941; Petunin 1948; Scoles and Ueti 2015). In the 1960s, the EP incidence in Russia decreased significantly. However, cases of EP are periodically observed in various regions of Siberia. In particular, in 2008, an outbreak of infection was noted in the Ust-Udinsky district of the Irkutsk province (Eastern Siberia), which led to the death of animals (Fedulina et al. 2014). Unfortunately, EP has not been practically studied in Russia in recent decades. There are only fragmentary data on cases of this disease, and the diagnosis in most cases was based only on the symptoms. Moreover, there are no data on the genetic diversity of the causative agent of EP from any regions of Russia.
This study was aimed to investigate the epidemiology of EP in three remote regions of Western and Eastern Siberia and to examine genetic variability of revealed EP agents.
Materials and methods
Sampling
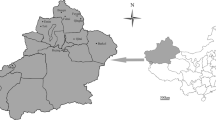
A total of 750 blood samples were collected in 2012–2022 from apparently healthy horses from randomly chosen farms, stables, and equestrian centers from two districts of Novosibirsk province (n = 20), 13 districts of Irkutsk province (n = 424), and eight districts of Altai region, including seven districts of the Republic of Altai (n = 286) and one district of the Altai Territory (n = 20) (Fig. 1). Blood samples (5 ml) were taken aseptically from each animal using VACUETTE® tubes containing EDTA (Greiner Bio-One, Austria), transported to the laboratory and stored at − 20 °C until DNA extraction.

Map of three regions of Russian Siberia showing the sites of sampling. The districts where horse blood samples were collected are marked by dark color. The districts were EP agents were identified were marked by
 —T. equi genotype E,
—T. equi genotype E,
 —T. equi genotype A,
—T. equi genotype A,
 —B. caballi
—B. caballi
Molecular genetic analysis
Total DNA was extracted from 200 μl of each blood sample using the Proba NK kit (DNA-Technology, Moscow, Russia) according to the manufacturer’s protocol. Piroplasmida DNA was detected by nested PCR for 18S rRNA gene using primer sets BS1/BS2 and PiroA/PiroC. For species determination, additional nested reactions were performed independently using primers Teq1 and Teq2, specific to T. equi, and primers Bcab1 and Bcab2, specific to B. caballi (Table 1). All PCR reactions were conducted under conditions previously described in Rar et al. (2011). The annealing temperature and the length of obtained PCR fragments are specified in Table 1. The amplified 18S rRNA gene fragments of all B. caballi and some T. equi specimens were sequenced. For subsequent sequencing of longer fragments, 1218–1252 bp fragments were amplified for a number of T. equi and B. caballi specimens using primer sets Bs3/Bs4 and Bs5/Bs6, respectively (Table 1).
The obtained PCR products were gel purified (0.6% SeaKem® GTG-agarose, Lonza, Haifa, Israel) and sequenced. Sanger reactions were conducted using the BigDye Terminator V. 3.1 Cycling Sequencing Kit (Applied Biosystems, CA, USA) and visualized with a 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). For sequence analysis, BlastN (http://www.ncbi.nlm.nih.gov/BLAST) and BioEdit (http://www.mbio.ncsu.edu/BioEdit/bioedit.html) programs were used. Phylogenetic trees were constructed using the maximum likelihood (ML) method based on the Tamura-Nei model in MEGA 7.0 with 1000 bootstrap replicates (Kumar et al. 2016) based on sequences from this study and sequences from the GenBank database available on 1 March 2024. The stability of the constructed trees was estimated by bootstrap analysis with 1000 replicates.
Differences in the prevalence of infectious agents were computed using the Pearson χ2 goodness-of-fit test (http://www.socscistatistics.com/tests/chisquare/). P < 0.05 was regarded as significant.
