
Abstract
MicroRNAs are critical gene regulators at the post-transcriptional level and play essential roles in numerous developmental processes in metazoan parasites including the causative agent of cystic echinococcosis, Echinococcus granulosus. The molecular basis of different patterns of E. granulosus development in the canine definitive host and in in vitro culture systems is poorly understood. In the present study, miRNA transcriptomes of the strobilated worms derived from experimental infection in the definitive host were compared with those from diphasic culture system after 60-day protoscoleces cultivation. Total RNA was extracted from in vivo- and in vitro-derived strobilated worms. Small RNA libraries were constructed, and deep sequencing was performed. Subsequently, differential miRNA expressions and target predictions were obtained, and pathway analysis was performed by gene ontology and KEGG. Seven miRNAs were differentially expressed between the in vivo- and in vitro-derived worms. In addition, we reported 13 novel miRNA candidates and 42 conserved miRNAs. Four out of five top miRNAs with the highest read counts were shared between the in vivo and in vitro-derived worms, i.e., egr-miR-10a-5p, egr-let-7-5p, egr-bantam-3p, and egr-miR-71-5p. Target prediction of the differential miRNAs between the two systems showed significant differences in the membrane-enclosed lumen, membrane part, and an intrinsic component of the membrane. Findings of KEGG analysis indicated that differentially expressed miRNAs were involved in hippo, MAPK, and WNT signaling pathways. The study demonstrated a significant difference in miRNA transcriptomes and related signaling pathways between the two systems, suggesting the importance of host–parasite interplay in the fate of protoscoleces development in in vivo and in vitro systems.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cystic echinococcosis (CE) caused by the small cyclophyllidean cestode, Echinococcus granulosus, is an important cosmopolitan cyclozoonosis. The World Health Organization (WHO) considered echinococcosis as one of the neglected zoonotic diseases. Global cases of echinococcosis has been estimated at 188,000 new cases per annum, 2 (Deplazes et al. 2017) with an annual human and livestock economic losses estimated at US$3 billion (https://www.who.int/news-room/fact-sheets/detail/echinococcosis). The parasite is perpetuated in a cycle involving canids as definitive hosts harboring the adult helminth in their small intestine and a broad range of herbivorous and omnivorous mammals as well as human as the intermediate hosts carrying the metacestodes in their visceral organs (Budke et al. 2006).
MicroRNAs (miRNAs) are short noncoding endogenous RNA molecules found in animals, plants, fungi, and some viruses (Michlewski and Cáceres 2019). Mature miRNAs are post-transcriptional regulators of gene expression through negative regulation of target mRNAs, cleavage-induced degradation, and subsequent expression inhibition of target genes (Skalsky and Cullen 2010). MicroRNAs regulate and fine-tune a broad variety of fundamental biological processes including cell proliferation, differentiation, survival, homeostasis, stress responses, and apoptosis particularly during developmental processes (Mens and Ghanbari 2018). They have significantly contributed to the advent of morphological complexity and phenotypic evolution in animals (Heimberg et al. 2008); therefore, defects involved in miRNA biogenesis may lead to embryonic morphogenesis defects in germline (Du and Zamore 2005). miRNAs are generally considered as highly conserved throughout the animal kingdom. Since the initial description of miRNA in the early 1990s, more than 38,000 miRNAs in 271 organisms have been deposited in miRBase (http://www.mirbase.org/cgi-bin/browse.pl).
The studies to understand immunomodulatory capabilities of metazoan parasites at the transcriptional level show that miRNA-mediated RNA interference is one of the important regulatory mechanism in helminth parasites. A long list of miRNAs, including let-7, miR-223, miR-134, and miR-199 with a potential role in immune modulation, had been discovered in helminthic parasites (Arora et al. 2017). Several studies indicate the important role of miRNAs in the development and differentiation of different platyhelminth species such as E. granulosus (Cucher et al. 2011; Bai et al. 2014), E. multilocularis (Kamenetzky et al. 2016), E. canadensis (Cucher et al. 2015; Macchiaroli et al. 2015), Schistosoma japonicum (Cai et al. 2011), and S. mansoni (de Souza Gomes et al. 2011).
Bai et al. (2014), in a genome-wide sequencing study of small RNAs, reported 42 mature miRNAs and 23 miRNA stars in three life stages of E. granulosus (protoscoleces, adult worms, and cyst wall) (Bai et al. 2014). A recent study in bidirectional culture systems (cultivation of the protoscoleces towards cysts and strobilated worms) concluded that miRNAs in the early stages of E. granulosus were differentially expressed in the two systems (Bai et al. 2020).
James Desmond Smyth first discovered the phenomenal capacity of bidirectional development in E. granulosus protoscoleces (PSCs) (Smyth 1971). PSCs could develop to either strobilated worms or hydatid cysts in diphasic and monophasic culture systems, respectively. Interestingly, however, strobilated worms derived from the in vitro system do not mature and produce eggs as it normally occurred in natural definitive hosts. To date, factors involved in this phenomenon have not been precisely understood at cellular and molecular levels. In addition, the complex life cycle, peculiar transmission routes and switches between different hosts and environments, requires high level of adaptation in the parasite cellular machinery (Weber et al. 2006).
