
Abstract
Hepatozoon canis is a tick-borne pathogen of canids, which is distributed worldwide. However, very little is known about this protozoan parasite in Pakistan. This study provides the first molecular evidence of H. canis from farm dogs from three agro-ecological zones of Punjab, Pakistan. A conventional PCR targeting the 18S rRNA gene was used to characterize H. canis from farm dogs from three districts, namely Kasur, Rawalpindi, and Muzaffargarh, in Punjab. Of 341 blood samples tested, 155 (45.5%) were positive for H. canis, 73 (61.3%) from Kasur, 46 (42.5%) from Rawalpindi, and 36 (31.5%) from Muzaffargarh. Phylogenetic analyses revealed that 18S rRNA sequences of H. canis from this study clustered in three clades with those of H. canis from previously published studies to the exclusion of all other Hepatozoon spp. included in the analysis. This study provides the first insight into H. canis from farm dogs in Pakistan. Furthermore, it lays a foundation for future studies of the parasite to assess the impact of canine hepatozoonosis in dogs from various agro-ecological zones in Pakistan where pet ownership of dogs is increasing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatozoon represents an important genus of apicomplexan protozoa and consists of more than 300 species, with at least 46 of them infecting mammals (Smith 1996). Hepatozoon canis and Hepatozoon americanum are important tick-borne pathogens which cause canine hepatozoonosis in domestic and wild canids (Gomes et al. 2016). Ixodid ticks are known to transmit H. canis in canids, including Ambylomma ovale in South America (Rubini et al. 2009), and Rhipicephalus sanguineus and Rhipicephalus turanicus in different parts of the world (Baneth et al. 2007; Giannelli et al. 2017), whereas Ambylomma maculatum is the only vector known for H. americanum in the USA (Mathew et al., 1998). The transfer of infection has also been reported by vertical transmission for H. canis (Murata et al. 1993) and by predation for H. americanum (Johnson et al. 2009).
H. canis and H. americanum infections may result in marked elevation of leukocytes, leading to clinical signs such as pyrexia, inappetence, hyperesthesia, muscle wasting, and weight loss (Baneth 2011). Most H. canis-infected dogs have subclinical infection or mild clinical signs, although a certain subset experience severe and potentially fatal disease (Baneth 2011).
As the name indicates, H. americanum has only been reported to cause clinical disease in dogs in the USA so far (Potter and Macintire 2010), whereas the occurrence of H. canis has been described from various tropical, subtropical, and temperate regions of the world, including Africa, Europe, Asia, and the USA (Aydin et al. 2015; Vojta et al. 2009). However, very limited information is available on the prevalence and characterization of canine hepatozoonosis from Pakistan (Qamar et al. 2017).
Pakistan is located in South Asia, between 30° 00′ North and 70° 00′ East with a total land area of 796,095 km2 (Farooqi et al. 2005; Maps 2017). Although 10 agro-ecological zones with different climatic conditions exist in the country, subtropical and partially temperate regions constitute the major proportion, extending from the Arabian Sea in the south to the Himalayas in the north (Farooqi et al. 2005). Due to such favorable climatic conditions for the transmission of tick-borne diseases, a high prevalence of H. canis would be expected in this region. However, there is paucity of information on the prevalence of H. canis from Pakistan, especially from farm dogs. Therefore, this study was designed to survey and genetically characterize H. canis from farm dogs in three different agro-ecological zones of Punjab, Pakistan.
Materials and methods
Study area and blood sampling
Blood samples (n = 341) were collected from the cephalic vein of apparently healthy farm dogs of both sexes in EDTA tubes and stored at − 20 °C until DNA extraction. The sample collection was performed between June and October 2016 from three different districts in the province of Punjab, including Kasur (31° 12′ 21″ N, 74° 45′ 81″ E; number of samples = 119), Muzaffargarh (30° 07′ 36″ N, 71° 18′ 05″ E; n = 114), and Rawalpindi (33° 59′ 84″ N, 73° 04′ 41″ E; n = 108) (Fig. 1). The average rainfall and temperature in these districts are summarized in Table 1 (PMD 2016). This study was approved by the animal ethics committee of the University of Veterinary and Animal Sciences, Lahore, Pakistan.