Results
Theileria equi and B. caballi prevalence in equine blood
Among 750 examined equine blood samples, 468 (62.4%) samples contained Piroplasmida DNA. In different regions, the portion of infected horses was similar and varied from 55.6% in Altai region to 67.5% in Irkutsk province (Table 2). However, the proportion of infected horses varied significantly between districts within the region. In Altai region, the prevalence of EP agents ranged from 0% (Ust-Kansky districts of the Republic of Altai and Charyshsky district of the Altai Territory) to 100% (Kosh-Agach districts of the Republic of Altai). In Irkutsk province, the portion of infected horses varied from 20% (Osinsky district) to 97.0% (Nukut district) (Table 2).
Theileria equi and B. caballi identification was carried out for all positive blood samples using species-specific PCR. Both EP agents were detected in all regions, with T. equi prevalence being significantly higher (P < 0.01) compared to B. caballi prevalence. Including cases of mixed infection, T. equi was detected in 55.0%, 66.3%, and 52.6% of horse blood samples collected in Novosibirsk province, Irkutsk province, and Altai region, respectively; the prevalence of B. caballi in different regions ranged from 5.0 to 10.1%. Notably, B. caballi significantly more frequently (P < 0.01) was found simultaneously with T. equi (5.2%) than as a mono-infection (2.0%) (Table 2).
Theileria equi genetic variability
Among 453 T. equi samples identified in equine blood, 281 samples were genetically characterized by the 18S rRNA gene. For all genotyped samples, 400–750 bp fragments were sequenced, including the V4 hypervariable region. In addition, 1190–1195 bp fragments were sequenced for 30 samples from different locations. Based on conducted phylogenetic analysis, all determined T. equi sequences belonged to two clusters corresponding to genotypes A and E (Fig. 2). Genotype E prevailed in all regions, being found in all genotyped samples from Novosibirsk province, 84.1% (122/145) samples from Irkutsk province, and 92.9% (118/127) samples from Altai region (Table 3). Moreover, genotype E had a wider distribution compared to genotype A; it was identified in two districts in Novosibirsk province, 13 districts in Irkutsk province, and five districts in Altai region. On the contrary, genotype A was found only in two districts in Irkutsk province and three districts in Altai region. Despite the significant predominance of genotype E in most of the studied areas, only genotype A of T. equi was detected in the Ust-Koksa district in the Republic of Altai (Fig. 1; Supplementary Table S1).

The phylogenetic tree constructed by the ML method based on sequences of the 1195 bp fragment of the 18S rRNA gene of T. equi. The specimens from this study from different regions are marked by ●—Novosibirsk province, ■—Irkutsk province, ♦—the Republic of Altai. Genotypes of T. equi (A–E) are given according to Bhoora et al. (2020). The scale bar indicates an evolutionary distance of 0.005 nucleotides per position in the sequence. Significant bootstrapping values (> 80%) are shown on the nodes
All sequences belonging to genotype E were identical to the widespread T. equi variant, which was found in equines from China (KF559357, MH651213, MT093500, MT093500), South Korea (HM229408), Mongolia (LC781881, LC781885, LC781912), and Kazakhstan (MN857677). Of 32 samples of T. equi belonging to genotype A, 30 samples were identical to each other and to a specimen identified in equine blood from China (MT093496), whereas specimens Irk-Ecab_80 and Alt-Ecab_1/7 differed from them by one and seven nucleotide substitutions, demonstrating 99.9 and 99.4% identity, respectively.
The Irk-Ecab_80 sequence differed from all known sequences in the GenBank database. The Alt-Ecab_1/7 sequence has one polymorphic site, indicating the presence of mixed infection caused by two T. equi sequence variants. Notably, both sequence variants corresponded to the sequences previously found in equine blood from different regions: South Africa (EU888906), USA (JX177670), Brazil (KJ573370, KU240064, KY464029, KY952230, MG052898), Chile (MT463609), Cuba (MT463333, KY111762), Israel (KX227620, MK392050, MK063842, MN611318), and Iran (MK615933) (Fig. 2).