Different behaviors of the parasite development in the definitive host and in vitro culture can be attributed to different miRNA configuration, as major gene regulators. This opened a new field of research on parasite differentiation and provided a window for in-depth investigations on the molecular aspects of host–parasite interactions. In the present study, a global miRNA profiling is performed by using next-generation sequencing (NGS) in order to understand the role of miRNAs in the developmental and metabolic processes of E. granulosus. MiRNA transcriptomes of the strobilated worms derived from experimental infection in the definitive host were compared with those from diphasic culture system after 60-day protoscoleces cultivation. The findings increase our knowledge about molecular pathways involved in E. granulosus development.
Material and methods
Ethical statement
The study protocol was supervised and reviewed by the Research Ethics Review Committee of Kerman University of Medical Sciences under ethical approval code IR.KMU.REC.1398.084.
Preparation of in vivo strobilated worms
One mixed breed, 3-month-old, male dog with ideal health condition was purchased from a local vendor. After adapting to the new environment for 5 days and DHLPP vaccination, the animal was treated with 10 mg/kg praziquantel forte (fenbendazole 150 mg, pyrantel pamoate 144 mg, and praziquantel 50 mg). All rules and conditions for keeping the dog were carried out following the ethics committee’s protocols. Stool samples were regularly checked for the presence of parasite eggs, and the absence of infection was confirmed. The dog was experimentally infected with E. granulosus fertile hydatid cysts from a sheep liver. Fifty-four days post-infection with a positive stool exam showing the presence of E. granulosus eggs, the animal was euthanized according to the instructions by the Animal Ethics Committee. The adult worms attached to the small intestinal epithelium were recovered, washed with phosphate-buffered saline (PBS), and immediately transferred to the molecular lab for total RNA extraction (Doyle et al. 2020).
Preparation of in vitro strobilated worms
The protoscoleces of E. granulosus sensu stricto, G1 genotype, derived from a sheep liver hydatid cyst, were cultivated in a diphasic culture medium to obtain strobilated worms as described by Smyth (1971) . Briefly, protoscoleces were aspirated by aseptic puncture of the hydatid cyst. The PSCs were washed three times with sterile PBS containing 100 U/ml penicillin and 100 μg/ml streptomycin. Then they were washed through two layers of sterile gauze and treated in 20% dog bile in CMRL 1066 for 24 h. The PSCs were then transferred to the diphasic medium containing CMRL 1066 and coagulated calf serum as the liquid and solid phases, respectively. Strobilated worms were harvested after 60 days of cultivation (Smyth and Davies 1974).
Total RNA isolation
To avoid degradation, RNA extraction was immediately performed using the protocol described by Mousavi et al. (Mousavi et al. 2019) with some modifications to increase miRNA yield. Briefly, 500 strobilated worms obtained from in vitro and in vivo systems were separately collected and homogenized in 600 µl of cold TRIizol followed by the addition of 200 µl chloroform. The samples were placed on ice for about 5 min and centrifuged at 12,000 g for 20 min at 4 °C, and the aqueous supernatant was carefully transferred to a clean tube. This step was performed again by adding 100 µl chloroform. After removal of the aqueous supernatant, an equal volume of cold isopropanol was added. The tube was stored at − 20 °C overnight and centrifuged at 13,000 g for 60 min at 4 °C, and the pellet was resuspended in 1 mL of 75% ethanol, centrifuged at 13,000 g for 20 min at 4 °C, and left to dry at room temperature. Finally, RNA was resuspended in 50 µl RNase-free water and stored at − 80 °C until use. The quantity and purity of the purified RNA were measured using 260/280 and 260/230 ratios with NanoDrop Spectrophotometer (ND 1000, Fisher Thermo, Wilmington, DE, USA), and RNA integrity was checked by 1.5% agarose gel electrophoresis.
Small RNA library construction and deep sequencing
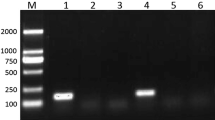
After performing quality control (QC), and calculation of RIN for each sample (Schroeder et al. 2006), the samples were proceeded to library construction. It is worth to note that similar to some insects and Platyhelminthes, RNA quality assessment is complicated in Echinococcus species, and RIN number cannot be considered as the sole criteria for RNA integrity because 28S rRNA is composed of two fragments which are bonded non-covalently (Winnebeck et al. 2010; Haçariz and Sayers 2013). RIN calculation is based on 28S and 18S ribosomal RNAs integrity, and in Echinococcus, 28S band is not visible, since it is fragmented into two parts and runs along with 18S band and thus looks degraded in gel electrophoresis (Mara Rosenzvit, personal communication). Failure to visualize 28S rRNA, in these species, may lead to an erroneous assumption of degraded RNA. On the fragment size ruler, the 18S band must be observed at approximately 1200 bp.
Small RNA libraries were constructed using the Illumina TruSeq Small RNA Library Prep Kit. The libraries were prepared by random fragmentation of the RNA samples, followed by 5′ and 3′ adapter ligation. After adapter ligation, reverse transcription and PCR amplification were performed. Finally, the cDNA libraries were size selected and sequenced (single-end reads 51 bp) on an Illumina Genome Analyzer platform. One biological replicate from each sample type/condition (in vivo worms or in vitro worms) was used for library construction. The library construction and sequencing reactions were performed at Macrogen, Korea.
