
Abstract
Giardia duodenalis is an intestinal protozoan that can infect both humans and animals, leading to public health issues and economic losses in the livestock industry. G. duodenalis has been reported to infect dairy cattle, but there is limited information available on large-scale dairy farms in Xinjiang, China. The study collected 749 fresh faecal samples from five large-scale cattle farms in Xinjiang, China. The study used a nested PCR assay of the small subunit ribosomal RNA (SSU rRNA*) gene to determine the presence of G. duodenalis. The results showed that 24.0% (180/749) of dairy cattle were positive for G. duodenalis, with the highest infection rate observed in pre-weaned calves (45.1%, 69/153). Among the 180 G. duodenalis positive samples, three assemblages were identified: assemblage E (n = 176), assemblage A (n = 3) and assemblage B (n = 1). Sixty-nine, 67 and 49 sequences were obtained for the beta-giardin (bg*) gene, the glutamate dehydrogenase (gdh*) gene and the triose phosphate isomerase (tpi*) gene, respectively. Thirteen novel sequences of assemblage E were identified, including five sequences from the bg* gene, four sequences from the gdh* gene and four sequences from the tpi* gene. This study found that 32 G. duodenalis assemblage E isolates formed 26 MLGs, indicating genetic variation and geographic isolation-based differentiation in bovine-derived G. duodenalis assemblage E. These findings provide fundamental insights into the genetic diversity of G. duodenalis in dairy cattle and can aid in the prevention and control of its occurrence in large-scale dairy cattle farms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Giardia duodenalis, also known as Giardia lamblia, or Giardia intestinalis, is a significant cause of diarrhoeal diseases globally and can infect a wide range of vertebrates, including humans (Ryan and Cacciò 2013; Rojas-López et al. 2022). In domestic animals, infection with G. duodenalis can lead to reduced reproductive performance in adults and developmental delays in young animals (O’Handley and Olson 2006). G. duodenalis is mainly transmitted through the faecal-oral route (Feng and Xiao 2011). The global prevalence of this disease is estimated to be between 2 and 7% in developed countries and up to 30% in low-income countries (Fantinatti et al. 2016). Giardiasis can cause diarrhoea and other intestinal symptoms. Additionally, it has been associated with arthritis and irritable bowel syndrome. The World Health Organisation (WHO) included giardiasis in its Neglected Diseases Initiative in 2006 (Painter et al. 2017; Nakao et al. 2017).
G. duodenalis has been classified into eight assemblages (A–H) based on host specificity and genetic differences. Assemblages A and B are considered typical zoonotic assemblages, while assemblages C–H have stricter host specificity (Heyworth 2016; Ryan et al. 2021). G. duodenalis assemblage E has been found to be the dominant assemblage in dairy cattle worldwide (Taghipour et al. 2022). Only a few studies have reported assemblage A/B as the dominant assemblage in dairy cattle (Coklin et al. 2007; Wang et al. 2014; Baroudi et al. 2017). Recently, multilocus genotyping (MLG) has been used to analyse the genetic diversity and zoonotic risk of G. duodenalis based on the bg*, gdh* and tpi* genes (Fu et al. 2022; Zhao et al. 2023b).
The National Bureau of Statistics reports a significant increase in the number of dairy cattle in Xinjiang, China, which is attributed to the implementation of scientifically informed large-scale breeding practices. However, large-scale animal husbandry can lead to the rapid spread of epidemics due to the close contact and similar living conditions of dairy cattle in high-density farming environments (Brooks-Pollock and Keeling 2009).
Compared to the data on Giardia duodenalis infections in cattle from central and eastern China, there is limited epidemiological data for G. duodenalis in large-scale dairy cattle farms in Xinjiang. The aim of this study was to investigate the infection rate and genetic characteristics of G. duodenalis in dairy cattle from large-scale farms in Xinjiang, China.
Materials and methods
Ethical standards
The protocol in the present study was not required to be reviewed and approved by the Animal Ethical Committee. Prior permission was obtained from the farm owner before collecting faecal samples. In the present study, faecal samples were collected from the rectum of each dairy cow without harm.