Babesia caballi genetic variability
Of 54 B. caballi specimens, 53 samples were genetically characterized by the 18S rRNA gene; for 28 specimens from different locations, 1146–1162 bp fragments were sequenced. Seven sequence variants (v1–v7) of B. caballi were identified; these variants differed between themselves by 1–13 mismatches (98.9–99.9% identity). Of them, variant v1 corresponded to B. caballi sequences from different regions, namely, China (MH651219, MN156287), Brazil (KY952236), Cuba (MT463341), and South Africa (EU642512) and from ixodid ticks from China (MN163022) and Kazakhstan (MN907450). Variant v4 was identical to B. caballi sequences from equine blood in China (MZ331329, MZ331331, MZ331349, and MZ331354). Sequences of other five variants differed from all known B. caballi sequences from the GenBank database. The distribution of B. caballi variants varied depending on geography. The greatest genetic diversity of B. caballi was observed in Altai region, where six variants (v1–v4, v6, and v7) were identified, while only three variants (v4, v5, and v7) were detected in Irkutsk province. In Novosibirsk province, the only B. caballi specimen corresponded to the widespread variant v1 (Table 3, Fig. 3). In some cases, the correlation has been observed between a particular B. caballi variant and location. Thus, the unique B. caballi variant v5 was found only in Bokhansky district of Irkutsk province, and in this district, variant v5 was found in four of five infected horses including cases of mixed infection (Supplementary Table S1).

The phylogenetic tree constructed by the ML method based on sequences of the 933 bp fragment of the 18S rRNA gene of B. caballi. The specimens from this study from different regions are marked by ●—Novosibirsk province, ■—Irkutsk province, ♦—the Republic of Altai. Genotypes of B. caballi (A and B) are given according to Bhoora et al. (2020). Babesia caballi sequences determined in this study were subdivided into branches 1–4. The scale bar indicates an evolutionary distance of 0.005 nucleotides per position in the sequence. Significant bootstrapping values (> 80%) are shown on the nodes. Babesia gibsoni was used as an outgroup
Cases of mixed infection with two B. caballi variants were established based on the presence of polymorphic sites at positions characteristic of the identified B. caballi variants. As a result, four variants of mixed infections (v1/v2, v2/v3, v4/v5, and v4/v7) were detected. Cases of mixed infection with two B. caballi variants were detected in 5 of 22 infected horses in Irkutsk province and in 12 of 30 infected horses in Altai region (Table 3). Considering mixed infections, B. caballi variant v4 appeared to be the most common and widespread; it was detected in 21 of 22 horse blood samples from six districts of the Irkutsk region and in 15 of 30 blood samples from four districts of the Republic of Altai (Table 3, Supplementary Table S1).
Phylogenetic analysis demonstrated that the determined B. caballi sequences belong to four distinct branches within genotype A (Fig. 3). The first branch was formed by B. caballi variant v1 altogether with previously described B. caballi sequences from different regions. The second branch was formed by B. caballi variants v4–v5 and sequences from equine blood from China. Two remaining branches contained only B. caballi sequences from this study, belonging to variants v2–v3 (a branch 3) and v6–v7 (a branch 4) (Fig. 3).
To establish phylogenetic positions of identified B. caballi sequences, we analyzed all available B. caballi sequences of the 18S rRNA gene with a length more than 900 bp from the GenBank database and B. caballi sequences from this study. The reconstructed phylogenetic tree contained five monophyletic clusters (designated A1–A5) within genotype A and three monophyletic clusters (B1–B3) within genotype B (Fig. 4). Cluster A1 was formed by 55 sequences from remote regions of Europe, Asia, Africa, and Americas, including B. caballi variant v1 from this study. Cluster A2 contained 53 sequences from Russian Siberia (variants v4 and v5) and China. Each of clusters A3 and A4 contained three sequences from this study (variants v2, v3 and v6, v7) and cluster A5 was formed by 11 sequences from Chinese pikas and a vole. A new cluster B3 contained two B. caballi sequences from Guinea ticks (Fig. 4).

