
Abstract
We recently described a targeted amplicon deep sequencing (TADS) strategy that utilizes a nested PCR targeting the 18S rDNA gene of blood-borne parasites. The assay facilitates selective digestion of host DNA by targeting enzyme restriction sites present in vertebrates but absent in parasites. This enriching of parasite-derived amplicon drastically reduces the proportion of host-derived reads during sequencing and results in the sensitive detection of several clinically important blood parasites including Plasmodium spp., Babesia spp., kinetoplastids, and filarial nematodes. Despite these promising results, high costs and the laborious nature of metagenomics sequencing are prohibitive to the routine use of this assay in most laboratories. We describe and evaluate a new metagenomic approach that utilizes a set of primers modified from our original assay that incorporates Illumina barcodes and adapters during the PCR steps. This modification makes amplicons immediately compatible with sequencing on the Illumina MiSeq platform, removing the need for a separate library preparation, which is expensive and time-consuming. We compared this modified assay to our previous nested TADS assay in terms of preparation speed, limit of detection (LOD), and cost. Our modifications reduced assay turnaround times from 7 to 5 days. The cost decreased from approximately $40 per sample to $11 per sample. The modified assay displayed comparable performance in the detection and differentiation of human-infecting Plasmodium spp., Babesia spp., kinetoplastids, and filarial nematodes in clinical samples. The LOD of this modified approach was determined for malaria parasites and remained similar to that previously reported for our earlier assay (0.58 Plasmodium falciparum parasites/µL of blood). These modifications markedly reduced costs and turnaround times, making the assay more amenable to routine diagnostic applications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
We recently described a universal parasite diagnostic assay (UPDx), a metagenomic assay targeting the eukaryotic 18S rDNA gene that was developed to characterize parasitic communities in blood samples (Flaherty et al. 2018). This first UPDx assay was later modified to include a nested amplification step that improved the assay’s limit of detection (LOD) (Flaherty et al. 2021). This nested version of UPDx was referred to as nUPDx (Flaherty et al. 2021). Unique among metagenomics assays, the nUPDx amplicon possesses restriction sites that exist only in vertebrates and not in parasites. Taking advantage of these vertebrate-specific restriction sites, nUPDx includes restriction enzyme digestion steps that are performed on the genomic DNA extract prior to amplification and on the PCR product from the first amplification. These digestion steps reduce the abundance of host-derived sequences in the final PCR product and therefore result in markedly improved sensitivity for the identification of any parasites in the samples (Flaherty et al. 2021).
The nUPDx assay has a limit of detection (LOD) similar to pathogen-specific real-time PCR assays and facilitated detection of DNA from malaria parasites (Plasmodium malariae, P. falciparum, P. vivax, and P. ovale), Babesia species (B. microti, B. divergens, and B. duncani), kinetoplastids (Leishmania spp., Trypanosoma cruzi, and Trypanosoma brucei), and the filarial nematodes Loa loa and Brugia malayi (Flaherty et al. 2021). In most cases, it was able to provide a species-level diagnosis, which can be critical for proper patient management, such as determining the appropriate drug treatment for malaria. Despite its utility, as with other metagenomic sequencing methods, the library preparation protocol required for nUPDx is laborious, cumbersome, and very expensive in terms of time (person hours) and monetary costs (Hess et al. 2020). These aspects represent a significant barrier to the routine diagnostic use of nUPDx and similar metagenomic assays in place of cheaper and widely available PCR-based diagnostics.
To counteract this barrier, Illumina published a guide describing how investigators can combine PCR amplification with library preparation to produce sequencing-ready amplicons, thereby reducing the need for library preparation (Anonymous 2022). Briefly, these guidelines describe how to modify user‐defined PCR primers, allowing amplification of target DNA while simultaneously preparing the amplicon for sequencing by incorporating the necessary Illumina adapter sequences. A subsequent amplification step is then performed with primers complementary to adapter sequences, to add multiplexing indices and sequencing adapters for MiSeq system sequencing. This protocol and its adaptations have already been successfully applied to metagenomics studies (Lee et al. 2019; Diaz-Torres et al. 2021), including an adaptation of nUPDx developed at the Wadsworth Center in the New York State Department of Health (Clemons et al. 2022).
Given the diagnostic utility displayed by nUPDx, we sought to build upon the work of the Wadsworth Center (Clemons et al. 2022) by improving nUPDx further while also removing the need for expensive library preparation. In the Wadsworth Center study, as samples still underwent a nested PCR with the addition of overhang adapters during the second amplification step, a third amplification step was required to add indices and sequencing adapters from Illumina. In this study, we shortened the procedure to only two amplification steps by adding overhang adapters in the first step and custom index-incorporating primers in the second step. This study sought to compare this improved adapter-incorporating UPDx method (Ad_UPDx) to the previously described nUPDx approach (Flaherty et al. 2021) in terms of preparation speed, LOD, and cost.
