
Abstract
During amoebiasis, colonization of the gut by Entamoeba histolytica can lead to alterations of the host microbiota. In this study, we have compared the gut microbiota of patients of amoebiasis with healthy controls using 16S rRNA gene variable regions, (V1-V3) and (V3-V5), of the bacterial genome. From this 16S rRNA gene amplicon data, one paired-end and two single-end datasets were selected and compared by the number of OTUs obtained, sequence count, and diversity analysis. Our results showed that the V1-V3-paired-end dataset gave the maximum number of OTUs in comparison to the two single-end datasets studied. The amoebiasis samples showed a significant drop in richness in the alpha diversity measurements and lower intra group similarity compared to the healthy controls. Bacteria of genus Prevotella, Sutterella, and Collinsella were more abundant in healthy controls whereas Escherichia, Klebsiella, and Ruminococcus were more abundant in the E. histolytica-positive patients. All the healthy controls harbored bacteria belonging to Faecalibacterium, Prevotella, Ruminococcus, Subdoligranulum, and Escherichia genera while all the E. histolytica-positive patient samples contained genus Enterobacter. The compositional changes in the gut microbiome observed in our study indicated a higher prevalence of pathogenic bacteria along with a depletion of beneficial bacteria in E. histolytica-infected individuals when compared with healthy controls. These results underline the interplay between E. histolytica and the human gut microbiome, giving important inputs for future studies and treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amoebiasis is caused by infection with the protozoan parasite Entamoeba histolytica. Around 50 million people over the world each year are infected with amoebiasis, and this disease causes around 40–100 thousand deaths each year (Haque et al. 2003). In 2015, it was the fourth leading cause of death from parasitic diseases and was responsible for 67,900 deaths worldwide (Wang et al. 2016). During amoebic infection, the parasite invades the human intestine. Initial infection with E. histolytica occurs after ingestion of cysts from fecally contaminated water or food. The cysts form trophozoites after excystation in the lumen of the small intestine. Extra intestinal amoebiasis occurs when the amoeba invades the intestinal mucosa and travels to the bloodstream (Haque et al. 2003, Burgess and Petri 2016). This manifests as amoebic liver abscess (ALA) and in rare cases causes infections of the brain.
The initial interaction of the amoeba with the host occurs with the indigenous gut microbiota present in the lumen of the large intestine (Moonah et al. 2013, Marie and Petri 2014). The human microbiota including the gut flora of the human gastrointestinal tract is a complex community of bacteria composed of at least several hundred species. The bacterial cells outnumber the eukaryotic cells in the human body. They share a symbiotic relationship with each other and can influence the physiology and disease progression (Burgess and Petri 2016, Sassone-Corsi and Raffatellu 2015). The gut microbiota determines Entamoeba virulence and colonization of the large intestine and the immune response of the human host during amoebiasis. The bacterial microbiota is an important influencing factor in the clinical outcome of the E. histolytica infections.
A recent study by Reyna-Fabián et al. (2016) has suggested that the host intestinal microbiota may influence the parasite during amoebic liver abscesses (ALA). Using 16S rRNA gene sequencing, they identified bacterial populations in ALA and pyogenic liver abscesses, suggesting that the ALA are coinfected with non-culturable bacteria from the intestinal microbiota as well as potentially pathogenic bacteria. The study concludes that extraintestinal infection by Entamoeba is accompanied by translocation of different ratios of intestinal bacteria to the liver. However, no specific bacteria are associated with any species or genotype of Entamoeba histolytica or Entamoeba dispar. In ALA, the Entamoeba-bacteria interaction seems to be entirely nonspecific. The bacterial groups associated were Klebsiella, Streptococcus, Prevotella, Fusobacterium, Sediminibacterium, Bacteroidetes, Pseudomonas, and Ralstonia.
A study from this laboratory also found coinfections with Bacteroidetes and Peptostreptococcus in ALA pus samples and observed a statistically significant reduction in the population of Lactobacillus in patients with amoebic colitis and ALA (Rani et al. 2006). Another study from this laboratory using qPCR technique has reported a significant decrease in the population of Bacteroidetes, Clostridium subgroups coccoides and leptum, Lactobacillus, Campylobacter, and Eubacterium from the gut bacterial microbiota during Entamoeba infections (Verma et al. 2012). As early as 1966, a study reported that amoebic lesions developed rapidly in the presence of Bacillus subtilis, Clostridium perfringens, Staphylococcus aureus, Streptococcus faecalis, and Lactobacillus acidophilus (Phillips and Gorstein 1966). In case of other parasitic infections like Giardiasis (Toro-Londono et al. 2019), a major change in the microbial population of the gut from a Bacteroidetes-enriched to Prevotella-enriched flora was observed.
This study attempts to map the alterations in the gut microbiota during amoebic infections and to identify specific bacterial genera that undergo a change in patients infected with E. histolytica. To this end, we collected 29 stool samples from a healthy population as controls (referred to as “[healthy controls]”) and 14 stool samples of patients testing positive for E. histolytica (referred to as “[Entamoeba positive patients]”) after procuring necessary ethical clearances. We have targeted 16 S rRNA (V1-V3) and (V3-V5) variable regions of bacteria for amplification to compare the bacterial population between the [healthy control] and [Entamoeba positive patients] groups. Paired-end and single-end datasets from these variable regions were studied using statistical parameters, such as abundance and diversity measures. The region giving the most robust data was identified and used subsequently for comparison between the [healthy controls] and the [Entamoeba-positive patients] groups.
Materials and methods
Sample collection
Whole stool samples were collected from healthy volunteers of a community not suffering from enteric diseases for use as controls after taking their informed consent. It was ensured that all the inclusion criteria were met and none of the subjects fell in the exclusion group. Inclusion criteria were laid down as follows: subjects selected were above 15 years of age, did not consume any antibiotic or anti-amoebic drugs or probiotics for the past one month, and were willing to participate in the study. Exclusion criteria being subjects under 15 years of age, without clear medical history, treated with an antibiotic or anti-amoebic or probiotic in the past month, or not willing to participate in study.
During the study period, around 400 stool samples were collected from patients with chronic/acute diarrhea as diagnosed by microbiologists and gastroenterologists from the Safdarjung Hospital, New Delhi (India). The sample size was calculated keeping in mind the current prevalence rate of Entamoeba infection (8–15%). The inclusion criteria of the patients were as follows: subjects with clinical diagnosis of amoebic colitis in addition to meeting the inclusion criteria for controls and not falling in the exclusions criteria (Ethical clearance for the study was obtained from both the participating institutes namely, JNU (IERB No. 2015/Faculty/82) and Safdarjung Hospital (No. IEC/SJH/VMMC/Project/September/2014/535)).
