
Abstract
Piroplasm including Babesia spp. and Theileria spp. in cattle can cause illness that affects livestock productivity, resulting in significant production losses, especially in tropical and subtropical regions such as Thailand. This study aimed to investigate the prevalence of bovine piroplasms and to identify these blood parasites based on the 18S ribosomal RNA gene in cattle in the northeastern part of Thailand. Piroplasmid infections among beef and dairy cattle were examined using nested PCR. Furthermore, amplicon DNA was sequenced and analyzed, and a phylogenetic tree was constructed to determine the genetic diversity and relationships of the parasite in each area. A total of 141 out of 215 (65.6%) cattle were positive for infection with Babesia or Theileria. DNA analysis revealed that infection by Babesia bigemina, Babesia bovis, Theileria orientalis, Theileria sinensis, and Theileria sp. were common piroplasms in cattle in this region, with a high sequence shared identity and similarity with each other and clustered with isolates from other countries. This study provides information on the molecular epidemiology and genetic identification of Babesia spp. and Theileria spp. in beef and dairy cattle to provide a better understanding of piroplasm infection in cattle in this region, which will help control these blood parasites. Moreover, this is the first report identifying T. sinensis circulating among Thai cattle.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In animal production, the emergence and spread of piroplasmosis caused by piroplasm have gained prominence as a significant threat that poses constant challenges to livestock health and productivity. Piroplasms are protozoan parasites belonging to phylum Apicomplexa, order Piroplasmida, and genera Babesia and Theileria, which have a complex life cycle that involves transmission through ticks and an obligate intracellular stage within the host’s blood cells, resulting in hematologic disorders (Uilenberg 2006; Lempereur et al. 2017; Jalovecka et al. 2018). In cattle, the Babesia parasites enter the bloodstream and invade their red blood cells primarily manifesting as hemolytic anemia, fever, jaundice, and hemoglobinuria (Hashem et al. 2018), while Theileria parasites enter and invade white blood cells, particularly leukocytes, and exceed in the erythrocytes. Theileria infection frequently presents as mild fever, hemorrhages on the mucosa and serosal surfaces, anemia, jaundice, and lymphocyte enlargement (Ma et al. 2020). Cattle become infected with Babesia or Theileria when they are bitten by infected ticks, predominantly ticks in the genus Hyalomma spp. (Aktas et al. 2004; Sang et al. 2021), Rhipicephalus spp. (Giglioti et al. 2018; Kakati et al 2015), and Haemaphysalis spp. (Marendy et al. 2020; Phipps et al. 2022).
Previously, Babesia and Theileria infections in cattle have been examined based on the morphological characteristics of these parasites using blood smears under a microscope. However, molecular techniques have assisted and facilitated more accurate species identification, along with the assessment of genetic diversity in these piroplasms. In previous studies in Thailand, molecular detection of piroplasms in cattle indicated the presence of endemic species, including Babesia bovis, Babesia bigemina (Cao et al. 2012; Simking et al. 2013; Srionrod et al. 2022), and Theileria orientalis (Jirapattharasate et al. 2016, 2017; Adjou Moumouni et al. 2023). For molecular detection, the small subunit ribosomal RNA gene (18S rRNA gene) is one of the most genes effectively utilized for parasite identification and also phylogenetic analysis of piroplasmids (Allsopp and Allsopp 2006; Lack et al. 2012; Kumar et al. 2022).
However, information on the prevalence of Babesia and Theileria infections in cattle, as well as inadequate information on their genetic characterization which is essential for the control and prevention of these piroplasms, is limited in Thailand and needs updating. In the present study, we demonstrated the molecular screening of Babesia or Theileria infection in beef and dairy cattle from the northeastern part of Thailand. We also identified the Babesia and Theileria species and conducted a phylogenetic analysis based on the 18S rRNA gene. This epidemiological study investigated the species and genetic diversity of Babesia and Theileria in cattle to enhance the understanding of these infections and to raise awareness among farmers and healthcare professionals for effective strategic control.