The compressed phylogenetic tree constructed by the ML method based on 933 bp fragment of the 18S rRNA gene of B. caballi. Phylogenetic analysis included all sequences available up to March 2024. Phylogenetic clusters determined in this study were designated as A1–A5 and B1–B3. The numbers in parentheses indicate the number of samples belonging to the certain cluster. The scale bar indicates an evolutionary distance of 0.005 nucleotides per position in the sequence. Significant bootstrapping values (> 65%) are shown on the nodes. Babesia gibsoni was used as an outgroup
Discussion
In this study, the prevalence and genetic diversity of EP pathogens in Russia were investigated using molecular methods for the first time. The study involved 750 horses from three remote regions of Siberia: Novosibirsk province, Irkutsk province, and Altai region, which includes the Republic of Altai and the Altai Territory (Fig. 1). The Republic of Altai is the territory with the largest number of horses among the Siberian provinces; in 2014, more than 130,000 horses were recorded here. In contrast, only 66,900, 36,000, and 33,500 horses were registered in the Altai Territory, Irkutsk province, and Novosibirsk province, respectively (Kniazev and Timchenko 2016). In all regions, horses are bred for sport, tourism, and recreation. In addition, local national minorities (Altaians and Buryats) breed horses for meat production in the Republic of Altai and Irkutsk province; in these regions, horses often graze freely. However, regardless of the purposes of breeding horses and the conditions of their keeping, all horses can have frequent contacts with ticks during grazing and, despite antiprotozoal treatment, can receive EP pathogens.
Sampling was carried out in different parts of the Republic of Altai and in one district of the Altai Territory adjacent to the Republic of Altai. In Irkutsk province, the samples were collected in its southeastern part, where the number of horses was the greatest (Fig. 1). In both regions, horses from private farms, equestrian centers, and racetracks were studied. In Novosibirsk province, all examined horses were from private stables. Despite the large distance between the sampling areas and the difference in their natural and climatic conditions, the prevalence of EP pathogens in horses was similar in different regions and exceeded 50%. However, some sampling areas were free of EP (Supplementary Table S1). In at least some cases, the absence of infected horses was related to the absence of specific vectors. Thus, at sampling points in the Charyshsky district of the Altai Territory, the population of ixodid ticks was represented predominantly by Ixodes spp., but not by Dermacentor spp., which are the most likely carriers of EP pathogens in Siberia (Budnik 1955; Ovchinnikov et al. 1941).
The obtained results demonstrated that significant parts of the Republic of Altai, Irkutsk province, and, possibly, Novosibirsk province are endemic for EP and therefore preventive measures are necessary. Particular attention should also be paid to locations where agents of EP have not been found, since a possible change in the species composition of ticks in EP-free areas can lead to an outbreak of EP. In all studied regions, two causative agents of EP were identified—T. equi and B. caballi, and the prevalence of T. equi was significantly higher compared to B. caballi (Table 2). Similar high prevalences of EP were observed in most endemic areas of South Africa, Cuba, Brazil, Mongolia, and China, and in most areas, horses were also more likely to be infected with T. equi than with B. caballi (Bhoora et al. 2020; Chen et al. 2022; Díaz-Sánchez et al. 2018; Otgonsuren et al. 2024; Qablan et al. 2013; Tirosh-Levy et al. 2020a). This may be due to the fact that T. equi can persist in the blood of horses throughout their lives, while B. caballi can only persist for several years (Scoles and Ueti 2015).
This study provides the first data concerning genetic diversity of both T. equi and B. caballi in Russia. Despite T. equi being shown to be a genetically variable species (Bhoora et al. 2009; Díaz-Sánchez et al. 2018; Tirosh-Levy et al. 2020a, b; Seo et al. 2013; Wang et al. 2019), the analyzed T. equi specimens from Siberia were highly conserved and belonged only to genotypes A and E among five previously described T. equi genotypes. Theileria equi genotype A is the most widespread in the world; however, in all studied regions of Siberia genotype E was predominant. Similarly, genotype E was predominant in equine blood in various regions of nearby China and Mongolia, where it was identified in 93–95% of infected horses (Chen et al. 2022; Otgonsuren et al. 2024).