The stool samples were collected in standard sterile stool collection vials, transported in ice, and stored at − 80 °C till processed. All the controls and patients were required to fill out a patient information form, which included details of their age, sex, and diet.
DNA isolation
DNA was isolated from stool samples using QIAamp DNA Stool Mini Kit following the manufacturer’s protocol. Before the DNA was isolated, the frozen samples were thawed in an ice bath before extraction. From each sample collected, 200 mg (± 5) was weighed out carefully for DNA isolation. A bead beating step was added to the manufacturer’s protocol to ensure better DNA isolation. Shortly, 200 mg (± 5) of stool sample was added to a sterile 2 ml microfuge tube, and to it, around 50 garnet beads of 1 mm diameter were added along with 1 ml Inhibitex buffer (from QIAamp DNA Stool Mini kit). The bead beating step was performed at 58 cycles per second for half a minute followed by a minute’s rest using a mini bead beater. This beading step was repeated three times until the stool sample was completely homogenized. After this, the rest of the steps were performed following the manufacturer’s protocol (Qiagen). The DNA isolated was then stored at − 20 °C.
The next step involved identification of the samples carrying the parasite. This was conducted by subjecting the DNA sample for PCR amplification using E. histolytica specific primers (Srivastava et al. 2005) shown in Table 1. This set of primers targeted mobile SINE elements (short interspersed nuclear elements), non-LTR retrotransposons, which are present in many copies in E. histolytica. EhSINE1, one such SINE element of which 500 copies are present in E. histolytica, was used to design a primer specific for E. histolytica. Corresponding copies of this element are absent in Entamoeba dispar (a morphologically similar intestinal commensal which is non-pathogenic and non-invasive). The PCR was carried out in a thermocycler, with the following conditions: initial denaturation was 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 1 min, and elongation at 72 °C for 30 s. Final elongation was at 72 °C for 7 min.
Online resource 1 shows details of the 29 stool samples from a healthy population [healthy controls] and the 14 stool samples of patients testing positive for E. histolytica [Entamoeba positive patients].
16S rRNA gene amplicon sequencing
Amplification and sequencing of V1-V5 regions (in two parts, namely, V1-V3 and V3-V5) of the 16S rRNA gene from the stool DNA of controls and patient samples positive for Entamoeba histolytica infections were done. The universal bacterial primers were used for PCR amplification (Lozupone et al. 2013). The PCR reaction volume of 25 μl was set up using NEB Taq polymerase. PCR conditions were as follows: initial denaturation at 95 °C for 30 s, followed by 30 cycles of 95 °C for 30 s; annealing at specific temperatures for 45 s; and 68 °C for 30 s. The final extension temperature was 72 °C for 5 min. Primer compositions and annealing temperatures used were as reported in Iyer et al. 2019. The PCR products were purified using the Pure link kit and were used to build a library for sequencing on Illumina HiSeq 2500 Rapid/MiSeq sequencing platforms (by AgriGenome Labs Pvt Ltd., Kochi, India). Libraries from each of the samples were tracked using a sequence barcode. Library quantification was done on Qubit 3.0. FastQ files were demultiplexed into total reads per sample. The reads were 250 base pair long and paired-end.
While merging the paired-end reads from the V3-V5 region, a significant number of reads remained unmerged due to lack of minimum overlap. This was attributed to the said region being longer than 500 nucleotides in majority of the gut bacteria (Johnson et al. 2019). Given this constraint, the data analyzed in this study were both paired-end and single-end sequence datasets from the V1-V3 region and only single-end sequences from V3-V5 (Table 2).
Amplicon-based analysis
This study was designed to get insights into compositional changes in gut microbiota of Entamoeba histolytica-positive patients with respect to the healthy control individuals. Short read sequences obtained from the NGS platform were analyzed by using the QIIME 2–2019.10 pipeline (Bolyen et al. 2019). The read quality of raw data was checked by FastQC (Andrews 2010) and MultiQC tool (Ewels et al., 2016). Adapter contents from reads were removed by Trimmomatic (Bolger et al. 2014). Raw sequences of all three datasets were imported into QIIME by Fastq manifest format. DADA2 plugin was used for denoising and feature table/operational taxonomic unit (OTU) table construction. Greengenes database was used as a reference to assign taxonomy to each representative sequence (DeSantis et al. 2006). For the V1-V3-PE dataset, reference sub-sequences were extracted using the forward and reverse primers listed in Table 3 and with minimum and maximum amplicon size of 400 and 490 bases respectively. Different primer sequences were used for the V3-SE dataset with minimum and maximum amplicon size of 400 and 700 bases, respectively. Classifier was trained with the extracted sequences from the database and was used to classify the OTUs. The classifier of the V1-V3-PE dataset was used for the classification of V1-SE dataset, as this region lies within the V1-V3-PE region.
Alpha rarefaction plots and various alpha and beta diversity metrics were computed for each dataset using the QIIME2 at dataset-specific sampling depth (19,000, 108,000, and 99,100 for V1-V3-PE, V1-SE, and V3-SE, respectively). The values for sampling depth were decided based on the distribution of sequence count of the processed dataset, such that we retain the maximum number of samples for analysis. Alpha rarefaction plots were generated to see how the richness varies between [healthy controls] and [Entamoeba-positive patients] at increasing value of sampling depth. The intra-group similarities for healthy and patient groups were based on unweighted and weighted UniFrac distance matrix obtained from QIIME2. The non-redundant pairwise distances for each group in unweighted UniFrac distance matrix were statistically compared by Welch two sample t-test using the R-3.6.3 suite.
Differentially abundant genera between two groups were identified using both ANCOM (analysis of composition of microbiome) and DESeq2 (Love et al. 2014) with default parameters. Differentially abundant taxa with DESeq2 were determined after controlling the effect of covariates such as diet, age, and sex of the host. ANCOM uses W-score to report whether a species is differentially abundant. W-score is essentially the count of the number of sub-hypotheses that have passed for a given species/OTU. In the background, ANCOM runs a bunch of pairwise tests, wherein the sub-hypothesis (for the 2 class case) is structured as below:
where μi(1) is the ith species abundance, μr(1) is the rth species abundance in the population 1, μi(2) is the ith species abundance, and μr(2) is the rth species abundance in the population 2. The number of times H0i is rejected for the ith species is represented by W-score (Mandal et al. 2015).