We applied Ad_UPDx to various clinical blood samples containing a range of blood parasites including the apicomplexan parasites Plasmodium spp. and Babesia spp., which are nationally notifiable in the USA (Hwang et al. 2009; Bishop et al. 2021). Parasites from these genera cause potentially lethal infections, and the incidence of malaria and babesiosis diagnoses is increasing in the USA due to increasing international travel and an increase in domestically acquired infections, respectively (Dye-Braumuller and Kanyangarara 2021; Mace et al. 2021; Menis et al. 2021). These trends highlight the need for modern diagnostic assays that accurately detect and differentiate morphologically similar blood-borne parasites.
Additionally, we tested blood samples containing kinetoplastid parasites and filarial nematodes. These parasites are encountered less frequently in the USA than Plasmodium spp. and Babesia spp., although detection and differentiation of rarer infections are also important functions of reference diagnostic laboratories where we anticipate that Ad_UPDx could be implemented. We also assessed the LOD of Ad_UPDx using serially diluted, quantified cultures of P. falciparum.
Methods
Source of samples
A total of 36 blood samples were analyzed in parallel using nUPDX and Ad_UPDx for direct comparison. Blood samples were confirmed positive or negative for parasites using pathogen-specific PCR and/or by light microscopic examination of stained blood smears, as described in Table 1. Thirteen samples had tested positive for a single parasite, which included Plasmodium falciparum, Plasmodium ovale, Plasmodium malariae, Plasmodium vivax, Babesia microti, Babesia divergens-like variant MO1, Leishmania sp., Brugia malayi, Loa Loa (n = 1 each), and Trypanosoma cruzi collected during acute infection (n = 2) and during chronic infection (n = 2). Three samples had tested positive for multiple malaria parasites, including P. falciparum/P. ovale, P. falciparum/P. vivax, and P. falciparum/P. malariae. To determine the LOD of Ad_UPDx, we tested a serial dilution of a Plasmodium falciparum strain 3D7 culture spiked into parasite-free blood in duplicate with concentrations ranging from 58,000 to 0.0058 parasite/μL (i.e., eight tenfold dilution steps). P. falciparum was chosen as the representative parasite for LOD estimation for Ad_UPDx because it had been used for this purpose for nUPDx previously (Flaherty et al. 2021). Parasite culture and serial dilutions were prepared as before (Flaherty et al. 2021) to produce two sets of serial dilutions (duplicates). For this study, the LOD was defined as the lowest concentration that generated positive results in at least one of the duplicates. Finally, four parasite-free samples from healthy blood donors were included in this study as negative controls.
DNA extraction
DNA was extracted using a QIAamp DNA Blood Mini QIACube Kit on a QIACube for automated extraction, according to the manufacturer’s instructions (QIAGEN, Germantown, MD, USA). The elution volume of Buffer AE was adjusted to 50 μL.
Ad_UPDx assay design
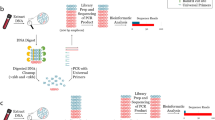
The Ad_UPDx assay was designed by combining the previously described nUPDx assay (Flaherty et al. 2021) and the NEBNext Ultra DNA Library Prep Kit protocol for Illumina sequencing (New England Biolabs, Ipswich, MA, USA). The first amplification targeted a ~ 200 bp fragment of the 18S rRNA genes using primers that possessed overhang adapters containing the priming sites for a second amplification (Fig. 1). The second amplification was then performed with primers complementary to the adapter overhang sequences incorporated during the first PCR, allowing nested amplification and addition of indices and flow cell adapters simultaneously (Fig. 1).

Combined DNA amplification and library preparation using the newly described Ad_UPDx protocol. First, specific and user‐defined forward and reverse primers targeting the region of interest are designed with overhang adapters compatible with Illumina sequencing and used for template amplification from DNA samples. Then, primers complementary to adapter sequences are used to attach multiplexing indices and Illumina flow cell adapters
Forward and reverse UPDx primers with overhang adapters
Overhang adapter sequences were added to the 5’ end of the inner nUPDx primers. Adapter sequences were modified from the NEBNext® Multiplex Oligos for Illumina® (96 Index Primers) manual for Illumina sequencing (New England Biolabs). Sequences of the newly designed forward and reverse Ad_UPDx primers are available in Table 2.