Sample collection
A total of 749 faecal samples were collected from dairy cattle between 2019 and 2020 from five large-scale farms (farm 1–farm 5) in Xinjiang. The farms were located in different positions in Alar city (farms 1–3), Hutubi city (farm 4), and Tiemenguan city (farm 5). The samples were collected from five age groups of dairy cattle. The study involved 153 pre-weaned calves (aged 0–3 months), 150 post-weaned calves (aged 3–6 months), 148 fattening dairy cattle (aged 6–12 months), 148 replacement heifers (aged 1–2 years and unproduced), and 150 adult heifers (over 2 years old and produced).
Faecal samples were collected directly from the rectums of the dairy cattle using disposable gloves. The amount of each samples ranged from 10 to 30 g. The samples were then placed in clean, self-sealing bags, labelled with the sampling information, and stored at 4 °C.
DNA extraction and PCR amplification
DNA was extracted from each faecal sample (approximately 200 mg) using an E.Z.N.A. Stool DNA Kit (Omega Bio-Tek Inc., Norcross/GA, USA) following the manufacturer’s suggested technique. The extracted DNA was stored at − 20 °C.
To determine the presence of G. duodenalis, the SSU rRNA* gene was targeted in all DNA samples (Appelbee et al. 2003). Each sample was analysed in duplicate using positive controls (dog-derived assemblage D DNA) and negative controls (sterile water). All samples positive for G. duodenalis were genotyped using multilocus analysis based on the bg* (Lalle et al. 2005), gdh* (Cacciò et al. 2008) and tpi* (Sulaiman et al. 2003) genes to determine G. duodenalis subtypes. The genotyping analysis included 291 bp for SSU rRNA*, 511 bp for bg*, 520 bp for gdh* and 530 bp for tpi*.
Sequence and phylogenetic analyses
The SSU rRNA*, tpi*, gdh* and bg* genes of positive secondary PCR products were sequenced by GENEWIZ (Suzhou, China). The obtained sequences were uploaded to SEQMAN in DNAStar (http://www.dnastar.com/) for DNA profile proofreading and were compared and identified using Clustal X2.1 (http://www.clustal.org/) and GenBank (https://www.ncbi.nlm.nih.gov/genbank/).
To investigate the relationships between different isolates of G. duodenalis and reveal its genetic diversity, we constructed a phylogenetic tree based on the maximum likelihood neighbour-joining method using Mega7.0 software. The substitution rate calculation used the general time-reversible model, and we used 1000 replicates in the bootstrapping analysis.
Statistical analysis
Differences in infection rates between regions and physiological states were analysed by the χ2 test using the statistical software SPSS Statistics 26 (SPSS Inc., Chicago, IL, USA), and differences were considered statistically significant when P < 0.05.
Nucleotide sequence accession numbers
The nucleotide sequences obtained in this study have been deposited in NCBI’s GenBank database under the following accession numbers: OR538008–OR538012 for the bg* gene, OR538013–OR538016 for the gdh* gene and OR538017–OR538020 for the tpi* gene.
Results
Giardia duodenalis infection rate and distribution based on age
The study analysed 749 faecal DNA samples from dairy cattle using the SSU rRNA* gene of G. duodenalis. Of these, 180 samples (24.0%, 95% CI 21.0–27.1%) were identified as G. duodenalis-positive (Table 1). The lowest infection rate was found on farm 2 (0.7%), which was significantly lower than the rates on the other farms (farms 1 and 3) in Alar, as well as farms 4 and 5 (P < 0.05) (Table 1).
The study analysed the infection rate of G. duodenalis in dairy cattle across different age groups. The highest infection rate was observed in pre-weaned calves (45.1%, 69/153), which was significantly higher than the other age groups (P < 0.05). Adult heifers had the lowest infection rate (12.0%, 18/150). The infection rates of G. duodenalis in post-weaned calves, fattening dairy cattle and replacement heifers were 22.0% (33/150), 18.9% (38/148) and 21.6% (32/148), respectively (Table 2).
Molecular identification and distribution of assemblages
In this study, 180 amplified sequences of G. duodenalis were analysed for the SSU rRNA* gene. Out of these, 176 sequences belonged to G. duodenalis assemblage E (97.8%, 176/180), three sequences belonged to G. duodenalis assemblage A (1.7%, 3/180) and one sequence belonged to G. duodenalis assemblage B (0.6%, 1/180). In farm 1, three samples tested positive for assemblage A, while in farm 4, one sample tested positive for assemblage B. Assemblage E was the only assemblage detected in farms 2, 3 and 5, and it was the dominant assemblage on all five farms (refer to Table 1).