Results
E. histolytica prevalence in New Delhi hospital samples
Out of the 400 patient stool samples collected from a hospital at New Delhi, 14 stool samples were PCR positive for E. histolytica and were used in the study (Online resource 1). Therefore, the percent prevalence of E. histolytica positive from New Delhi was 3.5%. The samples were collected from the patients before any treatment was started. Earlier, our laboratory had reported the prevalence of E. histolytica infections to be 9% (Srivastava et al. 2005). The age, diet habits, and sex of the 29 [healthy controls] and 14 [Entamoeba-positive patient] samples used in the study are represented graphically in Fig. 1.

Graphical summary of metadata. Sex, diet, and age information of all samples used in the study. The labels “Non-veg” and “Veg” represent non-vegetarian and vegetarian diets, respectively
OTU prediction in paired-end and single-end datasets
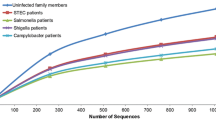
Bacterial populations were identified by 16S rRNA gene-based pipeline in both sets—control set consisting of 29 healthy controls and the test set consisting of 14 E. histolytica-positive samples. The paired-end reads from the V3-V5 region did not yield enough merged sequences on processing; therefore, the forward (single-end) reads were considered for analysis for V3-V5 data (see the “Materials and methods” section for details on dataset selection). The median number of processed sequences for the V1-V3 paired-end was ~ 92,000, and for the single-end (SE) datasets were ~ 310,000 and ~ 365,000 for V1-V3 and V3-V5, respectively (Fig. 2). These processed sequence sets are henceforth labeled as V1-V3-PE, V1-SE, and V3-SE, respectively as shown in Table 2. The average raw reads per sample were higher in our data as compared to similar studies and processed reads per sample were comparable (Milani et al. 2016; Morton et al. 2015; Dhakan et al. 2019; Liuet al. 2021) as shown in (Online resource 4).

Distribution of the number of raw and processed sequence data. The first and fourth box plots show the distribution of the number of raw reads in forty-three samples generated from the V1-V3 and V3-V5 regions, respectively. The distribution of the number of processed sequences is shown in the second, third, and fifth box plots for paired-end V1-V3, single-end V1-V3, and single-end V3-V5, respectively
The processed sequences of each dataset were clustered into OTUs. The OTUs in the V1-V3-PE set were higher than in the two single-end sets (Fig. 3a). This could be attributed to longer sequences having more unique or taxa-specific nucleotides. To examine the overlap among these three sets, the taxonomy of OTUs at the genus level was compared. About 58 genera were found to be common in all three datasets. Though genera in paired-end and single-end datasets from the same region of 16 s rRNA (V1-V3) were largely common, 5 and 6% of genera were uniquely present in V1-V3-PE and V1-SE, respectively. The comparison of genera between single-end datasets from different regions (V1-V3 and V3-V5) showed 48% overlap (Fig. 3b, Online resources 5).

a Number of operational taxonomic units (OTUs) observed in three processed sequence datasets. b Venn diagram showing the overlap in terms of the genus in three datasets
Abundance and prevalence of taxa
Taxonomic profiling at phylum level showed the higher relative frequency of four phyla, namely, Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria, compared to the rest as shown in Fig. 4 and Online resource 6. There were no phyla exclusive to any group. Profiles for lower taxonomic levels are shown in the Online resource 7, 8, 9, 10, and 11a.

Relative frequency of bacterial phyla present in the V1-V3-PE dataset. Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria were the most abundant phyla
We next examined the dominant genera in both groups. Faecalibacterium, Prevotella, Sutterella, Clostridiaceae 02d06, Subdoligranulum, Collinsella, and Bifidobacterium are some of the abundant genera in the [healthy controls] (Fig. 5a), whereas Faecalibacterium, Escherichia, Ruminococcus, Klebsiella, Enterobacter, and [Eubacterium] are the abundant ones in [Entamoeba-positive patients] (Fig. 5b). Group-wise average abundance of genera showed that Sutterella and Prevotella are more abundant in the [healthy controls] while Escherichia and Klebsiella are more abundant in the [Entamoeba-positive patients] (Fig. 6).

The top few abundant genera in [healthy controls] and [Entamoeba-positive patients]. a The seven most abundant genera in [healthy controls]. Median values of relative abundance of the genera in [healthy controls] and [Entamoeba-positive patients] are 11.02 and 9.03 for Faecalibacterium, 2.58 and 0.39 for Prevotella, 1.14 and 0.12 for Sutterella, 0.70 and 0.11 for Clostridiaceae 02d06, 0.51 and 0.24 for Subdoligranulum, 0.14 and 0.01 for Collinsella, and 0.14 and 0.07 for Bifidobacterium. b The five most abundant genera in [Entamoeba-positive patients] compared to [healthy controls], (excluding those already listed in plot a). Median-relative abundance values for each genus in [healthy controls] and [Entamoeba-positive patients] are 0.41 and 1.39 for Escherichia, 0.41 and 1.20 for Ruminococcus, 0.09 and 0.73 for Klebsiella, 0.003 and 0.20 for Enterobacter, and 0.03 and 0.1 for [Eubacterium]

Percent-relative abundance of top 35 genera (the fraction of OTUs with unassigned taxonomy was not plotted)
Among the top few abundant bacterial species at group level, Collinsella aerofaciens is more abundant in the controls, and Escherichia coli is more abundant in the patients as seen in Online resource 11b.
Among the most prevalent genera in each group, Prevotella, Faecalibacterium, Ruminococcus, Subdoligranulum, Escherichia, Bifidobacterium, Clostridiaceae 02d06, Sutterella, Coprococcus, and Collinsella are in greater prevalence in [healthy controls] as compared to the [Entamoeba-positive patients], whereas genus Klebsiella and Enterobacter are found more in patients (Fig. 7). These results are largely consistent with the other two single-end datasets leaving a few exceptions (V1-SE shown in Online resource 12 and14 and V3-SE in Online resource 13 and 15). The V3-SE dataset displayed more unique genera compared to the other two datasets, and those were among the prevalent and abundant genera (Figs. 5 and 7 and in Online resource 12, 13, 14, and 15). The abundant unique genera in the [healthy controls] are Megaspheara, Lactobacillus, Butyricoccus, and Dialister, and in the [Entamoeba positive patients], Oscillospora, Bifidobacterium, and Veillonella.