Preparation of primers for index and universal adapter-incorporating PCR
Primer sequences for the second amplification step were adapted from the NEBNext® Multiplex Oligos for Illumina® (96 Index Primers) manual for Illumina sequencing (New England Biolabs). A total of 96 forward index-incorporating primers were designed along with one reverse primer (Table 2, Supplementary information). To simplify reaction preparation, primer mixes were prepared in 96-well microplates. Each well contained 7 µL of one of the 10 μM forward index primers and 7 µL of 10 μM reverse primer (Univ_primerR).
First enzymatic digestion
Extracted DNA (7.5 μL) was subjected to the first enzymatic digestion in a final volume of 10 μL, including 0.5 μL of BamHI-HF (10 units), 1 μL of BsoBI (10 units), and 1 μL of 10X CutSmart Buffer. Samples were incubated for 1 h at 37 °C. All restriction enzymes were purchased from New England Biolabs.
First amplification step using UPDx primers with overhang adapters
Digested DNA (2 μL) was amplified using the AdUPDx_F1 and AdUPDx_R1 primers. Each reaction included 5 μL of 5X Q5 Reaction Buffer, 5 μL of 5X High GC Enhancer, 0.25 μL (500 units) of Q5 High-Fidelity DNA Polymerase, 0.5 μL of dNTPs (solution mix of 10 mM each), 9.5 μL PCR grade water, and 1.25 μL of each 10 μM primer, in a final volume of 25 μL. All PCR reagents (excluding primers and water) were purchased from New England Biolabs. Thermal cycling was performed as follows: 98.0 °C for 30 s, 30 cycles of 98.0 °C for 10 s, 67.0 °C for 30 s, 72.0 °C for 45 s, and 72.0 °C for 2 min.
Second enzymatic digestion
Restriction enzymes and buffer were directly added to the 25 μL of PCR products as follows: 0.5 μL of BamHI-HF (10 units), 1 μL of BsoBI (10 units), and 2.5 μL of 10X CutSmart Buffer. Samples were again incubated for 1 h at 37 °C.
Bead cleanup with size selection
The digested PCR product (29 μL) was processed according to NEBNext® Ultra™ II DNA Library Prep Kit manufacturer instructions (New England Biolabs) by performing a bead cleanup with size selection using Agencourt AMPure XP Beads (Beckman Coulter, Brea, CA, USA), accounting for an approximate insert size of 200 bp. Amplicons were eluted in 15 μL of 0.1X TE buffer.
Second amplification step for the addition of multiplexing indices and sequencing adapters
Cleaned amplicons (15 μL) were subjected to the second amplification step using the forward index and reverse universal primers (Table 2, Supplementary information). Reactions were prepared to contain 25 μL of NEBNext Ultra II Q5 Master Mix (New England Biolabs) and 10 μL of the primer mix previously prepared in the 96 microwell plates, including one forward index primer and the reverse Universal primer. Next, 15 μL of cleaned amplicon was added to the mix to a final volume of 50 μL. Thermal cycling was performed as follows: 98.0 °C for 30 s, 10 cycles of 98.0 °C for 10 s, 65.0 °C for 75 s, and 65.0 °C for 5 min.
nUPDx assay
The same DNA samples were processed with the comparison method, nUPDx, (Flaherty et al. 2021) in parallel to the Ad_UPDx method. During the library preparation index PCR step, index primers were selected that had not already been used with the Ad_UPDx method so that the same samples detected with both methods could be multiplexed in the same sequencing run.
Bead cleanup without size selection
A final bead cleanup without size selection was performed on sample libraries according to NEBNext® Ultra™ II DNA Library Prep Kit manufacturer instructions (New England Biolabs) using Agencourt AMPure XP Beads (Beckman Coulter). The amplicons were eluted in 40 μL of 0.1X TE buffer, and 5 μL of eluate from each sample was pooled for each method (Ad_UPDx and nUPDx) separately, resulting in two pooled libraries. Library fragment size estimation and DNA concentration were determined as described below.
Library fragment size and concentration
The concentration of the final pooled libraries for nUPDx and Ad_UPDx were individually determined using a Qubit 2.0 Fluorometer with the Qubit dsDNA High Sensitivity Assay Kit (Life Technologies, Grand Island, NY, USA). The average fragment size of each sample library pool was determined by the Agilent 2200 Tapestation System using Agilent D1000 ScreenTape reagents following manufacturer instructions (Agilent Technologies, Santa Clara, CA, USA).
Amplicon sequencing
Pooled libraries constructed using the Ad_UPDx and nUPDx methods were separately denatured and diluted to 10 pM. This normalization was performed to ensure that reads from each pooled library would be represented equally in the sequencing run. The pooled library was then spiked with the 10% PhiX control library (Illumina, San Diego, CA, USA), as per the manufacturer’s recommendations. The pooled libraries were sequenced using MiSeq® Reagent Nano Kit v2 (500 cycles) (Illumina) on an Illumina MiSeq Sequencing platform following the manufacturer’s instructions.