In each of the five age groups defined in this study, one case of assemblage A was found in post-weaned calves, replacement heifers and fattening cattle. Only one case of assemblage B was found in fattening cattle, and both pre-weaned calves and adult cattle. Assemblage E was the dominant assemblage in all age groups.
Polymorphisms of Giardia duodenalis isolates
Out of the 180 positive samples, 69 sequences were obtained for the bg* gene, with 68 belonging to assemblage E and 1 to assemblage A. For the gdh* gene, 67 sequences were obtained, with 66 belonging to assemblage E and 1 to assemblage A. Lastly, 49 sequences were obtained for the tpi* gene, with 48 belonging to assemblage E and 1 to assemblage A (refer to Table 1). The present study obtained 68 assemblage E sequences obtained for the bg* gene, which were classified into 13 subtypes, including 8 known subtypes and 5 novel subtypes, by comparison with MN833266 in GenBank (accession numbers OR538008–OR538012) (Table S1). Additionaliy, 66 assemblage E sequences were obtained for the gdh* gene and classified into 14 isoforms, including 10 known isoforms and 4 novel isoforms (accession numbers OR538013–OR538016) (Table S2) using the reference sequence MK645797. Using MG820468 as a reference sequence, 48 sequences of the tpi* gene were obtained and categorized into 15 subtypes, including 11 known subtypes and 4 novel subtypes (accession numbers OR538017–538020) (refer to Table S3).
For assemblage A, one sequence was obtained for each of the three genes, which belong to different samples. The sequence obtained for the bg* gene showed 100% similarity to MK610391, while the gdh* gene sequence was 100% similar to MN047217, and the tpi* gene sequence was 100% similar to MK639171.
Multilocus genotyping
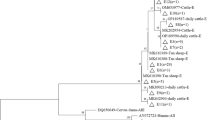
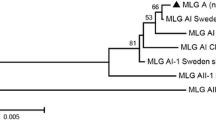
A total of 32 samples were identified as belonging to assemblage E based on being positive at all four genes (SSU rRNA*- bg*- gdh*- tpi*), forming 26 MLGs (named MLG-E1 to MLG-E26) (Table S4). Phylogenetic analyses of bg*- tpi*- gdh* tandem sequences showed that the MLGs obtained in this study were genetically distinct from the MLGs previously found in other regions (Fig. 1). However, they were similar to the MLGs found in Xinjiang, indicating a clear geographical segregation.

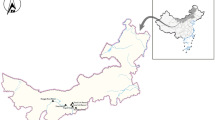
Geographic map of the sampling locations of Xinjiang, China. The authors originally designed the figure using ArcGIS 10.2 software. The original vector diagram imported into ArcGIS was adapted from Natural Earth (http://www.naturalearthdata.com)
Discussion
The study revealed that the infection rate of G. duodenalis in dairy cattle in certain areas of Xinjiang was 24.0% (180/149), which is higher than the 9.4% (52/556) reported in Jiangxi (Qi et al. 2016) and 19.8% (31/156) reported in Taiwan (Lam et al. 2021), but lower than of the 27.5% (144/524) reported in Yunnan (Heng et al. 2022) and 29.5% (149/505) reported in Inner Mongolia (Zhao et al. 2023a). The worldwide infection rate of G. duodenalis in dairy cattle ranges from 0 to 76.5%. The infection rate of G. duodenalis in dairy cattle based on molecular techniques was 21.8% (5845/26,767) (Taghipour et al. 2022), which is similar to the results of the present study. Variations in reported infection rates may be attributed to several factors, such as the testing method (microscopic or molecular), farming type (free-range or intensive), farm size, number of samples analysed, time of sample collection and environmental conditions.