Materials and methods
Study area and sample collection
A comprehensive investigation was conducted from July 2021 to July 2023 in six provinces in the northeastern part of Thailand: Udon Thani, Khon Kaen, Maha Sarakham, Roi Et, Ubon Ratchathani, and Chaiyaphum (Fig. 1). A total of 215 blood samples were collected from bovine species, comprising 134 samples from beef cattle and 81 samples from dairy cattle. Blood samples of approximately 1–2 mL were collected from the jugular vein or coccygeal vein and preserved in ethylenediaminetetraacetic acid (EDTA) anticoagulant tubes. Information regarding the sex, age, and production type of individual samples was systematically recorded and cataloged as part of the dataset under investigation. Blood samples were transported on ice to the laboratory at the Faculty of Veterinary Sciences of Mahasarakham University and stored at − 20 ℃ for long-term preservation until DNA extraction. All blood samples taken were approved by their owners and received approval from the ethics committee.

Map of the study area where cattle blood samples were collected across six provinces in the northeastern part of Thailand, consisting of Udon Thani (UD), Khon Kaen (KK), Chaiyaphum (CY), Maha Sarakham (MK), Roi Et (RE), and Ubon Rachathani (UB)
DNA extraction and nested PCR amplification of Babesia and Theileria 18S rRNA gene
Isolation of DNA from the collected blood samples used the GF-1 blood DNA extraction kit procedure (Vivatis, Malaysia), adhering to standardized protocols as prescribed by the manufacturer. Each extracted DNA sample underwent examination for Babesia and Theileria infection utilizing a nested PCR method. This method employed specific primers designed to target the 18S rRNA gene, approximately 1500 bp in length, of the parasite, as previously described (Masatani et al. 2017). The first PCR step utilized the primer pair, namely, BTH 18S 1st F (5′-GTGAAACTGCGAATGGCTCATTAC-3′) and BTH 18S 1st R (5′-AAGTGATAAGGTTCACAAAACTTCCC-3′), while the second step utilized the primer pair BTH 18S 2nd F (5′-GGCTCATTACAACAGTTATAGTTTATTTG-3′) and BTH 18S 2nd R (5′-CGGTCCGAATAATTCACCGGAT-3′). This nested PCR approach enabled the amplification of DNA from protozoa in the genera Babesia, Theileria, and Hepatozoon.
The nested PCR reactions were conducted in a final volume of 25 μL. Each reaction mixture comprised of 0.4 μM of each primer, 1 U of Taq polymerase (Vivatis, Malaysia), 1× PCR buffer, 1.5 mM MgSO4, 0.2 mM dNTPs, and 2 μL of template DNA (extracted DNA for the first PCR and PCR product from the primary amplification for the second PCR). Both PCR reactions comprised 35 cycles, which included denaturation at 95 °C for 45 s, annealing at 50 °C for 45 s, and extension at 72 °C for 90 s, followed by a final extension step at 72 °C for 5 min. Negative controls were prepared using PCR master mixes containing only the primers without any DNA template. PCR amplification was performed using a thermal cycler (Biometra GmbH, Germany). The resulting approximately 1500 base pairs from nested PCR were then visualized using 1% agarose gels stained with ViSafe Red Gel Stain (Vivantis, Malaysia) and examined under ultraviolet light using a gel documentation system from Bio-Rad, USA.
Descriptive statistics were employed to summarize the prevalence of Babesia or Theileria infection. Chi-squared tests were then employed to analyze the associations between infection rates and host factors, including breed, sex, and age with a significance level of p-value less than 0.05 (Social Science Statistics 2023).
DNA sequencing and phylogenetic analysis
We randomly selected 65 PCR amplicons from dispersed sampling sites, representing 50% of positive samples from each site, for purification and direct sequencing. The PCR products targeting the 18S rRNA genes underwent purification and sequencing at a commercial sequencing facility (1st Base, Malaysia). Electrograms of the sequences were meticulously examined for quality, appropriate length, and absence of double or multiple nucleotide peaks. The obtained DNA sequences were aligned and trimmed using the BioEdit sequence alignment editor program (Hall 1999). Subsequently, the nucleotide sequences were analyzed for similarity to sequences in the GenBank database using the BLAST program hosted by NCBI (https://www.ncbi.nlm.nih.gov/). Haplotype identification from the 18S rRNA sequences of Babesia and Theileria was conducted using the DnaSP6 program (Rozas et al. 2017).