Bioinformatic analysis
All raw sequencing reads have been made publicly available on the NCBI Sequence Read Archive under BioProject accession number PRJNA437674. Sequencing data were analyzed using a custom bioinformatic workflow. This workflow was adapted from open-source resources (Callahan et al. 2016; Lee 2019) and is available at the following GitHub repository: https://github.com/Mathilg/UPDx_workflow.git. Briefly, after primer removal using Cutadapt V2.10, reads were processed using the Dada2 R package (R version 4.0) and RStudio (version 1.4.1106). Reads were filtered based on quality, where sequences were truncated at the first instance of a quality score less than 2, and after truncating, sequences with an overall quality score less than 15 or with more than 2 potential erroneous base calls were discarded. Trimmed reads were filtered by length, where only reads ranging from 145 to 250 bases long were retained. Reads were then de-replicated (with a parameter of 100% sequence identity), and Amplicon Sequence Variants (ASVs) were determined using the core sample inference algorithm of Dada2 (Callahan et al. 2016). ASVs were then merged, with a minimum overlap region of 150 bp and 100% sequence identity. Finally, likely chimeras were identified and removed using the Dada2 chimera removal feature. Taxonomic assignment of the final sequences was performed by nucleotide similarity search using BLASTN.
To determine a “positivity” threshold (the number of parasite-matching reads required to validate the presence of that parasite in the sample), we utilized the same cutoff system as for the original UPDx method (Flaherty et al. 2018). Briefly, for a sample to be considered positive for any parasite taxon, the number of reads matching the 18S rDNA of this taxon must exceed either a minimum threshold of 20 reads or a dynamic threshold — whichever value was largest. The dynamic cutoff was computed based on the proportion of parasite-derived reads detected in the negative control samples sequenced in the same library due to index crosstalk, which is an artifact introduced during sample multiplexing (Flaherty et al. 2018). Computing the percentage of parasite-matching reads in a given sample was conducted in R (version 4.0) and RStudio (version 1.4.1106), where the number of reads matching any given parasite sequence was divided by the total number of reads obtained for that sample after cleaning and merging of the sequence data, multiplied by 100.
Results
Detection of various parasite taxa in blood
The diversity of parasite species detected using Ad_UPDx and nUPDx was the same based on the present comparison (Table 3, Fig. 2). The taxonomic assignment of sequences identified in blood samples and the proportion of parasite-derived reads obtained for each method is shown in Table 3 and Fig. 2.

Percentage of reads matching the target among parasite-positive samples processed with Ad_UPDx and nUPDx methods. After taxonomic assignments of the reads, for each sample infected with at least one parasite, we determined the percentage of reads matching the expected target using nUPDx (blue) or Ad_UPDx (red) methods. This percentage was computed as the number of reads matching any given parasite sequence divided by the total number of reads obtained for that sample after cleaning and merging the sequence data, multiplied by 100. Only one target has been detected in samples containing mixed infections of malaria species and is represented with a star (*)
Reads identical to a T. cruzi reference sequence from GenBank (KX007998.1) (Table 3) were detected in three of the four T. cruzi-positive samples using both nUPDx and Ad_UPDx. Similarly, reads identical to an 18S sequence from Babesia microti (KY649348.1) were detected in the B. microti-positive sample via both methods. For the B. divergens-positive blood sample, we detected reads possessing 100% identity to several Babesia species at the amplified region, including B. odocoilei, B. capreoli, B. divergens, B. venatorum, and Babesia sp. MO1 (KY805843.1, KY805834.1, MG344781.1, MG344777.1, AY048113.1) (Table 3). Reads possessing 100% identity to the expected 18S target for several members of the Onchocercidae family of nematodes and the Filarioidea superfamily of nematodes were detected in samples positive for Loa loa and Brugia malayi, respectively (Table 3), via both methods. For the Leishmania-positive blood sample, reads consistent with the presence of parasites from the Leishmaniinae subfamily were detected using both methods (Table 3).
Plasmodium malariae was detected using both methods, where reads identical to the expected amplicon for reference sequence KU510228.1 were observed. For blood samples containing either P. falciparum, P. vivax, or P. ovale, two different sequences were detected using Ad_UPDx. These sequences correspond to multiple paralogs of the 18S rDNA encoded in the genomes of these Plasmodium species (Steenkeste et al. 2009; Gruenberg et al. 2018). The nUPDx assay produced similar results, with the exception of the P. ovale sample, where nUPDx detected only one of the two 18S rDNA paralogs (Table 3).