Map of Pakistan showing different states and the study sites (indicated by black stars) within Punjab province
DNA extraction, PCR amplification, and sequencing
Genomic DNA was extracted from 200 μl of each blood sample using the GeneAll Exgene Genomic DNA micro extraction kit (Cambio Ltd., Cambridge, England) following the manufacturer’s guidelines. DNA was stored at − 20 °C until further use.
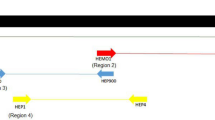
For the identification of H. canis, a 666-bp fragment of the 18S rRNA gene was amplified using conventional PCR by employing the HepF (5′-ATACATGAGCAAAATCTCAAC-3′) and HepR (5′-CTTATTATTCCATGCTGCAG-3′) primers (Inokuma et al. 2002). PCRs were performed in 25-μl volumes containing 10 mM Tris-HCl (pH 8.4), 50 mM KCl (Promega, Madison, WI, USA), 3.5 mM MgCl2, 250 μm of each deoxynucleotide triphosphate (dNTP), 25 pmol of each primer, and 1 U of GoTaq polymerase (Promega). PCR cycling conditions were initial denaturation at 95 °C for 12 min followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 57 °C for 30 s, and extension at 72 °C for 30 s, and the final extension was carried out at 72 °C for 7 min. PCRs were run using a T100 Thermal Cycler (BioRad, Hercules, CA, USA). Positive (H. canis) and negative (double-distilled water) controls were included in each PCR run. Following PCR, aliquots (5 μl) of individual amplicons were examined by agarose gel electrophoresis (1.5% gels in 0.5× TAE buffer [20 mM Tris, 10 mM acetic acid, 0.5 mM EDTA]). Gels were stained using GelRed Nucleic Acid Gel Stain (Biotium, Inc., Fremont, CA, USA), subjected to transillumination, and photographed using a gel documentation system (Kodak Gel Logic 1500 Imaging System, Eastman Kodak Company, Rochester, NY, USA).
Ten PCR amplicons from each district were randomly selected for DNA sequencing. Each amplicon was treated with shrimp alkaline phosphatase and exonuclease I (Werle et al., 1994) and then subjected to automated DNA sequencing using the 96-capillary 3730xl DNA Analyser (Applied Biosystems, Foster City, CA, USA) at Macrogen Incorporation, South Korea. Sequencing of the 18S rRNA gene was conducted using the PCR primers (HepF and HepR) in separate reactions. The quality of nucleotide sequences was appraised using the program Geneious R10 (Biomatters Ltd., Auckland, New Zealand) (Kearse et al. 2012), and polymorphic sites were designated using International Union of Pure and Applied Chemistry (IUPAC) codes.
DNA sequence and phylogenetic analyses
Consensus sequences from Geneious software were aligned with the MUSCLE V 3.8.31 program (Edgar 2004) and then adjusted manually using the Mesquite V 3.03 software (Maddison 2008). Sequence differences were calculated based on pairwise comparisons using the program BioEdit (Hall 2011).
Phylogenetic analyses were performed using Bayesian inference (BI) and neighbor-joining (NJ) methods. Previously published 18S rRNA sequences of Hepatozoon spp. (Alho et al. 2017; Aydin et al. 2015; Baneth et al. 2013; Criado-Fornelio et al. 2003; Inokuma et al. 2002; Kongklieng et al. 2015; Loftis et al. 2013; Maia et al. 2014; Mathew et al. 2000; Pawar et al. 2012; Qamar et al. 2017; Rubini et al. 2009; Vojta et al. 2009) available on GenBank and those determined herein were aligned (575 bp) and used for phylogenetic analyses. Toxoplasma gondii was used as the outgroup (Luton et al. 1995). The BI analysis was conducted, employing the Markov Chain Monte Carlo (MCMC) method in Mr Bayes 3.1.2 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003). The likelihood parameters for BI were based on the Akaike information criterion (AIC) and Bayesian inference criterion (BIC) tests in jModeltest v2.1.5 (Darriba et al. 2012). For all datasets, AIC revealed the Kimura three-parameter model of evolution, with gamma-distribution (TPM3uF+G) as the “best” model. Estimates of the base frequencies, the substitution rate model matrix, and the proportion of invariable sites were fixed. Posterior probabilities (pp) were calculated using 2,000,000 generations, employing four simultaneous tree-building chains, with every 100th tree being saved. A consensus tree (50% majority rule) was constructed based upon the remaining trees generated by BI. The NJ analyses were performed employing the online software, MEGA 7.026 (Tamura et al. 2013), and the nodes were tested for robustness with 10,000 bootstrap replicates. The phylogenetic trees produced from the BI and NJ analyses were compared for concordance in their topologies using the software FigTree 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
Results
A partial fragment of 18S rRNA was successfully amplified by PCR from 45.5% (155/341) of the samples. The prevalence of H. canis in three districts of Punjab is summarized in Table 1. Based on location, the highest prevalence of H. canis was found in Kasur (61.3%; 73/119) followed by Rawalpindi (42.5%; 46/108) and Muzaffargarh (31.5%; 36/114).