The highly prevalent genera in [healthy controls] and [Entamoeba-positive patients]. The genera unassigned by QIIME2 were not considered
Diversity in [Entamoeba-positive patients]
Within sample, alpha diversity measurements (Chao1 and observed OTUs) indicate a significant drop in richness in [Entamoeba-positive patients] compared to the [healthy controls] in V1-V3-PE data (p value: 0.008) (Fig. 8a). Other measures of alpha diversity that take into account the richness or evenness or both (such as Faith’s phylogenetic diversity and Shannon entropy) showed a similar trend but are statistically insignificant with (p values 0.12 and 0.34, respectively shown in Online resource 16. The alpha diversity values of single-end datasets also had a similar profile except for the Shannon’s diversity index of V3-SE as shown in Online resource 17.

Within and between sample diversity (alpha and beta) analysis of V1-V3-PE dataset. a The Chao1 index, which is a measure of richness within samples, showed a clear difference between two groups (p value 0.00021). b Rarefaction plot for [healthy controls] and [Entamoeba-positive patients] groups. The alpha diversity was measured in terms of observed OTUs. c Principal coordinate analysis (PCoA) plots based on weighted UniFrac distance. The green spheres indicate [healthy controls], and the red indicates [Entamoeba-positive patients]. d Hierarchical clusters using weighted UniFrac distance metric
Rarefaction analysis for both groups showed the difference in richness of two groups at a sampling depth of as low as 5,000 sequences (Fig. 8b). With increase in depth up to 22,000, the patient group had almost converged for observed OTUs, whereas the metric was still increasing for the healthy group, which indicated that the healthy group had much higher richness. Similar pattern is also observed in the two single-end datasets, though the sampling depths are slightly different as shown in Online resource 18.
Furthermore, the microbiome composition/structure was compared between the groups by estimating beta diversity. Principal coordinate analysis (PCoA) plots show two sparse clusters but none of the clusters correspond to the groups of [Entamoeba-positive patients] and [healthy controls] (Fig. 8c). An alternate beta diversity metric, namely, hierarchical clustering, of weighted UniFrac distances corroborated the above pattern (Fig. 8d). We further examined if E. histolytica infection had affected the intra-group similarity and found that the intra-group similarities significantly declined in the [Entamoeba-positive patients] compared to [healthy controls] (p value: 1.2E − 05) shown in Online resource 19. The single-end datasets also show a similar trend in the between-sample beta diversity patterns as shown in Online resource 20.
Differential abundance analysis
The comparison of taxonomic profiles did not show samples organizing as distinct groups (of [healthy controls] and [Entamoeba-positive patients]), so we further examined if abundance of any of the taxa was affected by Entamoeba histolytica infection. Differential abundance analysis was performed using ANCOM, which is suited for compositional data (Mandal et al. 2015), and with DESeq2, where confounding factors of age, sex, and diet were controlled.
The ANCOM analysis using the V1-V3-PE data suggests that the three phyla, namely, Firmicutes, Actinobacteria, Bacteroidetes, and Proteobacteria, have lower abundance (with W-score of ≥ 5), in the [Entamoeba-positive patients] compared to the [healthy controls] shown in Fig. 9a and Online resource 21). The DESeq2 results also show a significant decrease in abundance of Firmicutes, Bacteroidetes, and Proteobacteria in the [Entamoeba-positive patients] as shown in Online resource 22 a.

Differential abundance of taxa at following levels: a phylum, b class, c order, d family, e genus, and f species. The results are obtained from the ANCOM tool of QIIME2
At the class level in the same data, the abundance of Alphaproteobacteria, Betaproteobacteria, Coriobacteria, and Clostridia are significantly decreased in the [Entamoeba-positive patients] compared to the [healthy controls] by ANCOM (Fig. 9b, Online resource 21). In the DESeq2 results, Alphaproteobacteria, Clostridia, Betaproteobacteria, Bacteroidia, and Actinobacteria were also found to have significantly less abundance in [Entamoeba-positive patients] as compared to [healthy controls] as shown in Online resource 22 b.
At the order level, the abundance of Burkholderiales is reduced significantly in [Entamoeba-positive patients] as per ANCOM results (Fig. 9c, Online resource 21). In the DESeq2 results, levels of Clostridiales, Burkholderiales, Bacteroidales, and Bifidobacteriales decreased significantly in the [Entamoeba-positive patients] as shown in Online resource 22 c.
At the family level, Prevotellaceae and Alcaligenaceae are significantly less abundant in the patients, in the ANCOM results as shown in Fig. 9d and Online resource 21. The DESeq2 analysis indicate Prevotellaceae, Ruminococcaceae, Alcaligenaceae, Clostridiaceae, Bifidobacteriaceae, and Peptostreptococcaceae to be significantly less abundant in the patient group as shown in Online resource 22 d.
Among the differentially abundant genera inferred by ANCOM, Prevotella, Subdoligranulum, and Sutterella display significantly lower abundance in the patients compared to the healthy samples in the ANCOM analysis as shown in Fig. 9e and Online resource 21. The DESeq2 analysis between the two groups at genus level shows Collinsella, Sutterella, Subdoligranulum, Faecalibacterium, Prevotella, Clostridium 02d06, Bifidobacterium, and Butyricoccus significantly decrease in the patient samples as shown in Online resource 22 e. Ruminococcus, Succinovibrio, Escherichia, and Klebsiella are more abundant in the [Entamoeba-positive patients] as shown in Fig. 6; however, the difference is not significant.
A few of the highly abundant species are Faecalibacterium prausnitzii, Escherichia coli, Collinsella aerofaciens, Prevotella copri, Subdoligranulum variabile, Eubacterium biforme, and Bifidobacterium adolescentis as seen in Online resource 11 b. ANCOM analysis shows the Subdoligranulum variabile is significantly less abundant in the [Entamoeba-positive patients] compared to [healthy controls] (Fig. 9f). In the DESeq2 results with healthy controls as a base, Faecalibacterium prausnitzii, Collinsella aerofaciens, Subdoligranulum variabile, Clostridium SMB53, and Butyricoccus pullicaecorum were found significantly less abundant in the [Entamoeba-positive patients] as compared to the [healthy controls] as shown in Online resource 22 f.
In the ANCOM analysis for V1-SE data, the results are largely similar to the ones described for V1-V3 PE data as shown in Online resource 23. In case of [healthy controls], Coriobacteria at the order level and Coriobacteriaceae at the family level were additionally observed to be more abundant while the class Gammaproteobacteria were more abundant in the [Entamoeba-positive patients]. At the genus level, in case of [healthy controls], an increase in relative abundance of genus Catenibacterium is additionally observed, and in case of [Entamoeba positive patients], increased relative abundance of Shigella and Succinovibrio as shown in Online resource 12. Similar to that observed for V1-V3-PE, the genus Eubacterium and Ruminococcus also display higher relative abundance in the [Entamoeba-positive patients] group in this dataset.