For samples comprising naturally acquired mixed malaria species infections, only one of the two expected parasites was detected via both methods in all instances (Table 3). Only sequences corresponding to Plasmodium falciparum were detected in the Plasmodium falciparum/P. malariae and P. falciparum/P. vivax mixed samples. Only sequences corresponding to Plasmodium ovale were detected in the Plasmodium falciparum/P. ovale mixed sample (Table 3, Fig. 2).
One sample, Trypanosoma cruzi R2 (a sample collected from a chronic-phase Chagas disease patient and tested positive by real-time PCR), tested negative for parasites via both methods.
Assessing the limit of detection via serially diluted P. falciparum culture
We analyzed parasite-free human blood spiked with serial dilutions of cultured P. falciparum 3D7 parasites. No parasite-derived sequences were detected at dilutions below 0.58 parasites/μL for both methods (Table 4, Fig. 3). Reads corresponding to the two 18S paralogs expected for P. falciparum were detected in most duplicates, except for duplicate dilutions of 0.58 parasites/μL tested via nUPDx, where only one paralog was detected for one duplicate and the other duplicate was negative for any parasite-derived sequences. Both duplicates of the 0.58 parasites/μL dilution tested via Ad_UPDx were positive, and both expected P. falciparum 18S paralogs were detected in each (Table 4, Fig. 3). Thus, although both methods displayed an LOD of 0.58 parasites/μL; results were more consistent for Ad_UPDx at lower dilutions.

Mean percentage of total reads matching the target among serial dilutions of Plasmodium falciparum 3D7 culture, processed in duplicates with both Ad_UPDx and nUPDx. After cleaning, merging and taxonomic assignments of the reads, the mean percentage of reads matching the expected target (Plasmodium falciparum) was determined for nUPDx (blue) or Ad_UPDx (red) methods by dividing the matching reads by the total number of reads obtained for that sample, multiplied by 100. Mean and standard deviation were computed for each duplicated sample and displayed as error bars
One of two replicates of the second-highest P. falciparum dilution (5800 parasites/μL) analyzed using Ad_UPDx failed to produce a parasite-derived sequence, although the other replicate of this dilution did. Each of the two replicates generated for the other dilutions between 58,000 parasites/μL and 0.58 parasites/μL was positive using Ad_UPDx, suggestive of a technical error introduced during library preparation for that specific replicate only (Table 4, Fig. 3).
General comparison of nUPDx and Ad_UPDx
From DNA extraction to data analysis, Ad-UPDx took up to 5 days to complete, compared to close to 7 days for the nested UPDx method. Regarding the cost of each method, when considering the reagents for all digests, PCRs, ethanol washes, and bead cleanups during library preparation, Ad_UPDx was substantially cheaper at $11.24 per sample as compared to $40.40 for nUPDx. The cost of library preparation for nUPDx accounted for the bulk of the price difference ($29.79 per sample). These calculations assumed multiplexing of 80 samples within the same library. An overview of the two methods compared in this study is presented in Fig. 4.

Overview of the nUPDx method (Flaherty et al. 2021) and the newly described Ad_UPDx protocol. The comparison of the two approaches highlights the reduction of manipulation steps in Ad_UPDx, minimizing protocol complexity and leading to the reduction in turnaround time from 7 to 5 days and a cost decrease of analysis price per sample from $40.40 to $11.08
While both assays possessed a similar LOD, the proportion of total parasite-derived reads (for both paralogs) was higher for all replicates using Ad_UPDx compared to nUPDx (Table 4, Fig. 3). Furthermore, for clinical samples where the parasite load was expected to be low, e.g., the samples from patients with visceral leishmaniasis and chronic Chagas disease, Ad_UPDx generated higher percentages of parasite-derived reads, sometimes more than twice the proportions observed for nUPDx (Table 3, Fig. 2).
Discussion
We describe a new 18S metagenomic sequencing approach that simultaneously amplifies the 18S rRNA gene of eukaryotic pathogens while preparing the resultant amplicons for Miseq sequencing, similar to methods previously described for bacterial 16S metagenomics studies (Lee et al. 2019; Diaz-Torres et al. 2021). The introduction of an Illumina-compatible adapter and multiplexing index sequences to amplicons during PCR greatly reduced the complexity of preparing our pan-parasite TADS approach. The modifications to the original nUPDx method described here reduced turnaround times (from DNA extraction to generation of a result) by two days. Furthermore, the average cost per sample decreased from around $40 to $11, not considering the cost of human labor. Notably, relative to nUPDx, Ad_UPDx requires fewer “open-tube” steps and fewer reagents, reducing opportunities for contamination to occur.