A total of 30 PCR amplicons (i.e., 10 from each district) were randomly selected for DNA sequencing, and the sequence analysis revealed 15 different 18S rRNA sequences (578–626 bp). The nucleotide BLAST analysis revealed that all these sequences were most similar (i.e., sequence identity of > 97%) to those of H. canis available in public databases. An alignment of 15 sequences determined herein (GenBank accession nos. MG209580–MG209594) displayed two transitions either C ↔ T at position 10 or G ↔ A at positions 17, 41, 274, 429, and 470 (see supplementary Fig. 1). However, at five positions, three IUPAC ambiguity codes, including M (alignment position numbers 380, 409, 478), R (28), and X (315), were present (see supplementary Fig. 1). Pairwise comparisons of nucleotide sequences showed nucleotide differences ranging between 0.2 and 15.1% (Table 2). Furthermore, a comparison of partial 18S rRNA sequences determined herein and those of selected previously published sequences revealed that two sets of six sequences each were 98.8 to 99.6 identical to H. canis 18S rRNA sequences from Thailand (GenBank accession no. KF621082 vs MG209581, MG209583, MG209584, MG209588, MG209590, MG209591 found herein) and Japan (GenBank accession AF418558 vs MG209580, MG209582, MG209585, MG209586, MG209587, MG209593), respectively (Table 2). The remaining two sequences determined in this study (GenBank accession nos. MG209592, MG209594) were 94.2 to 96.4% identical to H. canis sequences from Rawalpindi district of Pakistan (KU535870) while our last sequence (GenBank accession no. MG209589) was 94.8% identical to H. canis sequence previously reported from the Lahore district of Pakistan (Table 2).
DNA sequences were aligned over 537 positions and subjected to phylogenetic analyses. The analyses revealed that the topologies of both trees produced separately using BI and NJ methods were similar (data not shown); hence, only the NJ tree is presented here (Fig. 2). Phylogenetic analyses of 18S rRNA data revealed that H. canis sequences determined herein clustered into three clades (Fig. 2). Clade 1 contained nine sequences (GenBank accession nos. MG209581, MG209583, MG209584, MG209588, MG209590, MG209591–MG209594) which grouped together with those from Pakistan (Rawalpindi), Portugal, Taiwan, Thailand, and the West Indies (Fig. 2). Clade 2 contained only one sequence (GenBank accession no. MG209582). Clade 3 comprised the remaining five sequences (MG209580, MG209585–MG209587, MG209589) which clustered with those from Brazil, Croatia, Japan, Pakistan (Lahore), and Spain (Fig. 2). Clade 4 comprised only a previously reported sequence from Turkey. Despite the variation observed in the 18S rRNA sequences of H. canis from farm dogs, all of them grouped together to the exclusion of all other Hepatozoon spp. included in the analyses (Fig. 2).