The obtained sequences of the partial 18S rRNA gene of Babesia and Theileria in this study were approximately 1405 bp in length (ranging from 1343 to 1458 bp). The resulting partial 18S rRNA gene sequences represented each Babesia and Theileria haplotype were then deposited into the GenBank database with accession numbers PP380178–PP380189. Phylogenetic relationships among the 18S rRNA haplotypes from this study and 27 related sequences from various geographical locations in GenBank were inferred using the maximum likelihood method in MEGA X (Kumar et al. 2018). Bootstrap analysis with 1000 replications was employed to assess the confidence of branching patterns in the trees.
Results
Prevalence of Babesia and Theileria in cattle
From a total of 215 samples, comprising 180 females and 35 males, spanning an age range from 2 months to 10 years, 65.6% (141/215) exhibited infection with Babesia or Theileria, as determined by nested PCR analysis. Specifically, among females, 65% (117/180) were found to be infected, whereas among males, the infection rate was slightly higher at 68.6% (24/35). Chi-squared analysis revealed that the observed differences in infection rates between sexes were not statistically significant. Furthermore, when considering the distribution of infection across different production types, it was observed that 66.4% (89/134) of beef cattle and 64.2% (52/81) of dairy cattle were infected. Chi-squared analysis indicated no significant difference in infection rates between beef and dairy cattle. Animals within the age range of 0–1 year showed an infection rate of 52.7% (20/38), adult animals aged between 1 and 6 years displayed an infection rate of 67.1% (104/155), and old animals aged over 6 years showed an infection rate of 77.3% (17/22). Despite these observed differences in infection rates across age groups, statistical analyses did not reveal any significant age-related associations with infection susceptibility within the studied population (Table 1).
Babesia and Theileria identification
Among the positive samples, a subset of 65 PCR products (representing 50% of positive samples from each province) was randomly selected for sequencing analysis and resulted in the successful sequencing of 64 specimens. Sequencing identified the presence of Babesia bovis, B. bigemina, Thileria sp., T. orientalis, and T. sinensis in cattle in the northeastern part of Thailand. Among the 64 sequences, 6 corresponded to Babesia species (comprising 2 sequences of B. bovis and 4 sequences of B. bigemina), and 58 sequences corresponded to Theileria species (comprising 28 samples of T. orientalis, 15 samples of Theileria sp., and 13 samples of T. sinensis).
Further analysis of the sequences revealed the presence of distinct haplotypes within both Theileria and Babesia species. Specifically, 58 sequences representing Theileria species were classified into 6 haplotypes, with 4 haplotypes associated with T. orientalis (accession numbers PP380178, PP380179, PP380181, and PP380182), one haplotype was Theileria sp. (accession number PP380180), and one haplotype was T. sinensis (accession number PP380183). The remaining 6 sequences corresponded to B. bovis for 2 haplotypes (accession nos. PP380184 and PP380185), while B. bigemina was differentiated into 4 distinct haplotypes (accession nos. PP380186–PP380189). Sequencing and DNA analysis revealed that infection of cattle with the piroplasms B. bigemina, B. bovis, T. orientalis, T. sinensis, and Theileria sp. was common in this region, with sequence similarities ranging between 99 and 100%, and sequences were homologous with sequences from other countries (Table 2).
Phylogenetic tree
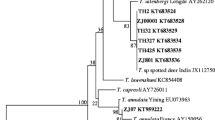
The phylogenetic analyses, utilizing the 18S rRNA gene, delineated distinct evolutionary lineages within Theileria and Babesia. Theileria was observed to segregate into three primary clades: T. orientalis, T. sinensis, and Theileria sp. In Thailand, T. orientalis was found to have four haplotypes, which grouped into two subgroups. Subgroup 1 included three haplotypes (PP380179, PP380181, PP380182) closely related to T. orientalis from dogs in Myanmar (LC602478.1), cattle in South Korea (MT889728.1), buffalo in India (OR068053.1), cattle in China (KU363043.1), and cattle in Pakistan (MG599099.1). Subgroup 2 contained one haplotype (PP380178) closely related to T. orientalis from cattle in Türkiye (OR211416.1) and ticks in China (MH208641.1). For T. sinensis, all isolates, including those from Thai cattle (PP380183) as well as cattle from Malaysia (MT271911.1) and China (KX115427.1, KF559355.1, EU274472, HM538203.1), formed a single cohesive group. Theileria sp. exhibited two distinct subgroups in the phylogenetic analysis: one subgroup included isolates from cattle in India (OR067892.1), while the other subgroup consisted of isolates from Thai cattle (PP380180), which clustered together with isolates from cattle in Myanmar (LC57817.1), cattle in China (MN252454.1), and buffaloes in China (DQ286801.1). From phylogenetic analysis, T. orientalis and T. sinensis are closely related, whereas Theileria sp. is more distantly related and grouped with T. annulata.