Differentially abundant taxa in V3-SE ANCOM data were phylum Bacteroidetes, classes Bacteroides and Clostridia, and order Bacteroidales being significantly more abundant in [healthy controls]. At the family level, Alcaligenaceae and, at the genus level, Sutterella were found to be significantly more abundant in [healthy controls] as shown in Online resource 24. Genus Faecalibacterium, Veillonella, Collinsella, and Bifidobacterium are observed to be more abundant in the [Entamoeba-positive patient] samples in the V3-SE dataset as shown in Online resource 13. The differences in the V3-SE ANCOM data from the other two ANCOM datasets could be due to sub regional bias in the identification of bacterial taxa as observed by Johnson et al. (2019). Online resource 25 summarizes the changes in bacterial diversity in the V1-V3-PE, V1-SE, and V3-SE datasets, in [Entamoeba-positive patients] when compared to [healthy controls] in the ANCOM analysis.
Discussion
Previous studies have examined associations of E. histolytica infection with the gut microbiome. Verma et al. (2012) found significant reduction in Bacteroides, two subgroups of Clostridium, Lactobacillus, Campylobacter, and increase in Bifidobacterium by absolute quantification using real-time PCR. Gilchrist et al. (2016) showed that expansion of Prevotella copri level was associated with symptomatic diarrhea due to E. histolytica infections in children aged 2 to 3 years in an urban slum in Dhaka, Bangladesh. Morton et al. (2015) identified ten most important bacterial taxa which showed changes in abundance during Entamoeba (both histolytica and dispar) infections and reported that colonization by Entamoeba can be predicted by an individual’s gut microbiota composition with 79% accuracy. These taxa included Prevotella copri. A recent study by Yanagawa et al. (2021) looked at the gut microbiome during asymptomatic and symptomatic Entamoeba histolytica infections among patients in Japan, by analyzing the V3-V4 region of the bacterial 16 S rRNA. Their analysis showed the asymptomatic cases to have more uniform gut microbiome as evidenced by beta diversity analysis. There was an increased proportion of Coriobacteriaceae, Ruminococcaceae, and Clostridiaceae, and Collinsella aerofaciens was significantly higher in the asymptomatic cases. Our study attempts to understand the effect of symptomatic Entamoeba histolytica infections on the gut microbiome by comparing it with that of healthy individuals.
Symptomatic Entamoeba infection perturbs the gut microbiome
Significant decline of alpha diversity in the [Entamoeba-positive patients] indicated perturbation in the gut microbial ecosystem. While this perturbation was highly significant in terms of reduction in richness, significant decline in evenness was also seen. This suggests that gut microbiome of [Entamoeba-positive patients] not only had relatively fewer microbes but also showed more variation in abundance across patient samples. On the other hand, the taxonomic profiles of [Entamoeba-positive patients] show a decline in intra-group similarity relative to [healthy controls], indicating higher variation in microbiome composition of patients. The absence of grouping of samples into their respective (patients and healthy) groups based on microbiome composition shows that the perturbation however did not affect a major fraction of the taxa, and alterations are possibly directional in only a few of the taxa. The individual taxonomic groups which showed perturbation (in terms of evenness) were few but belonged to all levels of classification. At genus level, Faecalibacterium, Prevotella, Sutterella, Subdoligranulum, Collinsella, and Bifidobacterium, significantly reduced in abundance among [Entamoeba-positive patients].
Reduction in abundance of two species of Prevotella, namely, P. copri and P. stercorea, was reported among rural African samples colonized with Entamoeba (Morton et al. 2015). Although we observed an overall reduction in abundance at genus level, the relative abundance of these two Prevotella species show more variation in [Entamoeba-positive patients], with a slight increase in the median value, though not significant as shown in Online resource 26. P. copri can exist as a normal component of the gut microbiome but is also referred to as a pathobiont and is associated with severe inflammation inrheumatoid arthritis (Hofer 2014). Moreover, a study among children with diarrhea in Bangladesh showed an increase in the levels of P. copri in patients with diarrheagenic Entamoeba infections (Gilchrist et al. 2016). Therefore, the present study is in consonance with previous studies indicating an increase in the P. copri levels in [Entamoeba-positive patients].
In our study, genera Subdoligranulum and Sutterella also display a lower relative abundance in the [Entamoeba-positive patients]. Several studies have associated Subdoligranulum with a healthy metabolic state (Leclercq et al. 2014; Qin et al. 2014; Holmstrøm et al. 2004; Vinolo et al. 2011; Van Immerseel et al. 2010; Kaakoush et al. 2012). However, Van Hul et al. (2020) in their study, showed that supplementation with S. variabile in diabetic mice did not have any significant beneficial effects and concluded that any correlation between a given bacteria and health as shown by metagenomic data require further validation with experiments. Our observations suggesting a loss or decline of Subdoligranulum genus during E. histolytica infections could indicate gut microbiome dysbiosis favoring diarrheal symptoms. However, this needs further validation.
Hiippala et al. (2016) have shown that members of genus Sutterella are widely prevalent commensals with mild pro-inflammatory capacity in the human gastrointestinal tract but their contribution to the dysregulation of epithelial homeostasis connected with the increase of Proteobacteria is insignificant. They suggest that the ability of Sutterella spp. to adhere to intestinal epithelial cells indicate an immunomodulatory role. However, the relationship between disease pathology and loss of abundance of Sutterella in [Entamoeba-positive patients] is not clear and needs to be further investigated.
Collinsella and Bifidobacterium are from the family Coriobacteriaceae. They have been shown as the major lactose utilizers among the human gastrointestinal microbiota with an in vitro model of the human colon. Collinsella species break down carbohydrates to produce acetate, formate, and lactate (Kovatcheva-Datchary 2010). Prominent lactate utilizing microbiota Firmicutes and Bacteroides produce short-chain fatty acids—butyrate or propionate from lactate (Duncan et al. 2004; Reichardt et al. 2014). Decrease in abundance of Collinsella and Bifidobacterium during amoebiasis may consequently reduce the positive effects of butyrate and propionate on the host (Wang et al. 2020). At the species level, our study found Collinsella aerofaciens to be significantly reduced in abundance in the patient samples. C. aerofaciens was also found to be significantly less abundant in symptomatic patients of amoebiasis in the study by Yanagawa et al. 2021. Butyrate-producing bacteria of genus Butyricoccus and species Butyricoccus Pullicaecorum were significantly less abundant in the [Entamoeba-positive patients]. These have been shown to reduce inflammatory pathogen load in the gut (Eeckhaut et al. 2016).