Genetic relationships of Hepatozoon canis from Pakistan (bold) with reference sequences selected from previous studies. The relationships were inferred based on phylogenetic analyses of 18S rRNA sequence data using the neighbor-joining (NJ) and Bayesian inference (BI) methods, and Toxoplasma gondii was used as an outgroup. The country of origin of each unique sequence of H. canis is given in parentheses. Nodal support values are indicated: bootstrap for NJ (first) and posterior probability for BI (second). The scale bar indicates distance
Discussion
This study provides the first insights into the genetic characterization of H. canis from farm dogs in three agro-ecological zones of Punjab (Pakistan). H. canis is a tick-borne protozoan parasite known to cause hepatozoonosis in dogs and presents a major clinically underestimated vector-borne disease in dogs across the tropical and subtropical regions globally, (Aydin et al. 2015; Baneth et al. 2011). However, there is paucity of information on the prevalence and infection intensities of H. canis in dogs from Asia. H. canis is frequently diagnosed using microscopic examination for the detection of gamonts in stained blood smears (Ibrahim et al. 1989). Tools such as enzyme-linked immunosorbent assay and indirect fluorescent antibody test have also been employed for the serological diagnosis of antibodies against H. canis (Baneth et al. 1998; Gonen et al. 2004). We used PCR for the detection of H. canis in dogs as this is the most reliable diagnostic tool for the detection of H. canis infection in dogs (Otranto et al. 2011; Vojta et al. 2009).
In this study, the prevalence of H. canis was higher (45.5%) than that of a recent study (11.9%) from Pakistan that also used PCR for the detection of the parasite (Qamar et al. 2017). These differences in the prevalence of H. canis could be due to (i) the difference in geographic locations of dogs (samples were collected from Islamabad, Lahore, and Multan districts in the previous study), (ii) differences in the environmental background of dogs sampled (farm dogs in this study versus pet dogs in the previous study), and (iii) differences in care for the dogs, including tick prophylaxis, i.e., the presence of more ticks on farm dogs as compared to pet dogs (Ahmad et al., unpublished data). Farm dogs are permitted to roam unsupervised and observed to interact more frequently with other farm and stray dogs when they accompany grazing animals. This increases their likelihood of acquiring H. canis infection. Contrarily, pet dogs are better cared for, are usually kept within the confines of the home and backyard, and are walked using a leash under strict supervision. Better tick management by urban dog owners could explain less tick-borne disease infestation and may be the likely cause of the lower prevalence of H. canis in a previous study from Pakistan (Qamar et al. 2017).
From a global perspective, the overall molecular prevalence (45.9%) of H. canis in dogs in this study falls within those reported in previous studies (0.9 to 67.7%) (Criado-Fornelio et al. 2009; Rubini et al. 2005). For example, the prevalence of H. canis (45.5%) in this study was comparable to that in a study from Spain (44%; Criado-Fornelio et al. 2007). However, this level of prevalence was relatively higher as compared to some reports from Brazil (3.8%; de Miranda et al. 2014), Croatia (11.8%; Vojta et al. 2009), Costa Rica (7.5%; Rojas et al. 2014), India (0.26 and 30%; Abd Rani et al. 2011; Singla et al. 2016), Iran (23%; Dalimi et al. 2017), Qatar (1.6%; Alho et al. 2017), Thailand (11.4%; Jittapalapong et al. 2006), and Turkey (3.6%; Aydin et al. 2015). Contrarily, some studies from Brazil have reported relatively higher prevalence (58.7 and 66.4%) of H. canis in dogs than that found in this study (Demoner et al. 2016; Spolidorio et al. 2009). The differences in infection rates could be due to various contributory factors such as the characteristics of the target dog population, sampling seasons, social factors (e.g., canine husbandry, use of tick preventers), geographical location, and climatic conditions which ultimately impact the abundance and distribution of vector tick species (de Miranda et al. 2014; Stich et al. 2014).