Sequence analysis confirmed the presence of Babesia 18S rRNA sequences, which were categorized into two main clades: B. bovis and B. bigemina. In this study, two haplotypes of B. bovis from Thailand were placed into different subgroups: the first haplotype (PP380184) grouped with B. bovis isolates from cattle in China (KY805831.1) in subgroup 1, while the second haplotype (PP380185) grouped with B. bovis isolates from cattle in South Africa (MH257728.1), Brazil (EF458212.1), and the USA (L31922) in subgroup 2. Additionally, four haplotypes of B. bigemina from Thailand were classified into two subgroups: the first haplotype (PP380189) clustered with B. bigemina isolates from ticks in Taiwan (OP604193.1) and cattle in China (JX495402.1), while the remaining haplotypes (PP380186–PP380188) formed a separate cohesive group (Fig. 2).

Phylogenetic analyses of Babesia and Theileria 18S rRNA sequences obtained from Thailand cattle and related sequences in GenBank using the maximum likelihood method. The sequences determined in this study are shown in bold font and the percentage of trees in which associated taxa clustered together is shown next to the branch
Discussion
Epidemiology research efforts are needed to enhance our understanding of bovine babesiosis and theileriosis and to develop effective strategies for disease control and prevention in Thai cattle populations. In the present study, we demonstrated the molecular detection based on 18S rRNA gene in samples from beef and dairy cattle in six provinces in the northeastern part of Thailand. This study documented the highest prevalence of piroplasmid infection (Babesia or Theileria) reported in Thailand to date at approximately 65.6% (95% CI: 58.8–71.9%). Furthermore, there was considerable variation in infection rates observed across sampled farms, ranging from 35 to 92.3%. This variability may be attributed to multiple factors, including herd size and farm management practices, particularly those about tick control strategies (Muhanguzi et al. 2010). In comparison to previous reports, which indicated prevalence rates of B. bovis and B. bigemina in cattle of 12% and 21% respectively in 2012 (Cao et al. 2012) and 11.1% and 12.5% respectively in 2017 (Jirapattharasate et al. 2017), this study demonstrates a substantial increase. Notably, recent studies in 2022–2023 have shown a decrease in Babesia prevalence, ranging from 1.2 to 5.8% (Koonyosying et al. 2022; Srionrod et al. 2022; Adjou Moumouni et al. 2023). Regarding Theileria prevalence, earlier research in 2017 reported a prevalence rate of 7.8% (Jirapattharasate et al. 2017), whereas a study conducted in 2022 recorded a higher prevalence of 36.5% (Koonyosying et al. 2022). Babesia and Theileria have a worldwide distributed with molecular prevalence of 25.3% in selected areas of China and Pakistan (Hassan et al. 2018), 36.1% in Kyrgyzstan (Aktaş et al. 2019), 52.8% in Nepal (Dhakal et al. 2023), and 87.3% in Nigeria (Famuyide et al. 2020). The varying prevalence in each regions underscores the influence of climatic and meteorological conditions which influence tick vector populations (M’ghirbi et al. 2008).