The data of small RNAseq from this study have been deposited in NCBI’s Sequence Read Archive (SRA) and are accessible through SRA Series project No. PRJNA703777 and BioSample accessions SAMN18024366 and SAMN18024367.
Source of genome assembly and annotations
The E. granulosus genome assembly was used as reference genome (PRJEB121), and the gene annotations of coding genes were downloaded from the WormBase ParaSite database version WS279 (Harris et al. 2003).
miRNA prediction
Illumina raw sequence reads produced by deep sequencing were evaluated using the FastQC software (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adapters and low-quality reads were removed by the BBmap software (https://sourceforge.net/projects/bbmap/) (Kechin et al. 2017). Also, only reads between 15 and 30 nt were retained and length distribution determined. Then, reads were BLASTN searched (include parameters) against an in-house database that included E. granulosus mRNAs, miscRNAs, LncRNA, snRNA, snoRNA, rRNA, tRNA, and repeat-associated RNAs. Reads that matched to any of these categories were discarded.. The miRDeep2 software package was used for miRNA prediction (Friedländer et al. 2012). Identical reads were collapsed into unique sequences and then mapped to the E. granulosus reference genome using the mapper module from miRDeep2 with the following parameters: -c -j -m -l 18. Mapped reads were used as input for miRNA prediction with the core algorithm of miRDeep2.
In order to generate a final set of high confidence miRNAs, the initial miRDeep2 output list of candidate miRNA precursors was manually curated using the following criteria: (i) miRDeep2 score > 5, (ii) significant randfold P value < 0.05, (iii) minimum mature read count > 10, and (iv) the minimum free energy (MFE) of precursor < − 20 kcal/mol (Kozomara and Griffiths-Jones 2011).
Novel candidates to miRNA precursors were BLAST searched (e value 0.01) against WormBase ParaSite, NCBI, and RNAcentral databases. Precursor sequences that overlapped exons of coding genes or non-coding RNAs (tRNAs, rRNAs, etc.) were discarded. Then, mature sequences were searched against miRBase 22 release using SSEARCH (e value 100). Mature sequences without (i) sense match, (ii) seed match (identical 2–8 nt in the 5′ end of the mature miRNA), and (iii) 70% of nucleotide identity with any known miRNA were considered novel miRNAs.
miRNA expression and differential expression analysis
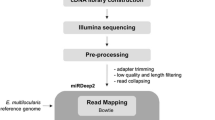
Expression levels of all known and novel miRNAs identified in each library were quantified using the quantifier module from miRDeep2. Differential expression analysis between samples/conditions was performed with the Bioconductor package EdgeR (Robinson et al. 2009; Dillies et al. 2013) in R (version 4.0) using raw read counts as input, which compared the read count values of miRNAs for the two conditions and statistically evaluated significant changes in expression (Fig. 1). Those miRNAs that showed log fold changes (log2 FC) ≥ │1.5│and false discovery rate (FDR) < 0.05 were considered differentially expressed.

The sequential order of data analysis used for global miRNA profiling of strobilated worms of Echinococcus granulosus derived from the in vivo and in vitro systems
miRNA target prediction
To obtain a set of putative three-prime untranslated regions (3’UTRs), genomic sequences of 300 nt in length located downstream the stop codon of all E. granulosus coding genes were retrieved from the WormBase ParaSite database (https://parasite.wormbase.org/index.html) using the BioMart tool. The following options were applied to the tool: Species: genome, Echinococcus granulosus; Gene: gene type, Protein coding, Output attributes: Retrieve sequences > Sequences: flank coding region, downstream flank (300), Count, Results. Then the results were exported in a FASTA file format.
The set of 3′ UTRs and the set of novel and differentially expressed miRNAs were used as input for miRNA target site prediction using miRanda (Enright et al. 2003) with default parameters except for (i) strict seed pairing and (ii) energy threshold: − 17 kcal/mol as previously described (Macchiaroli et al. 2015; Pérez et al. 2019) (Supplementary file 1, sheet 9 and 10).
Gene ontology and pathway analysis
A pathway analysis of the predicted miRNA targets was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kaas-bin/kaas_main) and the miRNAs’ interfered gene target pathways generated. In addition, a Gene Ontology (GO) functional enrichment analysis of the predicted miRNA targets was performed using the BLAST2GO software tool (http://www.BLAST2go.org/). Then, the WEGO tool (https://wego.genomics.cn/) was used for visualizing and plotting GO annotation results.
Results
Deep sequencing of two small RNA libraries from E. granulosus
To investigate the composition of miRNAs’ expression, two small RNA libraries from strobilated worms of E. granulosus derived from in vitro and in vivo systems were sequenced using Illumina sequencing technology (Fig. 2). After trimming adaptors and removing the low-quality sequences and RNAs smaller than 15 nt, we obtained 50,002,880 and 30,065,579 high-quality reads of small RNAs, sized 15–30 nt from in vivo and in vitro parasites, respectively (Supplementary file 1, sheet 1). Of these reads, 84% of in vivo adult worms and 54% of in vitro strobilated worms were 18–23 nt in length (Supplementary file 1: sheet 1, Supplementary file 2: Fig. S1), which is the typical size range for Dicer-derived products.