Previous studies have demonstrated that the infection rate of G. duodenalis is higher in pre-weaned dairy cattle than in post-weaned dairy cattle (Liu et al. 2015; Wang et al. 2019; Heng et al. 2022; Taghipour et al. 2022). These findings are consistent with the results of the present study. Table 2 shows that the infection rate of G. duodenalis was significantly higher in pre-weaned calves compared to dairy cattle of other ages. This may be due to the incomplete development of the immune system in pre-weaned calves making them more susceptible to pathogens. Most reports indicate that G. duodenalis infection is negatively correlated with the age of the animals (Santín et al. 2009; Wang et al. 2014; Huang et al. 2014). The results of the present study are consistent with this pattern, but the infection rate was higher in large-breed dairy cattle (21.6%) than in small-breed dairy cattle (18.9%). This may be due to the fact that large-breed dairy cattle enter the stage of social maturation and have more contact with other individuals. In large and crowded breeding environments, contact between animals can lead to the transmission of G. duodenalis, which increases the risk of infection.
G. duodenalis is classified into eight assemblages (A–H). Studies have consistently shown that assemblage E is the most prevalent in cows, followed by assemblages A, B and D (Taghipour et al. 2022; Meng et al. 2023). This study, assemblage E, A and B were detected using the SSU rRNA* gene, with assemblage E being the most dominant. Assemblage A and assemblage B are considered zoonotic assemblages in the traditional sense due to their wide range of hosts (Heyworth 2016). Assemblage E, on the other hand, is commonly found in hoofed animals such as dairy cattle and sheep (Taghipour et al. 2022; Geng et al. 2023) and is not typically considered zoonotic. However, it has been documented that assemblage E has caused human infections in various countries such as Brazil (Fantinatti et al. 2016), Australia (Zahedi et al. 2017) and New Zealand (Abdel-Moein and Saeed 2016). A study conducted in Egypt found that assemblage E was present in up to 62.5% of human samples (Abdel-Moein and Saeed 2016). Moreover, a growing body of research has demonstrated that G. duodenalis not only infects livestock in large-scale farms but also contaminates the surrounding environment(Tram et al. 2022; Zhao et al. 2023b). This contamination poses a risk not only to the health of other animals within the farms but also to humans who come into contact with these farms and may potentially be exposed to G. duodenalis (Robertson 2009; Berrilli et al. 2012). Therefore, further research is necessary to eatablish the public health implications of G. duodenalis in dairy cattle.
Twenty-six assemblage E MLG genotypes were identified in this study. The genetic differences of assemblage E were confirmed in various regions, with limited MLG intersection among samples. Each MLG was found in at most three dairy cattle, and only MLG-E1 was present simultaneously in both Alar and Tiemenguan (see Table S4). This relationship could be attributed to the substantial genetic diversity of G. duodenalis observed in Xinjiang’s dairy cattle. Figure 2 shows that the occurrence of E MLGs in the Tiemenguan and Alar areas was clustered on farms, while no substantial farm-based grouping was observed in Hutubi. This may be due to the low infection rate on Hutubi farms and the limited discovery of only two E MLGs, making the trial outcomes unrepresentative. Previous studies have found that G. duodenalis experiences geographical isolation (Cui et al. 2018). The E MLGs of G. duodenalis identified in this study were highly aggregated with those previously found in Xinjiang. However, most of them were significantly distinct from the E MLGs of G. duodenalis detected in dairy cows from Inner Mongolia (Zhao et al. 2023a), Guangdong (Cui et al. 2018), Sichuan (Dan et al. 2019), Yunnan (Heng et al. 2022), Jiangsu (Wang et al. 2019) and Shanghai (Wang et al. 2017). The study showed notable distinctions between the geographical distribution and genetic makeup of the E assemblage.

Phylogenetic evolutionary tree of G. duodenalis assemblages analysed by the maximum likelihood method based on bg*, tpi* and gdh* gene tandem sequences (bg*-tpi*-gdh*), with substitution rates calculated using a general time-reversible model. Bootstrap values greater than 50% from 1000 replicates are shown at the nodes. MLGs marked by the black triangles were the sequences obtained in this study
Conclusion
The study findings show a high infection rate of G. duodenalis in dairy cattle in Xinjiang, China, with a predominant infection of G. duodenalis assemblage E. The varying distribution of MLGs of G. duodenalis assemblage E in dairy cattle may indicate geographic isolation.
Data availability
The nucleotide sequences reported in this study have been deposited in the GenBank database of the National Center for Biotechnology Information under accession numbers OR538008–OR538020.