The nUPDx assay included forward and reverse primers with priming sites approximately 1.5 kilobases (kb) apart, flanking the original ~ 200 bp UPDx amplicon. In addition to redesigning the UPDx primers for the generation of sequencing-ready amplicons, Ad-UPDx excluded this 1.5 kb amplification step by incorporating priming sites for the second PCR amplification during the first amplification. In this way, we retained the nested aspect of nUPDx while substantially reducing the size of the first-step amplicon.
As expected, Ad_UPDx and nUPDx were equally effective at detecting malaria species, Babesia species, kinetoplastids, and filarial nematodes in clinical blood samples. The LOD, as determined using samples spiked with cultured P. falciparum, was the same for the two methods. However, Ad_UPDx generated higher proportions of parasite-derived reads for samples with low amounts of parasites, such as the serially diluted samples with 0.58 parasites/μL and 5.8 parasites/μL, and some of the clinical samples. This indicates that Ad_UPDx is more robust compared to nUPDx for the detection of low parasitemia samples, likely because of a reduction in the size of the first PCR amplicon (0.2 kb compared to 1.5 kb), and other factors, such as the implementation of fewer steps for Ad_UPDx providing fewer opportunities for DNA loss.
As described above, one of two replicates containing the second-highest P. falciparum dilution (5800 parasites/μL) failed to produce a positive result using Ad_UPDx. Given that the other replicate at this dilution returned a strong positive result and that all subsequent dilutions down to 0.58 parasites/μL returned a positive (and with greater coverage than nUPDx for the matching dilutions), we attribute this to human error during assay preparation.
Both Ad-UPDx and nUPDx detected T. cruzi in acutely infected Chagas disease patients but not in chronically infected. The acute phase of Chagas disease lasts from 6 days to approximately 2 months (Barratt et al. 2010), as T. cruzi trypomastigotes migrate through the blood and lymph, remaining detectable via PCR. However, during the chronic phase of infection, T. cruzi becomes largely intracellular as it invades host cells to become amastigotes that possess a tropism for cardiac myocytes. During the intracellular chronic phase of Chagas disease, T. cruzi DNA exists at lower concentrations in the blood and lymph, making PCR detection difficult. The two samples from chronically infected patients included in this study had been confirmed positive for T. cruzi by real-time PCR assays targeting multicopy molecular targets commonly used for diagnostic assays (Qvarnstrom et al. 2012). The lower copy number of the 18S rRNA gene in T. cruzi, compared to these other targets (i.e., the kinetoplast minicircles and minichromosomal repeats) (Gonzalez et al. 1984; Sturm et al. 1989; Hernandez et al. 1993), may account for the failure of Ad_UPDx and nUPDx to detect T. cruzi DNA in the blood of chronically infected patients in this study.
While Ad_UPDx and nUPDx each differentiated between all human-infecting Plasmodium species, taxonomic resolution was achieved only at the genus or family level for Babesia spp., filarial nematodes, and Leishmania spp. The 18S rRNA genes are highly conserved across parasite taxa, making it difficult to distinguish all parasites at the species level using our short ~ 200 base pair UPDx amplicon. Future iterations of the UPDx method could incorporate additional markers with higher diversity to improve taxonomic resolution. However, the detection of parasite taxa at the family or genus level still provides valuable information that may subsequently guide diagnostic efforts and patient management, especially in cases of atypical disease presentation. Ad_UPDx might also be a good asset in other situations where a broadly reactive, agnostic detection method is warranted, for example, to screen donated blood samples prior to transfusion.
Disappointingly, mixed Plasmodium species infections were not detected in this study using nUPDx or Ad_UPDx, in contrast to the success in doing so in previous evaluations of UPDx (Flaherty et al. 2018, 2021). Mixed Plasmodium species infections are typically dominated by one species that exists at a much higher parasitemia than observed for the lesser species within a natural mixed infection (McKenzie and Bossert 1997). Thus, PCR amplification greatly favors the dominant species, resulting in an abundance of reads derived from that species, rendering the minor species either difficult to detect or un-detectable. Evaluation of samples from a variety of mixed species infections is currently underway to clarify the ability of Ad_UPDx to detect the minor species in these situations.
Conclusion
We describe a metagenomic sequencing approach combining nested amplification and restriction-enzyme-based enrichment of parasite-derived 18S rDNA PCR product, which results in amplicons that are immediately ready for Miseq sequencing. This procedure avoids the need for expensive and laborious library preparation, saving time and money, and reduces opportunities for contamination by decreasing the number of required steps and reagents. This improved 18S metagenomic sequencing method (named Ad_UPDx) detected and differentiated the same diversity of parasite taxa as earlier iterations of UPDx but performed more consistently at the lower detection limit of the assay and generated a greater proportion of parasite-derived reads generally.