The frequency of dogs infected with H. canis was variable in different regions of Pakistan. The highest prevalence was found in Kasur district (61.3%) followed by Rawalpindi (42.5%) and Muzaffargarh (31.5%). These variations in infection rates could be due to differences in climatic conditions and geographical location of the three districts (see Table 1). For instance, Rawalpindi is located in the proximity of the foothills of the Himalayas and exhibits lower average temperatures (both summer and winter) and approximately five-times higher annual rainfall than the flat alluvial planes of Muzaffargarh (PMD 2016). However, despite the presence of more conducive environmental conditions for the transmission via ticks of hepatozoonosis in Rawalpindi, the prevalence of H. canis in this district was lower than that of Kasur (Dantas-Torres 2010; Ul-Hasan et al. 2012). This could possibly be due to stall feeding practices for livestock in Rawalpindi, which result in farm dogs being confined within the farm premises. Contrarily, farm dogs in Kasur are likely to interact more frequently with other farm and stray dogs during grazing of livestock. Under these conditions, dogs can get infested with ticks which could increase the likelihood of acquiring H. canis infection (de Miranda et al. 2014). The lowest prevalence in Muzaffargarh could be due to harsh environmental conditions (e.g., high temperature and low rainfall 0.77 mm during summer) in this region (PMD 2016); hence, a possible lower survival rate for tick vectors involved in the transmission of H. canis. As this is the first report of H. canis infection from farm dogs in Pakistan, almost all the above-listed factors warrant further investigation. Furthermore, farm dogs are likely to acquire infection by eating a tick vector (e.g., R. sanguineus) directly or potentially by ingesting a small rodent infected with the vector harboring the infective protozoan (Demoner et al. 2016). However, the role of these tick vectors and rodents in the transmission of H. canis in Pakistan remains poorly understood.
The 18S rRNA sequences H. canis clustered in three different clades, although with low statistical support for clade separation (see Fig. 2). Nine (out of 15; GenBank accession nos. MG209581, MG209583, MG209584, MG209588, MG209590, MG209591–MG209594) 18S rRNA sequences determined here showed a high similarity with those from previously published sequences from Pakistan (Rawalpindi), Portugal, Taiwan, Thailand, and West Indies. Similarly, five other sequences (GenBank accession nos. MG209580, MG209585–MG209587, MG209589) were closely related to those from Brazil, Japan, Pakistan (Lahore), and Spain while only one sequence grouped separately in between clades 1 and 3 (see Fig. 2). The weak statistical support for separation between clades in the phylogenetic tree suggests that H. canis genotypes represent a single species. However, the occurrence of 15 distinct genotypes of H. canis (found in this study) in Pakistan suggests a high genetic variability within this parasite population. Moreover, the distribution of our H. canis genotypes among these clades does not follow any specific pattern, i.e., the sequence variation is distributed among all three districts. Collectively, these data suggest a frequent minor strain variation and vast geographical spread of the protozoan parasite, not only in Pakistan but also throughout Asia. One possible explanation for the genetic diversity of H. canis could be the international movement of dogs from other countries (Ul-Hasan et al. 2012; Vojta et al. 2009). However, it is also possible that these genotypes might have diverged within the country and not have been introduced as such from other countries. The inter-continental movement of humans as well as animals due to natural disasters and warfare (e.g., World Wars) and social factors could also be considered as potential means for the spread of vectors and vector-borne diseases (Stich et al. 2014; Sutherst 2004).
Conclusion
The present study provides the first molecular characterization of H. canis using an 18S rRNA gene fragment from farm dogs in Pakistan. The prevalence of the parasite was much higher in all three agro-ecological zones studied as compared to a recent study using blood samples collected from Pakistani dogs living in urban settings. A molecular-phylogenetic analysis revealed that H. canis sequences grouped into three clades. Future studies should focus on testing H. canis in domestic and wild canids, covering various climatic regions of Pakistan and evaluating the effect of different factors on its prevalence. Furthermore, studies are required to characterize the tick vector(s) involved in the transmission of H. canis in Pakistan.