With two exceptions, the determined sequences of T. equi were identical within each genotype. Notably, the single identified sequence variant of genotype E was identical to widespread T. equi variant from neighboring China, Kazakhstan, Mongolia, and South Korea (Otgonsuren et al. 2024; Seo et al. 2013; Wang et al. 2019), whereas the predominant in Siberia sequence variant of genotype A was quite rare worldwide and corresponded to a single T. equi sequence from China (MT093496) (Fig. 2). The observed high genetic stability of T. equi isolates from Siberia suggests that T. equi infection in the studied regions was spread from a limited number of infected horses. Notably, an isolate Alt-Ecab_1/7, which significantly differed from other Siberian specimens, corresponded to a large number of T. equi sequences from geographically distant regions: South Africa, Americas, Israel, and Iran, but not from neighboring regions of China, Mongolia, or Kazakhstan (Bhoora et al. 2009; Díaz-Sánchez et al. 2018; Hall et al. 2013; Margalit Levi et al. 2018; Tirosh-Levy et al. 2020b). This discrepancy may be due to the fact that Alt-Ecab_1/7 sequence was found in a horse from an equestrian school (Gorno-Altaisk), which could have been imported from a region distant from Altai.
In contrast to T. equi, the examined isolates of B. caballi were significantly more variable. Seven sequence variants of B. caballi belonging to genotype A were discovered; five of them differed from previously known sequences (Bhoora et al. 2009; Chen et al. 2022; Díaz-Sánchez et al. 2018; Wang et al. 2019) and formed two well-supported branches on the phylogenetic tree (Fig. 3).
It is generally accepted that B. caballi sequences are subdivided into genotypes A, B1, and B2 (Bhoora et al. 2009, 2020; Tirosh-Levy et al. 2020a). Previously, the genetic heterogeneity of genotype A was demonstrated in a study of Nehra et al. (2021), in which, based on the analysis of 300 bp fragments, two B. caballi specimens from a horse and a tick from China were assigned to a separate clade. Later, a large number of new B. caballi sequences were submitted to the GenBank database (Chen et al. 2022; Kartashov et al. 2021). Therefore, to explore the phylogeny of B. caballi given the latest sequences, we performed a phylogenetic analysis based on all B. caballi 18S rRNA gene sequences (> 900 bp) available up to March 2024, including sequences determined in this study. The heterogeneity of genotype A turned out to be significantly higher than previously considered. It was first demonstrated that genotype A can be subdivided into five well-supported clusters; we propose to designate them as clusters A1–A5. Clusters A1 and A2 included the majority of known sequences (more than 50 sequences in each cluster), whereas each of other clusters includes 3–11 sequences. Notably, cluster A1 included sequences from different continents (Bhoora et al. 2009; Díaz-Sánchez et al. 2018; Wang et al. 2019), while cluster A2 contained only sequences from China (Chen et al. 2022) and Russian Siberia (this study). The minor clusters A3 and A4 were formed by sequences from Siberia, and cluster A5 contained B. caballi sequences from atypical hosts—Chinese pikas and voles (Fig. 4). Notably, based on analysis of shorter sequences, cluster A5 probably also includes Chinese isolates from a horse and a tick (Nehra et al. 2021). As for genotype B, in addition to the previously described clusters B1 and B2, it contains the third cluster, B3, formed by B. caballi sequences from Amblyomma variegatum and Rhipicephalus decoloratus from Guinea (Kartashov et al. 2021).
Conclusion
In this study, the genetic diversity of EP agents in Russian Siberia was first demonstrated. The identified T. equi sequences were highly conserved and belonged to genotypes A and E. The determined B. caballi sequences were more variable and belonged to four branches within genotype A. Seven sequence variants of B. caballi were found, and five of them were identified for the first time. The performed phylogenetic analysis of all available B. caballi sequences first demonstrated that genotype A of B. caballi can be reliably subdivided into five clusters, A1–A5. Thus, the conducted genetic study of B. caballi from Siberia significantly expanded the data on the genetic diversity of this agent.
Data availability
Sequence data that support the findings of this study were submitted to the GenBank database under accession numbers MG551915, OM475522-OM475525, and OR807510-OR807531 for T. equi and OR794981-OR794998 and PP471248-PP471249 for B. caballi.