Data availability
Raw reads analyzed in this study have been made publicly available by submission to NCBI Sequence Read Archive and can be accessed under BioProject accession number: PRJNA437674. BioSamples submitted to this BioProject that are relevant to the present study include the term “Ad_UPDx vs nUPDx” in their sample name..
References
Anonymous (2013) 16S Metagenomic Sequencing Library Preparation - Part # 15044223 Rev. B. https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf. Accessed 2022
Barratt JL, Harkness J, Marriott D, Ellis JT, Stark D (2010) Importance of nonenteric protozoan infections in immunocompromised people. Clin Microbiol Rev 23(4):795–836. https://doi.org/10.1128/CMR.00001-10
Bishop A, Wang HH, Grant WE (2021) Using data surveillance to understand the rising incidence of babesiosis in the United States, 2011–2018. Vector Borne Zoonotic Dis 21(5):391–395. https://doi.org/10.1089/vbz.2020.2754
Bonnet S, Jouglin M, Malandrin L, Becker C, Agoulon A, L’Hostis M et al (2007) Transstadial and transovarial persistence of Babesia divergens DNA in Ixodes ricinus ticks fed on infected blood in a new skin-feeding technique. Parasitology 134(Pt 2):197–207. https://doi.org/10.1017/S0031182006001545
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13(7):581–583. https://doi.org/10.1038/nmeth.3869
Clemons B, Barratt J, Lane M, Qvarnstrom Y, Teal AE, Zayas G et al (2022) Assessing an adaptation of the universal parasite diagnostic assay for bloodborne parasites in a US state public health laboratory. Am J Trop Med Hyg 106(2):671–677. https://doi.org/10.4269/ajtmh.21-0707
de Almeida ME, Steurer FJ, Koru O, Herwaldt BL, Pieniazek NJ, da Silva AJ (2011) Identification of Leishmania spp. by molecular amplification and DNA sequencing analysis of a fragment of rRNA internal transcribed spacer 2. J Clin Microbiol 49(9):3143–9. https://doi.org/10.1128/JCM.01177-11
Diaz-Torres O, de Anda J, Lugo-Melchor OY, Pacheco A, Orozco-Nunnelly DA, Shear H et al (2021) Rapid changes in the phytoplankton community of a subtropical, shallow, hypereutrophic lake during the rainy season. Front Microbiol 12:617151. https://doi.org/10.3389/fmicb.2021.617151
Dye-Braumuller KC, Kanyangarara M (2021) Malaria in the USA: how vulnerable are we to future outbreaks? Curr Trop Med Rep 8(1):43–51. https://doi.org/10.1007/s40475-020-00224-z
Flaherty BR, Talundzic E, Barratt J, Kines KJ, Olsen C, Lane M et al (2018) Restriction enzyme digestion of host DNA enhances universal detection of parasitic pathogens in blood via targeted amplicon deep sequencing. Microbiome 6(1):164. https://doi.org/10.1186/s40168-018-0540-2
Flaherty BR, Barratt J, Lane M, Talundzic E, Bradbury RS (2021) Sensitive universal detection of blood parasites by selective pathogen-DNA enrichment and deep amplicon sequencing. Microbiome 9(1):1. https://doi.org/10.1186/s40168-020-00939-1
Gonzalez A, Prediger E, Huecas ME, Nogueira N, Lizardi PM (1984) Minichromosomal repetitive DNA in Trypanosoma cruzi: its use in a high-sensitivity parasite detection assay. Proc Natl Acad Sci U S A 81(11):3356–3360
Gruenberg M, Moniz CA, Hofmann NE, Wampfler R, Koepfli C, Mueller I et al (2018) Plasmodium vivax molecular diagnostics in community surveys: pitfalls and solutions. Malar J 17(1):55. https://doi.org/10.1186/s12936-018-2201-0
Hernandez R, Martinez-Calvillo S, Hernandez-Rivas R, Gomez E (1993) Trypanosoma cruzi ribosomal RNA genes: a review. Biol Res 26(1–2):109–114
Hess JF, Kohl TA, Kotrova M, Ronsch K, Paprotka T, Mohr V et al (2020) Library preparation for next generation sequencing: a review of automation strategies. Biotechnol Adv 41:107537. https://doi.org/10.1016/j.biotechadv.2020.107537
Hwang J, McClintock S, Kachur SP, Slutsker L, Arguin P (2009) Comparison of national malaria surveillance system with the national notifiable diseases surveillance system in the United States. J Public Health Manag Pract 15(4):345–351. https://doi.org/10.1097/PHH.0b013e31819d816a
Lee SY, Mac Aogain M, Fam KD, Chia KL, Binte Mohamed Ali NA, Yap MMC et al (2019) Airway microbiome composition correlates with lung function and arterial stiffness in an age-dependent manner. PLoS One. 14(11):e0225636. https://doi.org/10.1371/journal.pone.0225636