References
Abd Rani PA, Irwin PJ, Coleman GT, Gatne M, Traub RJ (2011) A survey of canine tick-borne diseases in India. Parasit Vectors 4(1):141. https://doi.org/10.1186/1756-3305-4-141
Alho AM, Lima C, Latrofa MS, Colella V, Ravagnan S, Capelli G, Madeira de Carvalho L, Cardoso L, Otranto D (2017) Molecular detection of vector-borne pathogens in dogs and cats from Qatar. Parasit Vectors 10:298
Aydin MF, Sevinc F, Sevinc M (2015) Molecular detection and characterization of Hepatozoon spp. in dogs from the central part of Turkey. Ticks Tick Borne Dis 6:388–392
Baneth G (2011) Perspectives on canine and feline hepatozoonosis. Vet Parasitol 181(1):3–11. https://doi.org/10.1016/j.vetpar.2011.04.015
Baneth G, Samish M, Shkap V (2007) Life cycle of Hepatozoon canis (Apicomplexa: Adelorina: Hepatozonoidae) in the ticks Rhipicephalus sanguineus and domestic dogs (Canis familiaris). J Parasitol 93:283–299
Baneth G, Sheiner A, Eyal O, Hahn S, Beaufils JP, Anug Y (2013) Redescription of Hepatozoon felis (Apicomplexa: Hepatozoidae) based on phylogenetic analysis, tissue and blood form morphology, and possible transplacental transmission. Parasit Vectors 6(1):102. https://doi.org/10.1186/1756-3305-6-102
Baneth G, Shkap V, Samish M, Pipano E, Savitsky I (1998) Antibody response to Hepatozoon canis in experimentally infected dogs. Vet Parasitol 74:299–305
Criado-Fornelio A, Buling A, Cunha-Filho NA, Ruas JL, Farias NAR, Rey-Valeiron C, Pingret JL, Etievant M, Barba-Carretero JC (2007) Development and evaluation of a quantitative PCR assay for detection of Hepatozoon sp. Vet Parasitol 150(4):352–356. https://doi.org/10.1016/j.vetpar.2007.09.025
Criado-Fornelio A, Buling A, Pingret JL, Etievant M, Boucraut-Baralon C, Alongi A, Agnone A, Torina A (2009) Hemoprotozoa of domestic animals in France: prevalence and molecular characterization. Vet Parasitol 159(1):73–76. https://doi.org/10.1016/j.vetpar.2008.10.012
Criado-Fornelio A, Martinez-Marcos A, Buling-Saraña A, Barba-Carretero JC (2003) Molecular studies on Babesia, Theileria and Hepatozoon in southern Europe. Vet Parasitol 114(3):173–194. https://doi.org/10.1016/S0304-4017(03)00141-9
Dalimi A, Jameie F, Mohammadiha A, Barati M, Molaei S (2017) Molecular detection of Hepatozoon canis in dogs of Ardabil Province, northwest of Iran. Arch Razi Inst 72:197–201
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Meth 9(8):772–772. https://doi.org/10.1038/nmeth.2109
de Miranda RL, O'Dwyer LH, de Castro JR, Metzger B Rubini AS, Mundim AV, Eyal O, Talmi-Frank D, Cury MC, Baneth G (2014) Prevalence and molecular characterization of Hepatozoon canis in dogs from urban and rural areas in Southeast Brazil. Res Vet Sci 97(2):325–328. https://doi.org/10.1016/j.rvsc.2014.06.015
Demoner LDC, Magro NM, da Silva MRL, de Paula Antunes JMA, Calabuig CIP, O’Dwyer LH (2016) Hepatozoon spp. infections in wild rodents in an area of endemic canine hepatozoonosis in southeastern Brazil. Ticks Tick Borne Dis 7(5):859–864. https://doi.org/10.1016/j.ttbdis.2016.04.002
Dantas-Torres F (2010) Biology and ecology of the brown dog tick, Rhipicephalus sanguineus. Parasit Vectors 3(1):26–26. https://doi.org/10.1186/1756-3305-3-26
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. https://doi.org/10.1093/nar/gkh340
Farooqi AB, Khan AH, Mir H (2005) Climate change perspective in Pakistan. Pak J Meteorol 2
Giannelli A, Lia RP, Annoscia G, Buonavoglia C, Lorusso E, Dantas-Torres F, Baneth G, Otranto D (2017) Rhipicephalus turanicus, a new vector of Hepatozoon canis. Parasitology 144(06):730–737. https://doi.org/10.1017/S003118201600250X