Apart from the above-mentioned genera, there are a few key genera that were found to be in greater abundance in the [Entamoeba-positive patients], though not statistically significant. These are Escherichia, Klebsiella, Succinovibrio, and Ruminococcus. Both Escherichia and Klebsiella belong to the class Gammaproteobacter, and earlier studies have shown that the abundance of bacteria from the class Gammaproteobacter was also shown to be associated with enterocolitis in new born infants (Toro-Londono et al. 2019). An earlier study from this lab found members of Bifidobacterium to be more abundant during E. histolytica infections (Verma et al. 2012). Members of both Bifidobacterium and Ruminococcus ferment mucin and use it as a source of energy. They were found to colonize mucin in an in vitro study where colonization of gastric mucin was examined, using fecal samples (Leitch et al. 2007). E. histolytica binds to and breaks down mucus with mucin degrading enzymes; however, it also potently increases mucus secretion by intestinal goblet cells during symptomatic E. histolytica infections (Leon-Coria et al. 2020). E. histolytica and the gut microbes mutually benefit from each other. E. histolytica derive nutrient source from breakdown of complex carbohydrate by the gut microbes and also feed on them, while gut microbes benefit from increased mucus secretion.
Gut microbiome response to symptomatic Entamoeba infection was different from that during asymptomatic colonization
Morton et al. (2015) had earlier examined the effect of Entamoeba infection on the gut microbiome, but unlike the current study where samples were from active diarrheal cases, their samples primarily had Entamoeba colonization without any symptoms. Interestingly, the response of the gut microbiome to these two infectious stages was largely different. For instance, the diversity within samples was significantly higher in the E. histolytica-positive samples, as well as the relative abundance of the majority of the phyla including the dominant Firmicutes. This was in clear contrast to the findings of the current study where a declining trend was observed for both. These findings indicate that the E. histolytica does alter the gut microbiome in both stages, but in the diarrheal stage of infection, it not only shows larger perturbation but also alters abundance of some of the probiotic bacteria as discussed above and favors the colonization by pathogenic bacteria as the discussion follows.
Perturbation of pathogenic bacteria and their effects in [Entamoeba-positive patients]
Pathogenic bacteria Staphylococcus aureus and Shigella dysenteriae were reported to increase the virulence of E. histolytica by altering the host’s immune response, while Escherichia coli did not elicit a similar response (Galván-Moroyoqui et al. 2008). Shaulov et al. (2018) have reported that E. coli encoded malate dehydrogenase and its product oxaloacetate contributes to E. histolytica’s resistance to oxidative stress in the lumen of the large intestine. A study by Mousa et al. (2021) showed genus Klebsiella to be associated with pyogenic liver abscess and was isolated in the pus/blood culture of patients. The prevalence of genera belonging to Klebsiella and Enterobacter was observed to increase in [Entamoeba-positive patients] in our V1-V3-PE analysis while the prevalence of genus Streptococcus was greater in the [Entamoeba-positive patients] in the V1-SE and V3-SE datasets. Yanagawa et al. (2021) also found a greater abundance of two species of Streptococcaceae family in patients with symptomatic Entamoeba histolytica infections. Yamashiro et al. (1996) had found abnormal colonization by Streptococcus and Staphylococcus species in children with Kawasaki syndrome, and Esposito et al. (2019) in a review article quoted this and other studies to show that imbalance in Streptococcus during Kawasaki syndrome may interfere with their innate and adaptive immunity. In our study, we see a higher prevalence of the pathogenic bacteria of genus Streptococcus in [Entamoeba-positive patients] which could lead to changes in immunity and contribute to the enhanced invasiveness.
Conclusion
The compositional changes in gut microbiota of patients infected with Entamoeba histolytica as compared to that of healthy controls was studied using 16S rRNA gene sequencing of the (V1-V5) region. The results indicated that among the three datasets studied [V1-V3-PE (paired-end), V1-SE (single-end), and V3-SE], the paired-end dataset gave the maximum number of OTUs, and 58 genera were found to be common between the three datasets. A significant decline in the microbial alpha diversity in terms of richness and evenness was seen in the patients. Differential abundance analysis showed a significant reduction in abundance of Faecalibacterium, Prevotella, Sutterella, Subdoligranulum, and Colinsella at the genus level in the patients indicating dysbiosis. Pathogenic bacteria of genus Enterobacter and Klebsiella as well as Streptococcus increased in abundance during Entamoeba histolytica infection and may contribute to the increased invasiveness of the parasite by causing decreased host immunity. Entamoeba histolytica infections thus caused changes in the gut microbiome with a decrease in the richness and evenness of gut microbial diversity, a decreased abundance of genera that are associated with probiotic and immunomodulatory roles, and simultaneous increase in abundance of pathogenic bacteria. This study provides a deep understanding of the interplay between the parasite E. histolytica and human gut microbiome and provides important inputs for future studies and the treatments.
Data availability
The data generated and analyzed in this study have been submitted in the NCBI Bio Project database under project number PRJNA608066 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA608066).