Schematic representation of the study on global miRNA profiling of Echinococcus granulosus worms derived from the in vivo and in vitro systems. a Timeline (days) of in vivo and in vitro developmental stages of E. granulosus cultured in CMRL 1066 medium and host intestine for 55 days. b–d Workflow of miRNA-seq study from total RNA extraction to analysis of NGS data
After sequence mapping, 70.72% and 60.11% of clean reads of in vivo and in vitro worms, respectively, perfectly matched the E. granulosus genome (Table 1). In the in vivo adult worms’ dataset, 5.96% of the mapped sequences were related to rRNA and 4.04% to tRNA, whereas these figures for the in vitro strobilated worms dataset were 21.13% and 5.60% for rRNA and tRNA, respectively. The known non-target RNAs including degraded remnant of mRNAs, miscRNA, LncRNA, snRNA, snoRNA, rRNA, tRNA, and repeat-associated RNA were discarded, and the remaining mapped small RNA reads were used to find known and novel miRNAs.
Known and novel miRNAs of E. granulosus strobilated worms from in vivo and in vitro systems
After applying initial filtering, 76 miRNA candidates were identified (Supplementary file 1: sheet 2), of which we reported 13 novel miRNA candidates (Fig. 3) present in the two libraries, according to the criteria described in the methods (Table 2, Supplementary file 1: sheet 3). The genomic sequence flanking all these miRNAs can be folded into characteristic miRNA stem-loop secondary hairpin structures and have a 1–2 nt 3′overhang pattern generated by Dicer cleavage during mature miRNA generation. We found 42 conserved miRNAs present in the datasets, out of which, 25 were predicted as star miRNAs (Supplementary file 1: sheet 4).

Schematic representation of the secondary structure of novel miRNA 1–13 and highlight the consensus mature miRNA in the structure of precursor miRNAs
Out of 5-top high miRNAs’ read counts, four miRNAs were similar between the two systems (egr-miR-10a-5p, egr-let-7-5p, egr-bantam-3p, and egr-miR-71-5p). The fifth highest expressed miRNA was different between the two systems, i.e., egr-miR-2b-3p and egr-miR-125-5p were the fifth predominant miRNAs in the strobilated and adult worms, respectively (Supplementary file 1: sheet 7).
According to Table 3, we found seven miRNAs with significant differential expression between the in vivo- and in vitro-derived worms. All differentially expressed miRNAs were conserved miRNAs. While five miRNAs, egr-miR-10228-3p, egr-miR-10228-5p, egr-miR-31-3p, egr-miR-31-5p, and egr-miR-10257-5p, showed significantly higher read counts in the adult worms, 2 miRNAs, egr-miR-96-3p, and egr-miR-7b-5p shower higher expression in the strobilated worms (Supplementary file 1: sheet 8).
Target prediction of the differential and novel miRNAs
Sheets 9 and 10 in the Supplementary file 1 show the target genes for the novel and differential miRNAs, respectively. Target search of the 13 novel miRNAs showed 881 total genes for of which 296, 282, and 303 genes had GO terms related to biological process, cellular component, and molecular function, respectively (Supplementary file 2: Table S1, Fig. S1). GO terms of target genes predicted for the differentially expressed miRNAs targets showed 586 total genes for seven miRNAs, among them 190, 195, and 201 genes were related to biological process, cellular component, and molecular function, respectively (Supplementary file 2: Table S2, Fig. S2). Cellular component genes including organelle lumen (GO: 0,043,233), the membrane-enclosed lumen (GO: 0,031,974), membrane part (GO: 0,044,425), and intrinsic component of membrane (GO: 0,031,224) were significantly different target genes between the differential miRNAs and the reference genome; however only macromolecule localization (GO: 0,033,036) was significantly different for biological process genes (Supplementary file 2: Tables S3 and S4, Figs. S3 and S4). KEGG analysis showed high-level functions of biological pathways as shown in the Supplementary file 2 including Hippo signaling (Supplementary file 2: Fig. S5), mitogen-activated protein kinases (MAPK) (Supplementary file 2: Fig. S6), and WNT signaling pathways (Supplementary file 2: Fig. S7).
Discussion
Comparison of the worms derived from experimental in vivo infection of dogs and the strobilated worms from in vitro cultivation of E. granulosus provides significant clues in the understanding of the molecular basis of growth and development in the tapeworms. In this study, we compared miRNA profiles in the adult and strobilated worms of Echinococcus granulosus derived from experimental infection and culture media. Results of the present study provided experimental evidence of the presence of novel and conserved miRNAs found in the in vivo- and in vitro-derived helminths. In this study, miRNAs were found as the most abundant type of small RNAs related to the in vivo and in vitro worms showing 42,232,121 and 17,888,324 total clean reads, respectively (Table 1). This finding is in agreement with previous high-throughput analyses in the genus Echinococcus (Bai et al. 2014; Macchiaroli et al. 2015; Kamenetzky et al. 2016).