References
Abdel-Moein KA, Saeed H (2016) The zoonotic potential of Giardia intestinalis assemblage E in rural settings. Parasitol Res 115:3197–3202. https://doi.org/10.1007/s00436-016-5081-7
Appelbee AJ, Frederick LM, Heitman TL, Olson ME (2003) Prevalence and genotyping of Giardia duodenalis from beef calves in Alberta, Canada. Vet Parasitol 112:289–294. https://doi.org/10.1016/s0304-4017(02)00422-3
Baroudi D, Khelef D, Hakem A, Abdelaziz A, Chen X, Lysen C, Roellig D, Xiao L (2017) Molecular characterization of zoonotic pathogens Cryptosporidium spp., Giardia duodenalis and Enterocytozoon bieneusi in calves in Algeria. Veterinary Parasitology, Regional Studies and Reports 8:66–69. https://doi.org/10.1016/j.vprsr.2017.02.005
Berrilli F, D’Alfonso R, Giangaspero A, Marangi M, Brandonisio O, Kaboré Y, Glé C, Cianfanelli C, Lauro R, Di Cave D (2012) Giardia duodenalis genotypes and Cryptosporidium species in humans and domestic animals in Côte d’Ivoire: occurrence and evidence for environmental contamination. Trans R Soc Trop Med Hyg 106:191–195. https://doi.org/10.1016/j.trstmh.2011.12.005
Brooks-Pollock E, Keeling M (2009) Herd size and bovine tuberculosis persistence in cattle farms in Great Britain. Prev Vet Med 92:360–365. https://doi.org/10.1016/j.prevetmed.2009.08.022
Cacciò SM, Beck R, Lalle M, Marinculic A, Pozio E (2008) Multilocus genotyping of Giardia duodenalis reveals striking differences between assemblages A and B. Int J Parasitol 38:1523–1531. https://doi.org/10.1016/j.ijpara.2008.04.008
Coklin T, Farber J, Parrington L, Dixon B (2007) Prevalence and molecular characterization of Giardia duodenalis and Cryptosporidium spp. in dairy cattle in Ontario. Canada Vet Parasitol 150:297–305. https://doi.org/10.1016/j.vetpar.2007.09.014
Cui Z, Wang L, Cao L, Sun M, Liang N, Wang H, Chang Y, Lin X, Yu L, Wang R, Zhang S, Ning C, Zhang L (2018) Genetic characteristics and geographic segregation of Giardia duodenalis in dairy cattle from Guangdong Province, southern China. Infection, Infect Genet Evol 66:95–100. https://doi.org/10.1016/j.meegid.2018.09.019
Dan J, Zhang X, Ren Z, Wang L, Cao S, Shen L, Deng J, Zuo Z, Yu S, Wang Y, Ma X, Liu H, Zhou Z, Hu Y, Fu H, He C, Geng Y, Gu X, Peng G, Zhong Z (2019) Occurrence and multilocus genotyping of Giardia duodenalis from post-weaned dairy calves in Sichuan province. China Plos One 14:e0224627. https://doi.org/10.1371/journal.pone.0224627
Fantinatti M, Bello AR, Fernandes O, Da-Cruz AM (2016) Identification of Giardia lamblia Assemblage E in humans points to a new anthropozoonotic cycle. J Infect Dis 214:1256–1259. https://doi.org/10.1093/infdis/jiw361
Feng Y, Xiao L (2011) Zoonotic potential and molecular epidemiology of Giardia species and giardiasis. Clin Microbiol Rev 24:110–140. https://doi.org/10.1128/CMR.00033-10
Fu Y, Dong H, Bian X, Qin Z, Han H, Lang J, Zhang J, Zhao G, Li J, Zhang L (2022) Molecular characterizations of Giardia duodenalis based on multilocus genotyping in sheep, goats, and beef cattle in Southwest Inner Mongolia. China Parasite 29:33. https://doi.org/10.1051/parasite/2022036
Geng HL, Yan WL, Wang JM, Meng JX, Zhang M, Zhao JX, Shang KM, Liu J, Liu WH (2023) Meta-analysis of the prevalence of Giardia duodenalis in sheep and goats in China. Microb Pathog 179:106097. https://doi.org/10.1016/j.micpath.2023.106097
Heng ZJ, Yang JF, Xie XY, Xu CR, Chen JR, Ma J, He JJ, Mao HM (2022) Prevalence and multilocus genotyping of Giardia duodenalis in Holstein cattle in Yunnan. China Front Vet Sci 9:949462. https://doi.org/10.3389/fvets.2022.949462