Gomes LA, Moraes PHG, do Nascimento LCS, O’Dwyer LH, MRT N, AdRP R, DCF A, Gonçalves EC (2016) Molecular analysis reveals the diversity of Hepatozoon species naturally infecting domestic dogs in a northern region of Brazil. Ticks Tick Borne Dis 7(6):1061–1066. https://doi.org/10.1016/j.ttbdis.2016.09.008
Gonen L, Strauss-Ayali D, Shkap V, Vincent-Johnson N, Macintire DK, Baneth G (2004) An enzyme-linked immunosorbent assay for antibodies to Hepatozoon canis. Vet Parasitol 122(2):131–139. https://doi.org/10.1016/j.vetpar.2004.03.021
Hall T (2011) BioEdit: an important software for molecular biology. GERF Bull Biosci 2:60–61
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17(8):754–755. https://doi.org/10.1093/bioinformatics/17.8.754
Ibrahim NDG, Rahamathulla PM, Njoku CO (1989) Neutrophil myeloperoxidase deficiency associated with canine hepatozoonosis. Int J Parasitol 19(8):915–918. https://doi.org/10.1016/0020-7519(89)90119-7
Inokuma H, Okuda M, Ohno K, Shimoda K, Onishi T (2002) Analysis of the 18S rRNA gene sequence of a Hepatozoon detected in two Japanese dogs. Vet Parasitol 106:265–271
Jittapalapong S, Rungphisutthipongse O, Maruyama S, Schaefer JJ, Stich RW (2006) Detection of Hepatozoon canis in stray dogs and cats in Bangkok, Thailand. Ann N Y Acad Sci 1081(1):479–488. https://doi.org/10.1196/annals.1373.071
Johnson EM, Panciera RJ, Allen KE, Sheets ME, Beal JD, Ewing SA, Little SE (2009) Alternate pathway of infection with Hepatozoon americanum and the epidemiologic importance of predation. J Vet Int Med 23(6):1315-1318
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kongklieng A, Intapan PM, Boonmars T, Thanchomnang T, Janwan P, Sanpool O, Lulitanond V, Taweethavonsawat P, Chungpivat S, Maleewong W (2015) Detection of Babesia canis vogeli and Hepatozoon canis in canine blood by a single-tube real-time fluorescence resonance energy transfer polymerase chain reaction assay and melting curve analysis. J Vet Diagn Investig 27(2):191–195. https://doi.org/10.1177/1040638714567935
Loftis AD, Kelly PJ, Freeman MD, Fitzharris S, Beeler-Marfisi J, Wang C (2013) Tick-borne pathogens and disease in dogs on St. Kitts, West Indies. Vet Parasitol 196(1-2):44–49. https://doi.org/10.1016/j.vetpar.2013.01.024
Luton K, Gleeson M, Johnson AM (1995) rRNA gene sequence heterogeneity among Toxoplasma gondii strains. Parasitol Res 81:310–315
Maddison WP (2008) Mesquite: a modular system for evolutionary analysis. Evolution 62:1103–1118
Maia C, Ferreira A, Nunes M, Vieira ML, Campino L, Cardoso L (2014) Molecular detection of bacterial and parasitic pathogens in hard ticks from Portugal. Ticks Tick Borne Dis 5(4):409–414. https://doi.org/10.1016/j.ttbdis.2014.01.009
Maps (2017) Maps of world. https://www.mapsofworld.com/lat_long/pakistan-lat-long.html/ (Accessed 01 October 2017).
Mathew JS, Ewing SA, Panciera RJ, Woods JP (1998) Experimental transmission of Hepatozoon americanum Vincent-Johnson et al., 1997 to dogs by the Gulf Coast tick, Amblyomma maculatum Koch. Vet Parasitol 80(1):1–14
Mathew JS, Van Den Bussche RA, Ewing SA, Malayer JR, Latha BR, Panciera RJ (2000) Phylogenetics relationship of Hepatozoon (Apicomplexa: Adeleornia) based on molecular, morphologic, and life cycle character. J Parasitol 86:366–372
Murata T, Imoue M, Tateyama S, Taura Y, Nakama S (1993) Vertical transmission of Hepatozoon canis in dogs. J Vet Med Sci 55:867–868
Otranto D, Dantas-Torres F, Weigl S, Latrofa MS, Stanneck D, Decaprariis D, Capelli G, Baneth G (2011) Diagnosis of Hepatozoon canis in young dogs by cytology and PCR. Parasit Vectors 4:55
Pawar RM, Poornachandar A, Srinivas P, Rao KR, Lakshmikantan U, Shivaji S (2012) Molecular characterization of Hepatozoon spp. infection in endangered Indian wild felids and canids. Vet Parasitol 186:475–479
PMD 2016. Pakistan Metrological Department. http://www.pmd.gov.pk/ (accessed 01 October, 2017).