From the first attempts of in vitro cultivation of E. granulosus (Smyth et al. 1966), researchers realized that the in vitro-reared worms do not produce eggs as it usually occurred in the normal canine definitive hosts. Several attempts failed to succeed in the development of gravid proglottids with full-grown oncospheres in the in vitro strobilated worms. The molecular basis of this is not fully understood. Small RNAs may play a role in the differential development of E. granulosus in the two culture systems. In our study, target analysis for differentially expressed miRNAs showed that the membrane type genes are targeted by the miRNAs. These findings imply that miRNA read count variations in different systems may be associated with the differences in the environment. The worm contact to the epithelial cells of the intestine in the natural definitive host or to the supporting matrix in the in vitro culture system may trigger different internal molecular mechanisms.
Differential read counts were recorded in seven miRNAs. The two miRNAs, miR-31 and miR-10228, demonstrated the most significant differences in the read counts. It should be noted that the literature on miRNAs in E. granulosus are exceedingly poor. Therefore, there is no data on the expression profiles of the miRNA families in the strobilated compared to the adult worms. However, in recent years, a few studies are available on miR-31 for some platyhelminth species including Taenia crassiceps (Pérez et al. 2017) and Taenia hydatigena (Wu et al. 2019). No information is available for miR-10228 in the Platyhelminthes. There is no information on the target of these miRNAs in the Platyhelminthes; however, miRNA targets synonym to miR-31 have been attributed to tumor suppression, upregulation of human cervical cancer cells, inhibition of cell metastasis, and migration through downregulating BAP1, a tumor suppressor gene important to the development and prognosis of several types of cancers (Wang et al. 2017).
Very few data are available on the biological function of miR-10228, and further experimental research is needed to clarify the significance of this miRNA in the differentiation and development of invertebrate organisms. MiR-96, another differentially expressed miRNA in our study, may play a critical role in the development as well as progression of lung cancer (Pei et al. 2017), As an oncogene, it regulates cell growth and migration in a breast cancer model of epithelial–mesenchymal transition, reduces the size of tumors, and plays a role in maintaining a mesenchymal phenotype (Anderson and Guttilla Reed 2020).
Findings of KEGG analysis indicated that the differentially expressed miRNAs tend to target some of the genes involved in hippo signaling. Hippo signaling is an evolutionarily conserved pathway that controls organ size in a wide variety of organisms, resulting in cellular growth, the suppression of cell death, and tumor suppression (Kifle et al. 2017). Several pathways associated with the parasite membrane-bound proteins are known to be involved in the host–parasite interplay (Rosenzvit et al. 2006) or programmed cell death (apoptosis) with overactivity of caspase 3 involved in hydatid cyst infertility (Paredes et al. 2007), indicating that several factors including miRNAs are involved in the development of the protoscoleces into adult or strobilated worms.
Among the most abundant miRNAs in the two culture systems, miR-10a, let-7, bantam, and miR-71 are well-known in Echinococcus (Bai et al. 2014; Mortezaei et al. 2020) as well as in other tapeworms like Mesocestoides corti (Basika et al. 2016). These miRNAs are among the top most expressed miRNAs, indicating the significance of these miRNAs in the biological processes or molecular functions in the helminth parasites. Bantam miRNA hits the pro-apoptotic genes as a target for regulation and acts as an anti-apoptotic regulator and is classified as regulatory miRNA in Drosophila development (Brennecke et al. 2003). On the other hand, let-7 miRNA family shows a high level of evolutionary conservation among different species of Platyhelminthes (Faridi et al. 2020). It promotes differentiation during development and is known as a tumor suppressor and stem cell division regulator.
Perez et al. investigated the role of miR-71 through functional analysis by using anti-miRs (antisense oligonucleotides) and succeeded miR-71 knockdown in primary cell cultures of Echinococcus multilocularis. The findings showed that several miR-71 target genes were significantly upregulated. These genes are potentially involved in parasite development, host–parasite interaction, and some other unknown functions. The phenotype of miR-71-knockdown primary cell cultures was affected comparing to the controls, and they did not mature into fully developed metacestodes (Pérez et al. 2019).
To date, 111 precursors and 218 mature miRNA for E. granulosus have been identified in the miRBase (http://www.mirbase.org/). In this study, we present 13 new miRNAs. The number of new miRNAs reported in different helminths is relatively variable, probably due to the differences in the strategic command of the analysis pipelines. There is no robust data about the biological function of new miRNAs reported in the present study; nonetheless, GO term determined the hypothetical target of the miRNAs based on seed regions and revealed the targeted genes were mostly related to molecular function like cellular and metabolic processes. In a recent study of miRNAs in E. multilocularis using genome-wide data, 10 new miRNAs were proposed; however, bioinformatic analysis failed to assume any biological functions for the new miRNAs (Kamenetzky et al. 2016).
In accordance with other studies, we showed the top expressed miRNAs are similar among different species. In other words, they play critical key roles in regulating the machinery of the cells. To fully understand miRNA functions, further studies are required to knockdown particular miRNAs in the in vitro system, to determine the transcriptomic and phonotypic consequences within the cells.
Even after more than two decades of miRNA research, understanding miRNA biology and exact target analysis remained a challenge like looking for a needle in a haystack (Lai 2015). MiRNAs interfere in the gene regulation in a complex context, having that one miRNA typically targets many genes and one gene is potentially regulated by several miRNAs (Xu et al. 2020). This means that assuming one specific pathway to an individual miRNA could be erroneous and leads to incorrect conclusions. Whether miRNAs fine-tune large network of targets or suppress specific targets are questions remained to be answered. Blocking specific miRNAs and genome-wide approaches like RNAseq should be applied to understand the in vivo function of miRNAs in Echinococcus.