References
Abramov IV (1955) A new type of transmission by vector ticks of the causal agent of nuttalliasis of horses (Nuttallia equi, Laveran, 1901). Veterinariya 32:43–45 (from Russian)
Bhoora R, Franssen L, Oosthuizen MC, Guthrie AJ, Zweygarth E, Penzhorn BL, Jongejan F, Collins NE (2009) Sequence heterogeneity in the 18S rRNA gene within Theileria equi and Babesia caballi from horses in South Africa. Vet Parasitol 159:112–120. https://doi.org/10.1016/j.vetpar.2008.10.004
Bhoora RV, Collins NE, Schnittger L, Troskie C, Marumo R, Labuschagne K, Smith RM, Dalton DL, Mbizeni S (2020) Molecular genotyping and epidemiology of equine piroplasmids in South Africa. Ticks Tick Borne Dis 11:101358. https://doi.org/10.1016/j.ttbdis.2019.101358
Budnik VS (1955) New data on the mechanism of transmission of the causal agent of nuttalliasˆıs of horses by the tick Dermacentor marginatus Sulz. Veterinariya 32:36–43 (from Russian)
Chen K, Hu Z, Yang G, Guo W, Qi T, Liu D, Wang Y, Du C, Wang X (2022) Development of a duplex real-time PCR assay for simultaneous detection and differentiation of Theileria equi and Babesia caballi. Transbound Emerg Dis 69:e1338–e1349. https://doi.org/10.1111/tbed.14464
Coultous RM, McDonald M, Raftery AG, Shiels BR, Sutton DGM, Weir W (2020) Analysis of Theileria equi diversity in the Gambia using a novel genotyping method. Transbound Emerg Dis 67:1213–1221. https://doi.org/10.1111/tbed.13454
Dahmana H, Amanzougaghene N, Davoust B, Normand T, Carette O, Demoncheaux JP, Mulot B, Fabrizy B, Scandola P, Chik M, Fenollar F, Mediannikov O (2019) Great diversity of Piroplasmida in Equidae in Africa and Europe, including potential new species. Vet Parasitol Reg Stud Reports 18:100332. https://doi.org/10.1016/j.vprsr.2019.100332
de Sousa KCM, Fernandes MP, Herrera HM, Freschi CR, Machado RZ, André MR (2018) Diversity of piroplasmids among wild and domestic mammals and ectoparasites in Pantanal wetland, Brazil. Ticks Tick Borne Dis 9:245–253. https://doi.org/10.1016/j.ttbdis.2017.09.010
Díaz-Sánchez AA, Pires MS, Estrada CY, Cañizares EV, Del Castillo Domínguez SL, Cabezas-Cruz A, Rivero EL, da Fonseca AH, Massard CL, Corona-González B (2018) First molecular evidence of Babesia caballi and Theileria equi infections in horses in Cuba. Parasitol Res 117:3109–3118. https://doi.org/10.1007/s00436-018-6005-5
Dirks E, de Heus P, Joachim A, Cavalleri JV, Schwendenwein I, Melchert M, Fuehrer HP (2021) First case of autochthonous equine theileriosis in Austria. Pathogens 10:298. https://doi.org/10.3390/pathogens10030298
Elsawy BSM, Nassar AM, Alzan HF, Bhoora RV, Ozubek S, Mahmoud MS, Kandil OM, Mahdy OA (2021) Rapid detection of equine piroplasms using multiplex PCR and first genetic characterization of Theileria haneyi in Egypt. Pathogens 10:1414. https://doi.org/10.3390/pathogens10111414
Fedulina OO, Rar VA, Suncova OV, Kozlova IV (2014) Identification of Theileria equi in the blood of horses in the Irkutsk region. Bull East Siberian Sci Center SB RAMS 6:101104 (in Russian)
Hall CM, Busch JD, Scoles GA, Palma-Cagle KA, Ueti MW, Kappmeyer LS, Wagner DM (2013) Genetic characterization of Theileria equi infecting horses in North America: evidence for a limited source of U.S. introductions. Parasit Vectors 6:35. https://doi.org/10.1186/1756-3305-6-35