Lee M (2019) Happy Belly Bioinformatics: an open-source resource dedicated to helping biologists utilize bioinformatics. J Open Source Educ 2(19). https://doi.org/10.21105/jose.00053
Mace KE, Lucchi NW, Tan KR (2021) Malaria surveillance - United States, 2017. MMWR Surveill Summ 70(2):1–35. https://doi.org/10.15585/mmwr.ss7002a1
McKenzie FE, Bossert WH (1997) Mixed-species plasmodium infections of Anopheles (Diptera:Culicidae). J Med Entomol 34(4):417–425. https://doi.org/10.1093/jmedent/34.4.417
Menis M, Whitaker BI, Wernecke M, Jiao Y, Eder A, Kumar S et al (2021) Babesiosis occurrence among United States medicare beneficiaries, ages 65 and older, during 2006–2017: overall and by state and county of residence. Open Forum Infect Dis 8(2):608. https://doi.org/10.1093/ofid/ofaa608
Qvarnstrom Y, Schijman AG, Veron V, Aznar C, Steurer F, da Silva AJ (2012) Sensitive and specific detection of Trypanosoma cruzi DNA in clinical specimens using a multi-target real-time PCR approach. PLoS Negl Trop Dis 6(7):e1689. https://doi.org/10.1371/journal.pntd.0001689
Rougemont M, Van Saanen M, Sahli R, Hinrikson HP, Bille J, Jaton K (2004) Detection of four Plasmodium species in blood from humans by 18S rRNA gene subunit-based and species-specific real-time PCR assays. J Clin Microbiol 42(12):5636–5643. https://doi.org/10.1128/JCM.42.12.5636-5643.2004
Snounou G, Viriyakosol S, Zhu XP, Jarra W, Pinheiro L, do Rosario VE et al (1993) High sensitivity of detection of human malaria parasites by the use of nested polymerase chain reaction. Mol Biochem Parasitol 61(2):315–20. https://doi.org/10.1016/0166-6851(93)90077-b
Souza SS, Bishop HS, Sprinkle P, Qvarnstrom Y (2016) Comparison of babesia microti real-time polymerase chain reaction assays for confirmatory diagnosis of babesiosis. Am J Trop Med Hyg 95(6):1413–1416. https://doi.org/10.4269/ajtmh.16-0406
Steenkeste N, Incardona S, Chy S, Duval L, Ekala MT, Lim P et al (2009) Towards high-throughput molecular detection of Plasmodium: new approaches and molecular markers. Malar J 8:86. https://doi.org/10.1186/1475-2875-8-86
Sturm NR, Degrave W, Morel C, Simpson L (1989) Sensitive detection and schizodeme classification of Trypanosoma cruzi cells by amplification of kinetoplast minicircle DNA sequences: use in diagnosis of Chagas’ disease. Mol Biochem Parasitol 33(3):205–214. https://doi.org/10.1016/0166-6851(89)90082-0
Acknowledgements
The authors thank Brianna Flaherty, former ORISE fellow at CDC, Stella Chenet and Maria Isabel Jercic from the Institute of Public Health, Chile, and Andrew Moorhead from the University of Georgia for providing samples. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Funding
This study was made possible by support from the Centers for Disease Control and Prevention Advanced Molecular Detection (AMD) Initiative.
Author information
Authors and Affiliations
Contributions
MG optimized the experimental design, performed test experiments, analyzed the data, generated the figures, and drafted the manuscript. ML performed LOD, DNA library preparations, and Illumina sequencing experiments. JB assisted with the conception of the method, data analysis, and editing of the manuscript. ET assisted with the conception of the method and experimental design. YQ supervised the study and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Ethics approval for the use of anonymized or de-identified blood samples as exempt human research was granted by the Centers for Disease Control and Prevention Human Research Protection Office (protocol #6756).
Consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Handling Editor: Una Ryan
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gondard, M., Lane, M., Barratt, J. et al. Simultaneous targeted amplicon deep sequencing and library preparation for a time and cost-effective universal parasite diagnostic sequencing approach. Parasitol Res 122, 3243–3256 (2023). https://doi.org/10.1007/s00436-023-07991-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-023-07991-4