Through DNA sequencing analysis, our study identified T. orientalis as the dominant species of piroplasm, followed by Theileria sp., T. sinensis, B. bigemina, and B. bovis in cattle farms in Thailand. This observation is consistent with prior findings in central and northern Thailand, which also highlighted the prevalence of T. orientalis as the dominant species (Koonyosying et al. 2022). Furthermore, similar dominance of Theileria has been reported in other regions such as China (Zhou et al. 2019) and Kyrgyzstan (Aktaş et al. 2019). T. orientalis was classified as a non-transforming species and grouped among lower pathogenic organisms (Sivakumar et al. 2014). Classification of this piroplasm is typically based on sequencing of the small subunit ribosomal RNA and the major piroplasm surface protein (MPSP) (Özübek and Aktaş, 2019). To date, researchers have identified at least 11 distinct genotypes within the T. orientalis worldwide based on the MPSP sequence. Among these genotypes, types 1 (Chitose) and 2 (Ikeda) are notable for causing clinical oriental theileriosis in cattle, with high morbidity and mortality (Gebrekidan et al. 2020). For future investigations, there is a significant opportunity to focus on the MPSP gene which will provide insights into the virulence and clinical outcomes associated with different strains of T. orientalis. In addition, production type, age, and sex did not exert a significant influence on the likelihood of infection with Babesia or Theileria in the studied cattle population. This finding correlated with previous studies in Malaysia and Egypt which similarly demonstrated that sex was not correlated with infection. However, factors such as production type and age exhibited significant associations (p < 0.05) with the prevalence of T. orientalis (Ola-Fadunsin et al. 2020; Selim et al. 2022).
Previous studies reported that the 18S rRNA fragments are appropriate markers to determine the genetic diversity for blood parasites (Bawm et al. 2021; Nehra et al. 2022). In this study, analysis of 18S rRNA revealed a notable degree of sequence similarity within the Babesia and Theileria of this population and the GenBank database. Phylogenetic analysis showed that Theileria could be divided into three groups: T. orientalis, T. sinensis, and Theileria sp. Furthermore, our investigation identified T. orientalis and T. sinensis as genetically more similar to each other, forming a distinct cluster separated from Theileria sp. suggesting a closer evolutionary relationship between T. orientalis and T. sinensis, distinguishing them from Theileria sp. based on 18S rRNA gene. Previously, epidemiological data indicated the presence of T. orientalis and Theileria sp. in Thailand, while T. sinensis had not been documented in the country. This study has now confirmed the first presence of T. sinensis circulating within the cattle population in Thailand. In addition, T. sinensis is generally considered to have low pathogenicity compared to other transforming species like T. parva or T. annulata. It is transmitted by Haemaphysalis ticks and commonly infects cattle, with reports in China and Malaysia (Kho et al. 2017; Jia et al. 2020). The discovery of this new protozoa in Thailand could be attributed to the presence of tick hosts and the tropical climate, which provide favorable conditions for the development of the protozoa’s life cycle when new pathogens are introduced.
Conclusion
This study showed a notably heightened molecular prevalence of piroplasm infection in cattle in Thailand compared to preceding reports, accomplished through the utilization of primers targeting the 18S rRNA gene. Through this investigation, advancements in our contemporary comprehension of the distribution and genetic diversity of piroplasms in both beef and dairy cattle populations in Thailand have been achieved. T. orientalis emerges as the dominant species, followed by Theileria sp., T. sinensis, B. bigemina, and B. bovis. The identification of Babesia and Theileria presence underscores a public health concern, indicating the potential for these animals to act as reservoirs for parasites, serve as vectors for pathogen transmission, and facilitate disease dissemination.
Data availability
All data generated or analyzed during this study are included in this article. The newly generated sequences were deposited in the GenBank database under the accession numbers PP380178-PP380189.