References
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 27 Apr 2021
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37(8):852–857. https://doi.org/10.1038/s41587-019-0209-9
Burgess SL, Petri WA (2016) The intestinal bacterial microbiome and E. histolytica infection. Curr Trop Med Rep 3(3):71–74. https://doi.org/10.1007/s40475-016-0083-1
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72(7):5069–5072. https://doi.org/10.1128/AEM.03006-05
Dhakan DB, Maji A, Sharma AK, Saxena R, Pulikkan J, Grace T, Gomez A, Scaria J, Amato KR, Sharma VK (2019) The unique composition of Indian gut microbiome, gene catalogue, and associated fecal metabolome deciphered using multi-omics approaches. GigaScience 8(3):giz004. https://doi.org/10.1093/gigascience/giz004
Duncan SH, Louis P, Flint HJ (2004) Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl Environ Microbiol 70(10):5810–5817. https://doi.org/10.1128/AEM.70.10.5810-5817.2004
Eeckhaut V, Wang J, Van Parys A, Haesebrouck F, Joossens M, Falony G, Raes J, Ducatelle R, Van Immerseel F (2016) The probiotic Butyricicoccus pullicaecorum reduces feed conversion and protects from potentially harmful intestinal microorganisms and necrotic Enteritis in broilers. Front Microbiol 7:1416. https://doi.org/10.3389/fmicb.2016.01416
Esposito S, Polinori I, Rigante D (2019) The gut microbiota-host partnership as a potential driver of Kawasaki syndrome. Front Pediatr 7:124. https://doi.org/10.3389/fped.2019.00124
Ewels P, Magnusson M, Lundin S, Käller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32(19):3047–3048. https://doi.org/10.1093/bioinformatics/btw354
Galván-Moroyoqui JM, Domínguez-Robles MDC, Franco E, Meza I (2008) The interplay between Entamoeba and enteropathogenic bacteria modulates epithelial cell damage. PLoS Negl Trop Dis 2(7):e266. https://doi.org/10.1371/journal.pntd.0000266
Gilchrist CA, Petri SE, Schneider BN, Reichman DJ, Jiang N, Begum S, Petri WA Jr (2016) Role of the gut microbiota of children in diarrhea due to the protozoan parasite Entamoeba histolytica. J Infect Dis 213(10):1579–1585. https://doi.org/10.1093/infdis/jiv772
Haque R, Huston CD, Hughes M, Houpt E, Petri WA Jr (2003) Amebiasis. N Engl J Med 348(16):1565–1573. https://doi.org/10.1056/NEJMra022710
Hiippala K, Kainulainen V, Kalliomäki M, Arkkila P, Satokari R (2016) Mucosal prevalence and interactions with the epithelium indicate commensalism of Sutterella spp. Front Microbiol 7:1706. https://doi.org/10.3389/fmicb.2016.01706
Hofer U (2014) Pro-inflammatory Prevotella? Nat Rev Microbiol 12(1):5–5. https://doi.org/10.1038/nrmicro3180
Holmstrøm K, Collins MD, Møller T, Falsen E, Lawson PA (2004) Subdoligranulum variabile gen. nov., sp. nov. from human feces. Anaerobe 10(3):197–203. https://doi.org/10.1016/j.anaerobe.2004.01.004
Iyer LR, Verma AK, Paul J, Bhattacharya A (2019) Phagocytosis of gut bacteria by Entamoeba histolytica. Front Cell Infect Microbiol 9:34. https://doi.org/10.3389/fcimb.2019.00034
Johnson JS, Spakowicz DJ, Hong BY, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M, Sodergren E, Weinstock GM (2019) Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Comm 10(1):1–11. https://doi.org/10.1038/s41467-019-13036-1
Kaakoush NO, Day AS, Huinao KD, Leach ST, Lemberg DA, Dowd SE, Mitchell HM (2012) Microbial dysbiosis in pediatric patients with Crohn’s disease. Jr Clin Microbiol 50(10):3258–3266. https://doi.org/10.1128/JCM.01396-12
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glockner FO (2013) Evaluation of the general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41(1):e1. https://doi.org/10.1093/nar/gks808
Kovatcheva-Datchary P (2010) Analyzing the functionality of the human intestinal microbiota by stable isotope probing. Wageningen University and Research
Leclercq S, Matamoros S, Cani PD, Neyrinck AM, Jamar F, Stärkel P, Windey K, Tremaroli V, Bäckhed F, Verbeke K, de Timary P, Delzenne NM (2014) Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc Nat Acad Sc 111(42):E4485–E4493. https://doi.org/10.1073/pnas.1415174111
Leitch ECM, Walker AW, Duncan SH, Holtrop G, Flint HJ (2007) Selective colonization of insoluble substrates by human faecal bacteria. Env Microbiol 9(3):667–679. https://doi.org/10.1111/j.1462-2920.2006.01186.x
Leon-Coria A, Kumar M, Chadee K (2020) The delicate balance between Entamoeba histolytica, mucus and microbiota. Gut Microbes 11(1):118–125. https://doi.org/10.1080/19490976.2019.1614363
Liu Y, Li X, Yang Y, Liu Y, Wang S, Ji B, Wei Y (2021) Exploring gut microbiota in patients with colorectal disease based on 16S rRNA gene amplicon and shallow metagenomic sequencing. Front MolBiosc 8.https://doi.org/10.3389/fmolb.2021.703638
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12):1–21. https://doi.org/10.1186/s13059-014-0550-8
Lozupone CA, Stombaugh J, Gonzalez A, Ackermann G, Wendel D, Vázquez-Baeza Y, Jansson JK, Gordon JI, Knight R (2013) Meta-analyses of studies of the human microbiota. Genome Res 23(10):1704–1714. https://doi.org/10.1101/gr.151803.112
Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD (2015) Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health and Dis 26(1):27663
Marie C, Petri WA Jr (2014) Regulation of virulence of Entamoeba histolytica. Ann Rev Microbiol 68:493. https://doi.org/10.1146/annurev-micro-091313-103550
Milani C, Ticinesi A, Gerritsen J, Nouvenne A, Lugli GA, Mancabelli L, Turronin F, Duranti S, Mangifesta M, Viappiani A, Ferrario C, Maggio M, Lauretani F, Vos WD, Sinderen DV, Meschi T, Ventura M (2016) Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Sci Rep 6(1):1–12. https://doi.org/10.1038/srep25945
Moonah SN, Jiang NM, Petri WA Jr (2013) Host immune response to intestinal amebiasis. PLoS Pathogens 9(8):e1003489. https://doi.org/10.1371/journal.ppat.1003489
Morton ER, Lynch J, Froment A, Lafosse S, Heyer E, Przeworski M, Blekhman R, Ségurel L (2015) Variation in rural African gut microbiota is strongly correlated with colonization by Entamoeba and subsistence. PLoS Genet 11(11):e1005658. https://doi.org/10.1371/journal.pgen.1005658
Mousa H, Al-Bluwi GSM, Drini A, Mohammed ZF, Gasmelseed HI, Alkoteesh JA, Babiker ZOE (2021) Importation of Entamoeba histolytica and predominance of Klebsiella pneumoniae in liver abscesses: a 7-year retrospective cohort study from the United Arab Emirates. Trop Dis Tr Med Vaccines 7(1):1–10. https://doi.org/10.1186/s40794-021-00140-8