Heyworth MF (2016) Giardia duodenalis genetic assemblages and hosts. Parasite 23:13. https://doi.org/10.1051/parasite/2016013
Huang J, Yue D, Qi M, Wang R, Zhao J, Li J, Shi K, Wang M, Zhang L (2014) Prevalence and molecular characterization of Cryptosporidium spp and Giardia duodenalis in dairy cattle in Ningxia, northwestern China. BMC Vet Res 10:292. https://doi.org/10.1186/s12917-014-0292-6
Lalle M, Pozio E, Capelli G, Bruschi F, Crotti D, Cacciò SM (2005) Genetic heterogeneity at the bata-giardin locus among human and animal isolates of Giardia duodenalis and identification of potentially zoonotic subgenotypes. Int J Parasitol 35:207–213. https://doi.org/10.1016/j.ijpara.2004.10.022
Lam HYP, Chen TT, Tseng YC, Chang KC, Yang TH, Peng SY (2021) Detection and genotyping of Giardia duodenalis from cattle and pigs in Hualien country, Eastern Taiwan. J Microbiol Immunol Infec 54:718–727. https://doi.org/10.1016/j.jmii.2020.05.009
Liu G, Su Y, Zhou M, Zhao J, Zhang T, Ahmad W, Lu H, Jiang N, Chen Q, Xiang M, Yin J (2015) Prevalence and molecular characterization of Giardia duodenalis isolates from dairy cattle in northeast China. Exp Parasitol 154:20–24. https://doi.org/10.1016/j.exppara.2015.03.020
Meng XZ, Kang C, Wei J, Ma H, Liu G, Zhao JP, Zhang HS, Yang XB, Wang XY, Yang LH, Geng HL, Cao H (2023) Meta-analysis of the prevalence of Giardia duodenalis in cattle in China. Foodborne Pathog Dis 20:17–31. https://doi.org/10.1089/fpd.2022.0052
Nakao JH, Collier SA, Gargano JW (2017) Giardiasis and subsequent irritable bowel syndrome: a longitudinal cohort study using health insurance data. J Infect Dis 215:798–805. https://doi.org/10.1093/infdis/jiw621
O’Handley RM, Olson ME (2006) Giardiasis and cryptosporidiosis in ruminants. Vet Clin North Am Food Anim Pract 22:623–643. https://doi.org/10.1016/j.cvfa.2006.07.002
Painter JE, Collier SA, Gargano JW (2017) Association between Giardia and arthritis or joint pain in a large health insurance cohort: could it be reactive arthritis? Epidemiol Infect 145:471–477. https://doi.org/10.1017/S0950268816002120
Qi M, Wang H, Jing B, Wang R, Jian F, Ning C, Zhang L (2016) Prevalence and multilocus genotyping of Giardia duodenalis in dairy calves in Xinjiang. Northwestern China Parasite Vectors 9:546. https://doi.org/10.1186/s13071-016-1828-3
Robertson LJ (2009) Giardia and Cryptosporidium infections in sheep and goats: a review of the potential for transmission to humans via environmental contamination. Epidemiol Infect 137:913–921. https://doi.org/10.1017/S0950268809002295
Rojas-López L, Marques RC, Svärd SG (2022) Giardia duodenalis. Trends Parasitol 38:605–606. https://doi.org/10.1016/j.pt.2022.01.001
Ryan U, Cacciò SM (2013) Zoonotic potential of Giardia. Int J Parasitol 43:943–956. https://doi.org/10.1016/j.ijpara.2013.06.001
Ryan UM, Feng Y, Fayer R, Xiao L (2021) Taxonomy and molecular epidemiology of Cryptosporidium and Giardia—a 50 year perspective (1971–2021). Int J Parasitol 51:1099–1119. https://doi.org/10.1016/j.ijpara.2021.08.007
Santín M, Trout JM, Fayer R (2009) A longitudinal study of Giardia duodenalis genotypes in dairy cows from birth to 2 years of age. Vet Parasitol 162:40–45. https://doi.org/10.1016/j.vetpar.2009.02.008
Sulaiman IM, Fayer R, Bern C, Gilman RH, Trout JM, Schantz PM, Das P, Lal AA, Xiao L (2003) Triosephosphate isomerase gene characterization and potential zoonotic transmission of Giardia duodenalis. Emerg Infect Dis 9:1444–1452. https://doi.org/10.3201/eid0911.030084