References
Adjou Moumouni PF, Galon EM, Tumwebaze MA, Byamukama B, Ngasaman R, Tiwananthagorn S, Kamyingkird K, Inpankaew T, Xuan X (2023) Tick-borne pathogen detection and its association with alterations in packed cell volume of dairy cattle in Thailand. Animals 13:2844. https://doi.org/10.3390/ani13182844
Aktas M, Dumanli N, Angin M (2004) Cattle infestation by Hyalomma ticks and prevalence of Theileria in Hyalomma species in the east of Turkey. Vet Parasitol 119(1):1–8. https://doi.org/10.1016/j.vetpar.2003.10.013
Aktaş M, Kısadere İ, Özübek S, Cihan H, Salıkov R, Cirak VY (2019) First molecular survey of piroplasm species in cattle from Kyrgyzstan. Parasitol Res 118:2431–2435. https://doi.org/10.1007/s00436-019-06370-2
Allsopp MT, Allsopp BA (2006) Molecular sequence evidence for the reclassification of some Babesia species. Ann N Y Acad Sci 1081:509–517. https://doi.org/10.1196/annals.1373.076
Bawm S, Sagara R, Kakisaka K, Thu MJ, Hmoon MM, Htun LL, Win MM, Nonaka N, Nakao R, Suzuki H, Katakura K (2021) Identification, genetic variation, and structural analysis of 18S rRNA of Theileria orientalis and Theileria velifera-like isolates from Myanmar. Parasitol Int 82:102299. https://doi.org/10.1016/j.parint.2021.102299
Cao S, Aboge GO, Terkawi MA, Yu L, Kamyingkird K, Luo Y, Li Y, Goo YK, Yamagishi J, Nishikawa Y, Yokoyama N (2012) Molecular detection and identification of Babesia bovis and Babesia bigemina in cattle in northern Thailand. Parasitol Res 111:1259–1266. https://doi.org/10.1007/s00436-012-2960-4
Dhakal M, Gompo TR, Devkota P, Kafle SC, Subedi JR, Gong H, Arima H, Culleton R, Asada M, Pandey K (2023) Molecular detection and identification of piroplasm in cattle from Kathmandu Valley. Nepal Pathogens 12(8):1045. https://doi.org/10.3390/pathogens12081045
Famuyide IM, Takeet MI, Talabi AO, Otesile EB (2020) Molecular detection and identification of piroplasms in semi-intensively managed cattle from Abeokuta. Nigeria Folia Vet 64(4):1–8. https://doi.org/10.2478/fv-2020-0031
Gebrekidan H, Perera PK, Ghafar A, Abbas T, Gasser RB, Jabbar A (2020) An appraisal of oriental theileriosis and the Theileria orientalis complex, with an emphasis on diagnosis and genetic characterisation. Parasitol Res 119:11–22. https://doi.org/10.1007/s00436-019-06557-7
Giglioti R, de Oliveira HN, Okino CH, de Sena Oliveira MC (2018) qPCR estimates of Babesia bovis and Babesia bigemina infection levels in beef cattle and Rhipicephalus microplus larvae. Exp Appl Acarol 75:235–240. https://doi.org/10.1007/s10493-018-0260-0
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hashem M, Neamat-Allah AN, Gheith MA (2018) A study on bovine babesiosis and treatment with reference to hematobiochemical and molecular diagnosis. Slov Vet Res 55:165–73. https://doi.org/10.26873/SVR-643-2018
Hassan MA, Liu J, Rashid M, Iqbal N, Guan G, Yin H, Luo J (2018) Molecular survey of piroplasm species from selected areas of China and Pakistan. Parasit Vectors 1–7. https://doi.org/10.1186/s13071-018-3035-x
Jalovecka M, Hajdusek O, Sojka D, Kopacek P, Malandrin L (2018) The complexity of piroplasms life cycles. Front Cell Infect Microbiol 23(8):248. https://doi.org/10.3389/fcimb.2018.00248
Jia L, Zhao S, Xie S, Li H, Wang H, Zhang S (2020) Molecular prevalence of Theileria infections in cattle in Yanbian, north-eastern China. Parasite 27(19):1–7. https://doi.org/10.1051/parasite/2020017
Jirapattharasate C, Moumouni PF, Cao S, Iguchi A, Liu M, Wang G, Zhou M, Vudriko P, Changbunjong T, Sungpradit S, Ratanakorn P (2016) Molecular epidemiology of bovine Babesia spp. and Theileria orientalis parasites in beef cattle from northern and northeastern Thailand. Parasitol Int 65:62–9. https://doi.org/10.1016/j.parint.2015.10.005