Significant developmental plasticity has been documented in Echinococcus species. Depending on the environmental stimuli, PSCs have the unusual ability to differentiate in two different directions in the in vitro setting. The nature of these stimuli is not completely understood; however, it is postulated that host–parasite interactions directly influence protoscoleces development (Debarba et al. 2015). Findings of our miRNAome study indicate that the in vitro- and in vivo-reared E. granulosus worms shared the same remarkable miRNA makeup. Therefore, it seems the in vitro culture of the strobilated worms is a suitable model for experimental studies on the physiology of Echinococcus growth and development, and it could appropriately replace the problematic experimental studies on dogs. However, failure of fertilization of the in vitro-reared worms provides some clues to understand the physiology of sexual maturity in the Platyhelminthes including lack of an appropriate matrix stimuli such as definitive host chewing, contact with dog gut environment, and digestive enzymes, as well as dog bile secretion (Debarba et al. 2020). Trigger stimuli play a significant role for parasite strobilar development and in designing suitable culture conditions for E. granulosus protoscoleces.
The physiology of fertilization in Cyclophyllidean tapeworms is poorly understood. Both self- and cross-fertilization occurred; however, cross-fertilization is considered as the main mode of fertilization in Echinococcus species. Host gut environment provides suitable physicochemical stimuli to facilitate cross-fertilization. Omics studies will clarify the molecular bases of these phenomena. More than 100-megabase genome size of E. granulosus arranged in more than 10,000 genes in nine chromosomes displays considerable gene losses for de novo synthesis of pyrimidines, purines, cholesterol, fatty acids, and most amino acids (Berriman et al. 2013). This means the organism relies on the host (or nutritional supplements in culture media) for obtaining these essential components. Therefore, PSC maturation is a multi-factorial complex matter with many unknown molecular pathways likely to affect the strobilar/sexual development to E. granulosus.
Due to some limitations, we inevitably used bioinformatic tools to describe miRNAome of experimentally developed E. granulosus isolates from two different environments, and we predicted potential miRNA targets; however, further in-depth wet lab studies are required to clarify and validate the role of the novel and differentially expressed miRNAs in the development and differentiation of E. granulosus. In the absence of comprehensive studies on miRNA comparisons between in vitro and in vivo systems and limited knowledge about their targets, the present study highlighted the critical role of miRNAs, as master regulators of gene expression, in differentiation and development of the E. granulosus protoscoleces towards strobilation, sexual maturity, and adulthood. The potential significance of miRNAs to subtly alter many targets based on a network model indicates the many intervening factors playing roles in the complex life cycle of E. granulosus and the capability of the protoscoleces to develop into adult or strobilated worms.
Conclusions
This study demonstrated a significant difference in miRNA transcriptomes and related signaling pathways between the two systems, signifying the importance of host–parasite interplay in the fate of protoscoleces development in in vivo and in vitro systems. More in-depth studies are required to improve our understanding of the molecular basis and the exact role of miRNAs in tapeworm’s growth and development.
Data availability
The data of small RNAseq from this study have been deposited in NCBI’s Sequence Read Archive (SRA) and are accessible through SRA Series project No. PRJNA703777, BioSample accessions: SAMN18024366 and SAMN18024367.
Code availability
Not applicable.
References
Anderson O, Guttilla Reed IK (2020) Regulation of cell growth and migration by miR-96 and miR-183 in a breast cancer model of epithelial-mesenchymal transition. PLoS ONE 15:e0233187. https://doi.org/10.1371/journal.pone.0233187
Arora N, Tripathi S, Singh AK et al (2017) Micromanagement of immune system: role of miRNAs in helminthic infections. Front Microbiol 8:586. https://doi.org/10.3389/fmicb.2017.00586
Bai Y, Zhang Z, Jin L et al (2014) Genome-wide sequencing of small RNAs reveals a tissue-specific loss of conserved microRNA families in Echinococcus granulosus. BMC Genomics 15:736. https://doi.org/10.1186/1471-2164-15-736
Bai Y, Zhang Z, Jin L et al (2020) Dynamic changes in the global transcriptome and microRNAome reveal complex miRNA-mRNA regulation in early stages of the bi-directional development of Echinococcus granulosus Protoscoleces. Front Microbiol 11:654. https://doi.org/10.3389/fmicb.2020.00654