Taghipour A, Sharbatkhori M, Tohidi F, Ghanbari MR, Karanis P, Olfatifar M, Majidiani H, Khazaei S, Bahadory S, Javanmard E (2022) Global prevalence of Giardia duodenalis in cattle: a systematic review and meta-analysis. Prev Vet Med 203:105632. https://doi.org/10.1016/j.prevetmed.2022.105632
Tram NT, Phuc PD, Phi NH, Trang LT, Nga TT, Ha HTT, Cam PD, Canh TQ, Karanis P (2022) Cryptosporidium and Giardia in biogas wastewater: management of manure livestock and hygiene aspects using influent, effluent, sewage canal samples, vegetable, and soil samples. Pathogens 11:174. https://doi.org/10.3390/pathogens11020174
Wang H, Zhao G, Chen G, Jian F, Zhang S, Feng C, Wang R, Zhu J, Dong H, Hua J, Wang M, Zhang L (2014) Multilocus genotyping of Giardia duodenalis in dairy cattle in Henan. China Plos One 9:e100453. https://doi.org/10.1371/journal.pone.0100453
Wang X, Cai M, Jiang W, Wang Y, Jin Y, Li N, Guo Y, Feng Y, Xiao L (2017) High genetic diversity of Giardia duodenalis assemblage E in pre-weaned dairy calves in Shanghai, China, revealed by multilocus genotyping. Parasitol Res 116:2101–2110. https://doi.org/10.1007/s00436-017-5509-8
Wang R, Li N, Jiang W, Guo Y, Wang X, Jin Y, Feng Y, Xiao L (2019) Infection patterns, clinical significance, and genetic characteristics of Enterocytozoon bieneusi and Giardia duodenalis in dairy cattle in Jiangsu, China. Parasitol Res 118:3053–3060. https://doi.org/10.1007/s00436-019-06426-3
Zahedi A, Field D, Ryan U (2017) Molecular typing of Giardia duodenalis in humans in Queensland—first report of Assemblage E. Parasitology 144:1154–1161. https://doi.org/10.1017/S0031182017000439
Zhao L, Zhang ZS, Han WX, Yang B, Chai HL, Wang MY, Wang Y, Zhang S, Zhao WH, Ma YM, Zhan YJ, Wang LF, Ding YL, Wang JL, Liu YH (2023a) Prevalence and molecular characterization of Giardia duodenalis in dairy cattle in Central Inner Mongolia. Northern China Sci Rep 13:13960. https://doi.org/10.1038/s41598-023-40987-9
Zhao Q, Lu C, Pei Z, Gong P, Li J, Jian F, Jing B, Qi M, Ning C (2023b) Giardia duodenalis in Hu sheep: occurrence and environmental contamination on large-scale housing farms. Parasite 30:2. https://doi.org/10.1051/parasite/2023004
Funding
This work was supported by the National Natural Science Foundation of China (31960704) and the Key Technologies R&D Programme of Xinjiang Production & Construction Corps (2020AB025).
Author information
Authors and Affiliations
Contributions
Bo Jing and Zhenjie Zhang contributed to the conception and design of the experiments. Meigui Huang performed the experiments. Bin Yang helped in interpretation of data. Qianming Zhao, Meng Qi and Chunyan Xu interpreted the results and drafted the manuscript. All of the authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Consent to participate
Not applicable.
Consent for publication
All the authors consent to publication of this article.
Competing interests
The authors declare no competing interests.
Additional information
Section Editor: Lihua Xiao
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, Q., Yang, B., Huang, M. et al. Molecular detection and genetic characteristics of Giardia duodenalis in dairy cattle from large-scale breeding farms in Xinjiang, China. Parasitol Res 123, 106 (2024). https://doi.org/10.1007/s00436-024-08123-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00436-024-08123-2