Potter TM, Macintire DK (2010) Hepatozoon americanum: an emerging disease in the south-central/southeastern United States. J Vet Emerg Crit Care 20:70–76
Qamar M, Malik MI, Latif M, Ain Q, Aktas M, Shaikh RS, Iqbal F (2017) Molecular detection and prevalence of Hepatozoon canis in dogs from Punjab (Pakistan) and hematological profile of infected dogs. Vector Borne Zoonotic Dis 17:179–184
Rojas A, Rojas D, Montenegro V, Gutiérrez R, Yasur-Landau D, Baneth G (2014) Vector-borne pathogens in dogs from Costa Rica: first molecular description of Babesia vogeli and Hepatozoon canis infections with a high prevalence of monocytic ehrlichiosis and the manifestations of co-infection. Vet Parasitol 199:121–128
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Rubini AS, dos Santos PK, Cavalcante GG, Ribolla PEM, O’Dwyer LH (2005) Molecular identification and characterization of canine Hepatozoon species from Brazil. Parasitol Res 97:91–93
Rubini AS, Paduan KS, Martins TF, Labruna MB, O’Dwyer LH (2009) Acquisition and transmission of Hepatozoon canis (Apicomplexa: Hepatozoidae) by the tick Amblyomma ovale (Acari: Ixodidae). Vet Parasitol 164:324–327
Singla LD, Sumbria D, Mandhotra A, Bal MS, Kaur P (2016) Critical analysis of vector-borne infections in dogs: Babesia vogeli, Babesia gibsoni, Ehrlichia canis and Hepatozoon canis in Punjab, India. Acta Parasitol 61:697–706
Smith TG (1996) The genus Hepatozoon (Apicomplexa: Adeleina). J. Parasitol 82:565–585
Spolidorio MG, Labruna MB, Zago AM, Donatele DM, Caliari KM, Yoshinari NH (2009) Hepatozoon canis infecting dogs in the State of Espírito Santo, southeastern Brazil. Vet Parasitol 163:357–361
Stich RW, Blagburn BL, Bowman DD, Carpenter C, Cortinas MR, Ewing SA, Foley D, Foley JE, Gaff H, Hickling GJ, Lash RR, Little SE, Lund C, Lund R, Mather TN, Needham GR, Nicholson WL, Sharp J, Varela-Stokes A, Wang D (2014) Quantitative factors proposed to influence the prevalence of canine tick-borne disease agents in the United States. Parasit Vectors 7:417
Sutherst RW (2004) Global change and human vulnerability to vector-borne diseases. Clin Microbiol Rev. 17:136–173
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 4.0. Mol Biol Evol 30:2725–2729
Ul-Hasan M, Abubakar M, Muhammad G, Khan MN, Hussain M (2012) Prevalence of tick infestation (Rhipicephalus sanguineus and Hyalomma anatolicum anatolicum) in dogs in Punjab, Pakistan. Vet Italia 48:95–98
Vojta L, Mrljak V, Ćurković S, Živičnjak T, Marinculić A, Beck R (2009) Molecular epizootiology of canine hepatozoonosis in Croatia. Int J Parasitol 39:1129–1136
Werle E, Schneider C, Renner M, Völker M, Fiehn W (1994) Convenient single-step, one tube purification of PCR products for direct sequencing. Nucleic Acids Res 22(20):4354
Acknowledgements
Abdullah S. Ahmad is grateful to the Higher Education Commission of Pakistan for the financial assistance under the International Research Support Initiative Program (IRSIP). We would like to thank Dr. Muhammad Luqman Sohail (The Islamia University, Bahawalpur, Pakistan) for his assistance in sample collection.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was approved by the animal ethics committee of the University of Veterinary and Animal Sciences, Lahore, Pakistan.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Section Editor: Xing-Quan Zhu
Electronic supplementary material
Supplementary fig. 1
(DOCX 19 kb)
Rights and permissions
About this article

Cite this article
Ahmad, A.S., Saeed, M.A., Rashid, I. et al. Molecular characterization of Hepatozoon canis from farm dogs in Pakistan. Parasitol Res 117, 1131–1138 (2018). https://doi.org/10.1007/s00436-018-5790-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-018-5790-1