Basika T, Macchiaroli N, Cucher M et al (2016) Identification and profiling of microRNAs in two developmental stages of the model cestode parasite Mesocestoides corti. Mol Biochem Parasitol 210:37–49. https://doi.org/10.1016/j.molbiopara.2016.08.004
Berriman TI, Zarowiecki M et al (2013) F1000Prime recommendations of: the genomes of four tapeworm species reveal adaptations to parasitism. Nature 496:57–63
Brennecke J, Hipfner DR, Stark A et al (2003) Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113:25–36
Budke CM, Deplazes P, Torgerson PR (2006) Global socioeconomic impact of cystic echinococcosis. Emerg Infect Dis 12:296–303. https://doi.org/10.3201/eid1202.050499
Cai P, Hou N, Piao X et al (2011) Profiles of small non-coding RNAs in Schistosoma japonicum during development. PLoS Negl Trop Dis 5:e1256. https://doi.org/10.1371/journal.pntd.0001256
Cucher M, Macchiaroli N, Kamenetzky L et al (2015) High-throughput characterization of Echinococcus spp. metacestode miRNomes. Int J Parasitol 45:253–267. https://doi.org/10.1016/j.ijpara.2014.12.003
Cucher M, Prada L, Mourglia-Ettlin G et al (2011) Identification of Echinococcus granulosus microRNAs and their expression in different life cycle stages and parasite genotypes. Int J Parasitol 41:439–448. https://doi.org/10.1016/j.ijpara.2010.11.010
de Souza Gomes M, Muniyappa MK, Carvalho SG et al (2011) Genome-wide identification of novel microRNAs and their target genes in the human parasite Schistosoma mansoni. Genomics 98:96–111. https://doi.org/10.1016/j.ygeno.2011.05.007
Debarba JA, Monteiro KM, Moura H et al (2015) Identification of newly synthesized proteins by Echinococcus granulosus protoscoleces upon induction of strobilation. PLoS Negl Trop Dis 9:e0004085. https://doi.org/10.1371/journal.pntd.0004085
Debarba JA, Sehabiague MPC, Monteiro KM et al (2020) Transcriptomic analysis of the early strobilar development of Echinococcus granulosus. Pathogens 9:1–12. https://doi.org/10.3390/pathogens9060465
Deplazes P, Rinaldi L, Alvarez Rojas C et al (2017). Global distribution of alveolar and cystic echinococcosis. Adv Parasitol 95:315–493. https://doi.org/10.1016/bs.apar.2016.11.001
Dillies MA, Rau A, Aubert J et al (2013) A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform 14:671–683. https://doi.org/10.1093/bib/bbs046
Doyle SR, Tracey A, Laing R et al (2020) Genomic and transcriptomic variation defines the chromosome-scale assembly of Haemonchus contortus, a model gastrointestinal worm. Commun Biol 3:656. https://doi.org/10.1038/s42003-020-01377-3
Du T, Zamore PD (2005) microPrimer: the biogenesis and function of microRNA. Development 132:4645–4652. https://doi.org/10.1242/dev.02070
Enright AJ, John B, Gaul U et al (2003) MicroRNA targets in Drosophila. Genome Biol 5:1–27. https://doi.org/10.1186/gb-2003-5-1-r1
Faridi A, Afgar A, Mousavi SM et al (2020) Intestinal expression of miR-130b, miR-410b, and miR-98a in experimental canine echinococcosis by Stem-Loop RT-qPCR. Front Vet Sci 7:507. https://doi.org/10.3389/fvets.2020.00507
Friedländer MR, MacKowiak SD, Li N et al (2012) MiRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40:37–52. https://doi.org/10.1093/nar/gkr688
Haçariz O, Sayers G (2013) Fasciola hepatica - where is 28S ribosomal RNA? Exp Parasitol 135:426–429. https://doi.org/10.1016/j.exppara.2013.07.026
Harris TW, Lee R, Schwarz E et al (2003) WormBase: a cross-species database for comparative genomics. Nucleic Acids Res 31:133–137. https://doi.org/10.1093/nar/gkg053
Heimberg AM, Sempere LF, Moy VN et al (2008) MicroRNAs and the advent of vertebrate morphological complexity. Proc Natl Acad Sci U S A 105:2946–2950. https://doi.org/10.1073/pnas.0712259105
Kamenetzky L, Stegmayer G, Maldonado L et al (2016) MicroRNA discovery in the human parasite Echinococcus multilocularis from genome-wide data. Genomics 107:274–280. https://doi.org/10.1016/j.ygeno.2016.04.002
Kechin A, Boyarskikh U, Kel A, Filipenko M (2017) CutPrimers: a new tool for accurate cutting of primers from reads of targeted next generation sequencing. J Comput Biol 24:1138–1143. https://doi.org/10.1089/cmb.2017.0096
Kifle DW, Sotillo J, Pearson MS, Loukas A (2017) Extracellular vesicles as a target for the development of anti-helminth vaccines. Emerg Top Life Sci 1:659–665. https://doi.org/10.1042/etls20170095
Kozomara A, Griffiths-Jones S (2011) MiRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39:D152–D157. https://doi.org/10.1093/nar/gkq1027
Lai EC (2015) Two decades of miRNA biology: lessons and challenges. RNA 21:675–677. https://doi.org/10.1261/rna.051193.115
Macchiaroli N, Cucher M, Zarowiecki M et al (2015) MicroRNA profiling in the zoonotic parasite Echinococcus canadensis using a high-throughput approach. Parasit Vectors 8:83. https://doi.org/10.1186/s13071-015-0686-8
Mens MMJ, Ghanbari M (2018) Cell cycle regulation of stem cells by microRNAs. Stem Cell Rev Reports 14:309–322. https://doi.org/10.1007/s12015-018-9808-y
Michlewski G, Cáceres JF (2019) Post-Transcriptional Control of miRNA Biogenesis. Rna 25:1–16. https://doi.org/10.1261/rna.068692.118