Jirapattharasate C, Adjou Moumouni PF, Cao S, Iguchi A, Liu M, Wang G, Zhou M, Vudriko P, Efstratiou A, Changbunjong T, Sungpradit S (2017) Molecular detection and genetic diversity of bovine Babesia spp., Theileria orientalis, and Anaplasma marginale in beef cattle in Thailand. Parasitol Res 116:751–762. https://doi.org/10.1007/s00436-016-5345-2
Kakati P, Sarmah PC, Ray D, Bhattacharjee K, Sharma RK, Barkalita LM, Sarma DK, Baishya BC, Borah P, Stanley B (2015) Emergence of oriental theileriosis in cattle and its transmission through Rhipicephalus (Boophilus) microplus in Assam. India. Vet World 8:1099. https://doi.org/10.14202/vetworld.2015.1099-1104
Kho KL, Amarajothi AD, Koh FX, Panchadcharam C, Nizam QN, Tay ST (2017) The first molecular survey of theileriosis in Malaysian cattle, sheep and goats. Vet Parasitol Reg Stud Reports 10:149–153. https://doi.org/10.1016/j.vprsr.2017.08.003
Koonyosying P, Rittipornlertrak A, Chomjit P, Sangkakam K, Muenthaisong A, Nambooppha B, Srisawat W, Apinda N, Singhla T, Sthitmatee N (2022) Incidence of hemoparasitic infections in cattle from central and northern Thailand. PeerJ 10:e13835. https://doi.org/10.7717/peerj.13835
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Kumar B, Maharana BR, Thakre B, Brahmbhatt NN, Joseph JP (2022) 18S rRNA gene-based Piroplasmid PCR: an assay for rapid and precise molecular screening of Theileria and Babesia species in animals. Acta Parasitol 67:1697–1707. https://doi.org/10.1007/s11686-022-00625-2
Lack JB, Reichard MV, Van Den Bussche RA (2012) Phylogeny and evolution of the Piroplasmida as inferred from 18S rRNA sequences. Int J Parasitol 42:353–363. https://doi.org/10.1016/j.ijpara.2012.02.005
Lempereur L, Beck R, Fonseca I, Marques C, Duarte A, Santos M, Zúquete S, Gomes J, Walder G, Domingos A, Antunes S (2017) Guidelines for the detection of Babesia and Theileria parasites. Vector Borne Zoonotic Dis 17:51–65. https://doi.org/10.1089/vbz.2016.1955
M’ghirbi Y, Hurtado A, Brandika J, Khlif K, Ketata Z, Bouattour A, (2008) A molecular survey of Theileria and Babesia parasites in cattle, with a note on the distribution of ticks in Tunisia. Parasitol Res 103:435–442. https://doi.org/10.1007/s00436-008-0995-3
Ma Q, Liu J, Li Z, Xiang Q, Wang J, Liu A, Li Y, Yin H, Guan G, Luo J (2020) Clinical and pathological studies on cattle experimentally infected with Theileria annulata in China. Pathogens 9:727. https://doi.org/10.3390/pathogens9090727
Marendy D, Baker K, Emery D, Rolls P, Stutchbury R (2020) Haemaphysalis longicornis: the life-cycle on dogs and cattle, with confirmation of its vector status for Theileria orientalis in Australia. Vet Parasitol 277:100022. https://doi.org/10.1016/j.vpoa.2019.100022
Masatani T, Hayashi K, Andoh M, Tateno M, Endo Y, Asada M, Kusakisako K, Tanaka T, Gokuden M, Hozumi N, Nakadohzono F (2017) Detection and molecular characterization of Babesia, Theileria, and Hepatozoon species in hard ticks collected from Kagoshima, the southern region in Japan. Ticks Tick Borne Dis 8:581–587. https://doi.org/10.1016/j.ttbdis.2017.03.007
Muhanguzi D, Matovu E, Waiswa C (2010) Prevalence and characterization of Theileria and Babesia species in cattle under different husbandry systems in Western Uganda. Int J Anim Vet Adv 2(2):51–58
Nehra AK, Kumari A, Kundave VR, Vohra S, Ram H (2022) Molecular insights into the population structure and haplotype network of Theileria annulata based on the small-subunit ribosomal RNA (18S rRNA) gene. Infect Genet Evol 99:105252. https://doi.org/10.1016/j.meegid.2022.105252
Ola-Fadunsin SD, Sharma RS, Abdullah DA, Gimba FI, Jesse FF, Sani RA (2020) Molecular detection, prevalence and risk factors of Theileria orientalis infection among cattle in Peninsular Malaysia. Prev Vet Med 180:105027. https://doi.org/10.1016/j.prevetmed.2020.105027
Özübek S, Aktaş M (2019) Genetic diversity of Theileria orientalis from cattle in Turkey. Comp Immunol Microbiol Infect Dis 1(65):132–136. https://doi.org/10.1016/j.cimid.2019.05.015