Ionita M, Nicorescu IM, Pfister K, Mitrea IL (2018) Parasitological and molecular diagnostic of a clinical Babesia caballi outbreak in Southern Romania. Parasitol Res 117:2333–2339. https://doi.org/10.1007/s00436-018-5899-2
Kartashov MY, Naidenova EV, Zakharov KS, Yakovlev SA, Skarnovich MO, Boumbaly S, Nikiforov KA, Plekhanov NA, Kritzkiy AA, Ternovoi VA, Boiro MY, Loktev VB (2021) Detection of Babesia caballi, Theileria mutans and Th. velifera in ixodid ticks collected from cattle in Guinea in 2017–2018. Vet Parasitol Reg Stud Rep 24:100564. https://doi.org/10.1016/j.vprsr.2021.100564
Kniazev SP, Timchenko AM (2016) Dynamics of the horse population and modern situation in Siberia. Vestn NGAU 1(38):139–145 (from Russian)
Knowles DP, Kappmeyer LS, Haney D, Herndon DR, Fry LM, Munro JB, Sears K, Ueti MW, Wise LN, Silva M, Schneider DA, Grause J, White SN, Tretina K, Bishop RP, Odongo DO, Pelzel-McCluskey AM, Scoles GA, Mealey RH, Silva JC (2018) Discovery of a novel species, Theileria haneyi n. sp., infective to equids, highlights exceptional genomic diversity within the genus Theileria: implications for apicomplexan parasite surveillance. Int J Parasitol 48:679–690. https://doi.org/10.1016/j.ijpara.2018.03.010
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33:1870–1874. https://doi.org/10.1093/molbev/msw054
Manna G, Cersini A, Nardini R, Bartolomé Del Pino LE, Antognetti V, Zini M, Conti R, Lorenzetti R, Veneziano V, Autorino GL, Scicluna MT (2018) Genetic diversity of Theileria equi and Babesia caballi infecting horses of Central-Southern Italy and preliminary results of its correlation with clinical and serological status. Ticks Tick Borne Dis 9:1212–1220. https://doi.org/10.1016/j.ttbdis.2018.05.005
Margalit Levi M, Tirosh-Levy S, Dahan R, Berlin D, Steinman A, Edery N, Savitski I, Lebovich B, Knowles D, Suarez CE, Baneth G, Mazuz ML (2018) First detection of diffuse and cerebral Theileria equi infection in neonatal filly. J Equine Vet Sci 60:23–28. https://doi.org/10.1016/j.jevs.2017.10.016
Nehra AK, Kumari A, Moudgil AD, Vohra S (2021) Phylogenetic analysis, genetic diversity and geographical distribution of Babesia caballi based on 18S rRNA gene. Ticks Tick Borne 12:101776. https://doi.org/10.1016/j.ttbdis.2021.101776
Onyiche TE, Suganuma K, Igarashi I, Yokoyama N, Xuan X, Thekisoe OA (2019) Review on equine piroplasmosis: epidemiology, vector ecology, risk factors, host immunity, diagnosis and control. Int J Environ Res Public Health 16:1736. https://doi.org/10.3390/ijerph16101736
Otgonsuren D, Amgalanbaatar T, Narantsatsral S, Enkhtaivan B, Munkhgerel D, Zoljargal M, Davkharbayar B, Myagmarsuren P, Battur B, Battsetseg B, Sivakumar T, Yokoyama N (2024) Epidemiology and genetic diversity of Theileria equi and Babesia caballi in Mongolian horses. Infect Genet Evol 119:105571. https://doi.org/10.1016/j.meegid.2024.105571
Ovchinnikov PA, Nikitenko GI, Zhiltsov PA, Zabelin VA (1941) Dermacentor nuttalli ticks as a vector of equine piroplasmosis and nuttalliosis. Veterinariya 2:15–16 (in Russian)
Petunin FA (1948) Hyalomma scupense P. Sch—a vector of nuttalliasis of horses. Veterinariya 25:14 (from Russian)
Qablan MA, Oborník M, Petrželková KJ, Sloboda M, Shudiefat MF, Hořín P, Lukeš J, Modrý D (2013) Infections by Babesia caballi and Theileria equi in Jordanian equids: epidemiology and genetic diversity. Parasitology 140:1096–1103. https://doi.org/10.1017/S0031182013000486