Mortezaei S, Afgar A, Sadeghi B et al (2020) Comparative analysis of miRNA expressions in different developmental stages of Echinococcus granulosus in mono-phasic and di-phasic culture systems. Infect Disord Drug Targets 21:1–9 https://doi.org/10.2174/1871526520999201103192518
Mousavi SM, Afgar A, Mohammadi MA et al (2019) Calmodulin-specific small interfering RNA induces consistent expression suppression and morphological changes in Echinococcus granulosus. Sci Rep 9:1–9. https://doi.org/10.1038/s41598-019-40656-w
Paredes R, Jiménez V, Cabrera G et al (2007) Apoptosis as a possible mechanism of infertility in Echinococcus granulosus hydatid cysts. J Cell Biochem 100:1200–1209. https://doi.org/10.1002/jcb.21108
Pei L, Zhou M, Tang Z et al (2017) MicroRNA-3646 promotes cell proliferation, migration and invasion by targeting RhoA in breast cancer. Int J Clin Exp Pathol 10:61–71
Pérez MG, Macchiaroli N, Lichtenstein G et al (2017) microRNA analysis of Taenia crassiceps cysticerci under praziquantel treatment and genome-wide identification of Taenia solium miRNAs. Int J Parasitol 47:643–653. https://doi.org/10.1016/j.ijpara.2017.04.002
Pérez MG, Spiliotis M, Rego N et al (2019) Deciphering the role of miR-71 in Echinococcus multilocularis early development in vitro. PLoS Negl Trop Dis 13:1–21. https://doi.org/10.1371/JOURNAL.PNTD.0007932
Robinson MD, McCarthy DJ, Smyth GK (2009) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Rosenzvit MC, Camicia F, Kamenetzky L et al (2006) Identification and intra-specific variability analysis of secreted and membrane-bound proteins from Echinococcus granulosus. Parasitol Int 55:S63–S67. https://doi.org/10.1016/j.parint.2005.11.009
Schroeder A, Mueller O, Stocker S et al (2006) The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol 7:1–14. https://doi.org/10.1186/1471-2199-7-3
Skalsky RL, Cullen BR (2010) Viruses, microRNAs, and host interactions. Annu Rev Microbiol 64:123–141. https://doi.org/10.1146/annurev.micro.112408.134243
Smyth JD (1971) Development of monozoic forms of Echinococcus granulosus during in vitro culture. Int J Parasitol 1:121–124. https://doi.org/10.1016/0020-7519(71)90004-X
Smyth JD, Davies Z (1974) In vitro culture of the strobilar stage of Echinococcus granulosus (sheep strain): a review of basic problems and results. Int J Parasitol 4:631–644. https://doi.org/10.1016/0020-7519(74)90028-9
Smyth JD, Howkins AB, Barton M (1966) Factors controlling the differentiation of the hydatid organism, Echinococcus granulosus, into cystic or strobilar stages in vitro. Nature 211:1374–1377. https://doi.org/10.1038/2111374a0
Wang N, Li Y, Zhou J (2017) miR-31 functions as an oncomir which promotes epithelial-mesenchymal transition via regulating BAP1 in cervical cancer. Biomed Res Int 2017:6361420. https://doi.org/10.1155/2017/6361420
Weber C, Guigon G, Bouchier C et al (2006) Stress by heat shock induces massive down regulation of genes and allows differential allelic expression of the Gal/GalNAc lectin in Entamoeba histolytica. Eukaryot Cell 5:871–875. https://doi.org/10.1128/EC.5.5.871-875.2006
Winnebeck EC, Millar CD, Warman GR (2010) Why does insect RNA look degraded? J Insect Sci 10:1–7. https://doi.org/10.1673/031.010.14119
Wu J, Yang J, He G et al (2019) High-throughput identification of microRNAs in Taenia hydatigena, a cestode threatening livestock breeding industry. Infect Genet Evol 75:103985. https://doi.org/10.1016/j.meegid.2019.103985
Xu P, Wu Q, Yu J et al (2020) A systematic way to infer the regulation relations of miRNAs on target genes and critical miRNAs in cancers. Front Genet 11:278. https://doi.org/10.3389/fgene.2020.00278
Acknowledgements
The authors wish to thank Mr. Saman Faridi for his kind technical help on graphical design.
Funding
The study was financially supported the Vice-Chancellor for Research and Technology, Kerman University of Medical Sciences, Grant No. 97000221.
Author information
Authors and Affiliations
Contributions
AF and MFH: conceptualization, study design, data validation, and writing—original draft preparation. AF, AA, and SMM: data curation. AF, MM, NM, and MFH: data analysis. MFH: funding acquisition. AF and SMM: laboratory experiments. AF, AA, SMM, MCR, MM, and MFH: revising and final approval of the manuscript. All co-authors have seen and agree with the contents of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
The study protocol was supervised and reviewed by the Research Ethics Review Committee of Kerman University of Medical Sciences under ethical approval code IR.KMU.REC.1398.084.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Section Editor: Bruno Gottstein
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Faridi, A., Mansouri, M., Macchiaroli, N. et al. MicroRNA profile of the strobilated worms of Echinococcus granulosus derived from in vivo and in vitro systems by using high-throughput approach. Parasitol Res 120, 3203–3214 (2021). https://doi.org/10.1007/s00436-021-07251-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-021-07251-3