Phillips BP, Gorstein F (1966) Effects of different species of bacteria on the pathology of enteric amebiasis in monocontaminated guinea pigs. Am Jr Trop Med Hyg 15(6, Pt. 1):863–8. https://doi.org/10.4269/ajtmh.1966.15.863
Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Chatelier EL, Yao J, Wu L, Zhou J, Ni S, Liu L, Pons N, Batto JM, Kennedy SP, Leonard P, Yuan C, Ding W, Chen Y, Hu X, Zheng B, Qian G, Xu W, Ehrlich SD, Zheng S, Li L (2014) Alterations of the human gut microbiome in liver cirrhosis. Nature 513:59–64. https://doi.org/10.1038/nature13568
Rani R, Murthy RS, Bhattacharya S, Ahuja V, Rizvi MA, Paul J (2006) Changes in bacterial profile during amebiasis: demonstration of anaerobic bacteria in ALA pus samples. Am Jr Trop Med Hyg 75(5):880–885
Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, Flint HJ, Louis P (2014) Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME Jr 8(6):1323–1335. https://doi.org/10.1038/ismej.2014.14
Reyna-Fabián ME, Zermeño V, Ximénez C, Flores J, Romero MF, Diaz D, Argueta J, Moran P, Valadez A, Cerritos R (2016) Analysis of the bacterial diversity in liver abscess: differences between pyogenic and amebic abscesses. Am Jr Trop Med Hyg 94(1):147. https://doi.org/10.4269/ajtmh.15-0458
Sassone-Corsi M, Raffatellu M (2015) No vacancy: how beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. Jr Immunol 194(9):4081–4087. https://doi.org/10.4049/jimmunol.1403169
Shaulov Y, Shimokawa C, Trebicz-Geffen M, Nagaraja S, Methling K, Lalk M, Weiss-Cerem L, Lamm AT, Hisaeda H, Ankri S (2018) Escherichia coli mediated resistance of Entamoeba histolytica to oxidative stress is triggered by oxaloacetate. PLoS Pathog 14(10):e1007295. https://doi.org/10.1371/journal.ppat.1007295
Srivastava S, Bhattacharya S, Paul J (2005) Species-and strain-specific probes derived from repetitive DNA for distinguishing Entamoeba histolytica and Entamoeba dispar. Exp Parasitol 110(3):303–308. https://doi.org/10.1016/j.exppara.2005.02.020
Toro-Londono MA, Bedoya-Urrego K, Garcia-Montoya GM, Galvan-Diaz AL, Alzate JF (2019) Intestinal parasitic infection alters bacterial gut microbiota in children. PeerJ 7:e6200. https://doi.org/10.7717/peerj.6200
Van Hul M, Le Roy T, Prifti E, Dao MC, Paquot A, Zucker JD, Delzenne NM, Muccioli GG, Clément K, Cani PD (2020) From correlation to causality: the case of Subdoligranulum. Gut Microbes 12(1):1849998. https://doi.org/10.1080/19490976.2020.1849998
Van Immerseel F, Ducatelle R, De Vos M, Boon N, Van De Wiele T, Verbeke K, Rutgeerts P, Sas P, Louis P, Flint HJ (2010) Butyric acid-producing anaerobic bacteria as a novel probiotic treatment approach for inflammatory bowel disease. Jr Med Microbiol 59(2):141–143. https://doi.org/10.1099/jmm.0.017541-0
Verma AK, Verma R, Ahuja V, Paul J (2012) Real-time analysis of gut flora in Entamoeba histolytica infected patients of Northern India. BMC Microbiol 12(1):1–11. https://doi.org/10.1186/1471-2180-12-183
Vinolo MA, Rodrigues HG, Nachbar RT, Curi R (2011) Regulation of inflammation by short chain fatty acids. Nutrients 3(10):858–876. https://doi.org/10.3390/nu3100858
Wang H, Naghavi M, Allen C, Barber RM, Bhutta ZA, Carter A et al (2016) Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388(10053):1459–1544. https://doi.org/10.1016/S0140-6736(16)31012-1
Wang SP, Rubio LA, Duncan SH, Donachie GE, Holtrop G, Lo G, Farquharson FM, Wagner J, Parkhill J, Louis P, Walker AW, Flint HJ (2020) Pivotal roles for pH, lactate, and lactate-utilizing bacteria in the stability of a human colonic microbial ecosystem. mSystems 5(5):e00645-20. https://doi.org/10.1128/mSystems.00645-20
Yamashiro Y, Nagata S, Ohtsuka Y, Oguchi S, Shimizu T (1996) Microbiologic studies on the small intestine in Kawasaki disease. Ped Res 39(4):622–624. https://doi.org/10.1203/00006450-199604000-00010
Yanagawa Y, Nagata N, Yagita K, Watanabe K, Okubo H, Kikuchi Y, Gatanaga H, Oka S, Watanabe K (2021) Clinical features and gut microbiome of asymptomatic Entamoeba histolytica infection. Clin Infect Dis 73(9):e3163–e3171. https://doi.org/10.1093/cid/ciaa820
Acknowledgements
We acknowledge the High Performance Computing facility at CMSD, University of Hyderabad.
Funding
This research was supported by a grant from the Department of Biotechnology, Government of India, and European Union under Indo-European collaboration on biotechnology with Indian investigators (order no.: BT/IN/Infect-Era/AB/2015). The authors thank the Department of Biotechnology, Government of India, for research associate fellowships to Lakshmi Rani Iyer, Anil Kumar Verma, and Vivek Thakur and Ramalingaswami fellowship to Vivek Thakur, Department of Science and Technology, Government of India, for JC Bose Fellowship to Alok Bhattacharya, and FIST grant to the School of Life Sciences, Jawaharlal Nehru University, New Delhi.
Author information
Authors and Affiliations
Contributions
Conceptualization: Alok Bhattacharya and Jaishree Paul; methodology: Anil Kumar Verma, Lakshmi Rani Iyer, and Vivek Thakur; formal analysis and investigation: Lakshmi Rani Iyer, Nisha Chandel, and Anil Kumar Verma; writing—original draft preparation: Lakshmi Rani Iyer, Nisha Chandel, and Vivek Thakur; writing—review and editing: Jaishree Paul and Alok Bhattacharya; funding acquisition: Alok Bhattacharya and Jaishree Paul; resources: Alok Bhattacharya, Jaishree Paul, and Ashish Kumar Mandal; supervision: Alok Bhattacharya, Jaishree Paul, and Vivek Thakur.
Corresponding author
Ethics declarations
Ethics approval
Ethical clearance for the study was obtained from both the participating institutes, namely, JNU (IERB No. 2015/Faculty/82) and Safdarjung Hospital (No. IEC/SJH/VMMC/Project/September/2014/535).
Consent to participate
Informed consent was taken of all participants included in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Section Editor: Yaoyu Feng
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Iyer, L.R., Chandel, N., Verma, A.K. et al. Effect of Entamoeba histolytica infection on gut microbial diversity and composition in diarrheal patients from New Delhi. Parasitol Res 122, 285–298 (2023). https://doi.org/10.1007/s00436-022-07728-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-022-07728-9