Rar VA, Epikhina TI, Livanova NN, Panov VV (2011) Genetic diversity of Babesia in Ixodes persulcatus and small mammals from North Ural and West Siberia, Russia. Parasitology 138:175–182. https://doi.org/10.1017/S0031182010001162
Scoles GA, Ueti MW (2015) Vector ecology of equine piroplasmosis. Annu Rev Entomol 60:561–580. https://doi.org/10.1146/annurev-ento-010814-021110
Seo MG, Yun SH, Choi SK, Cho GJ, Park YS, Cho KH, Kwon OD, Kwak D (2013) Molecular and phylogenetic analysis of equine piroplasms in the Republic of Korea. Res Vet Sci 94:579–583. https://doi.org/10.1016/j.rvsc.2013.01.014
Short MA, Clark CK, Harvey JW, Wenzlow N, Hawkins IK, Allred DR, Knowles DP, Corn JL, Grause JF, Hennager SG, Kitchen DL, Traub-Dargatz JL (2012) Outbreak of equine piroplasmosis in Florida. J Am Vet Med Assoc 240:588–595. https://doi.org/10.2460/javma.240.5.588
Tirosh-Levy S, Gottlieb Y, Fry LM, Knowles DP, Steinman A (2020a) Twenty years of equine piroplasmosis research: global distribution, molecular diagnosis, and phylogeny. Pathogens 9:926. https://doi.org/10.3390/pathogens9110926
Tirosh-Levy S, Steinman A, Levy H, Katz Y, Shtilman M, Gottlieb Y (2020b) Parasite load and genotype are associated with clinical outcome of piroplasm-infected equines in Israel. Parasit Vectors 13:267. https://doi.org/10.1186/s13071-020-04133-y
Wang J, Liu J, Yang J, Wang X, Li Z, Jianlin X, Li X, Xiang Q, Li Y, Liu Z, Luo J, Guan G, Yin H (2019) The first molecular detection and genetic diversity of Babesia caballi and Theileria equi in horses of Gansu province, China. Ticks Tick Borne Dis 10:528–532. https://doi.org/10.1016/j.ttbdis.2019.01.003
Wise LN, Kappmeyer LS, Mealey RH, Knowles DP (2013) Review of equine piroplasmosis. J Vet Intern Med 27:1334–1346. https://doi.org/10.1111/jvim.12168
Funding
This work was supported by the Russian state-funded projects for ICBFM SB RAS (grant number 121031300043–8), SC FHHRP (grant number 1021060107125–0-1.6.2), and Federal Altai Scientific Center for Agrobiotechnology (grant number 0534–2021-0005).
Author information
Authors and Affiliations
Contributions
VR: conceptualization, methodology, investigation, formal analysis, writing original draft. VM: sample collection, data analysis, funding acquisition. OS: sample collection, investigation, visualization. TE: investigation, data analysis. AT: investigation, formal analysis. IM: sample collection. VF: investigation. YI: investigation. IK: methodology, resources, funding acquisition. NT: conceptualization, funding acquisition, review and editing manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
All experiments with animals were conducted according to the guidelines for experiments with laboratory animals (Supplement to the Order of the Russian Ministry of Health, no. 755, of August 12, 1977). Animal use and experimental procedures were approved by the Bioethical Committee of the Scientific Center of Family Health Problems and Human Reproduction, Irkutsk, Russia (Protocol No. 5, 26.10.2012 and Protocol No. 2, 18.02.2020).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Section Editor: Dana Mordue
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rar, V., Marchenko, V., Suntsova, O. et al. The first study of the prevalence and genetic diversity of Theileria equi and Babesia caballi in horses in Russia. Parasitol Res 123, 279 (2024). https://doi.org/10.1007/s00436-024-08300-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00436-024-08300-3