Phipps LP, Hansford KM, Hernández-Triana LM, Golding M, McGinley L, Folly AJ, Vaux AG, de Marco MF, Carter DP, Medlock JM, Johnson N (2022) Detection of Borrelia and Babesia species in Haemaphysalis punctata ticks sampled in Southern England. Ticks Tick Borne Dis 13:101902. https://doi.org/10.1016/j.ttbdis.2022.101902
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sánchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Sang C, Yang M, Xu B, Liu G, Yang Y, Kairullayev K, Bauyrzhan O, Hazihan W, Hornok S, Wang Y (2021) Tick distribution and detection of Babesia and Theileria species in Eastern and Southern Kazakhstan. Ticks Tick Borne Dis 12(6):101817. https://doi.org/10.1016/j.ttbdis.2021.101817
Selim A, Attia K, AlKahtani MD, Albohairy FM, Shoulah S (2022) Molecular epidemiology and genetic characterization of Theileria orientalis in cattle. Trop Anim Health Prod 54(3):178. https://doi.org/10.1007/s11250-022-03176-w
Simking P, Saengow S, Bangphoomi K, Sarataphan N, Wongnarkpet S, Inpankaew T, Jittapalapong S, Munkhjargal T, Sivakumar T, Yokoyama N, Igarashi I (2013) The molecular prevalence and MSA-2b gene-based genetic diversity of Babesia bovis in dairy cattle in Thailand. Vet Parasitol 197:642–648. https://doi.org/10.1016/j.vetpar.2013.07.015
Sivakumar T, Hayashida K, Sugimoto C, Yokoyama N (2014) Evolution and genetic diversity of Theileria. Infect Genet Evol 1(27):250–263. https://doi.org/10.1016/j.meegid.2014.07.013
Social Science Statistics (2023) Chi-square test calculator. Social Science Statistics, Jeremy Stangroom, https://www.socscistatistics.com/tests/chisquare2/default2.aspx.
Srionrod N, Nooroong P, Poolsawat N, Minsakorn S, Watthanadirek A, Junsiri W, Sangchuai S, Chawengkirttikul R, Anuracpreeda P (2022) Molecular characterization and genetic diversity of Babesia bovis and Babesia bigemina of cattle in Thailand. Front Cell Infect Microbiol 12:1065963. https://doi.org/10.3389/fcimb.2022.1065963
Uilenberg G (2006) Babesia—a historical overview. Vet Parasitol 138:3–10. https://doi.org/10.1016/j.vetpar.2006.01.035
Zhou Z, Li K, Sun Y, Shi J, Li H, Chen Y, Yang H, Li X, Wu B, Li X, Wang Z (2019) Molecular epidemiology and risk factors of Anaplasma spp., Babesia spp. and Theileria spp. infection in cattle in Chongqing, China. PloS One 14:e0215585. https://doi.org/10.1371/journal.pone.0221359
Funding
This research project was financially supported by Thailand Science Research and Innovation (TSRI) Grant No. 660602/2566.
Author information
Authors and Affiliations
Contributions
Conceptualization: Tossapol Seerintra, Tongjit Thanchomnang, and Supawadee Piratae; methodology: Tossapol Seerintra, Wongwiwat Krinsoongnern, Tongjit Thanchomnang, and Supawadee Piratae; formal analysis and investigation: Tossapol Seerintra, Wongwiwat Krinsoongnern, Tongjit Thanchomnang, and Supawadee Piratae; writing—original draft preparation: Tossapol Seerintra and Supawadee Piratae; writing—review and editing: Tossapol Seerintra, Tongjit Thanchomnang, and Supawadee Piratae; funding acquisition: Supawadee Piratae; all authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
All experimental procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of Mahasarakham University (IACUC-MSU-3/2023).
Consent to participate
Written informed consent was obtained from the cattle owner.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Section Editor: Dana Mordue.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Section editor: Dana Mordue.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Seerintra, T., Krinsoongnern, W., Thanchomnang, T. et al. Molecular occurrence and genetic identification of Babesia spp. and Theileria spp. in naturally infected cattle from Thailand. Parasitol Res 123, 287 (2024). https://doi.org/10.1007/s00436-024-08299-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00436-024-08299-7