
Abstract
The two ruminant parasites, Paramphistomum cervi and Carmyerius gregarius, were collected from fresh-slaughtered native cattle at local abattoirs in Sadat district, Menoufia province and identified morphologically, then molecularly by sequencing the nucleotides of 18S ribosomal RNA gene (18S rRNA). The nucleotide sequences of the two isolates were 456 (P. cervi) and 401 bases for (C. gregarius). The data were used along with those of several other helminth species from the GenBank to identify these two species genetically. The nucleotide sequences were aligned using multiple sequence alignments of nucleotides by Clustal W 12.1 V and construct their relationship. Neighbor-joining analytical method was used showing sister relationship between C. gregarius from Sadat district and Gastrodiscoides hominis (EF027096) with relative identity of (98%) due to the presence of single nucleotides polymorphisms (SNPs) in the form of indels as nine nucleotides positions. But when clustering of P. cervi Sadat isolate with Paramphistomoidea sp. S4 isolate P5 (GU735643), this relationship shows complete identity (99%) between them. The homology and diversity was done using Bayesian analyses in MrBayes v3.1. This work will give a useful guide for other researchers for the molecular taxonomic position of Paramphistomatidae spp. in Sadat district among the different species around the world.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Paramphistomosis is a disease of domestic and wild ruminants caused by digenetic trematodes of the superfamily Paramphistomoidea. Several species belonging to this superfamily differs in morphological features and the required snail intermediate hosts. The availability of PCR technique and genotyping technology combined with new software tools that can scan raw sequences of genes and identify clues facilitate more accurate identification of the parasite species on a genomic level. This will provide the basis for future development of safe and effective strategies for prevention and control of this disease (Shaheen and Bazh 2012; Shaheen et al. 2013).
Moreover, the obtained gene sequence of Paramphistomatidae spp. gives effective method for subspecific differentiation by using a phylogenic tree of these parasites (Lotfy et al. 2010). The molecular phylogenetic tree for paramphistomes helps to assess their diversity. Based on the reported difficulties of morphological identification of paramphistomes, the development of alternative approaches to better delineate species is occurred such as Yang et al. (2016a) who described one paramphistomes genetically (Gastrothylax crumenifer) from cattle rumen, and also, Yang et al. (2016b) described another paramphistomes (Homalogaster paloniae) from black goat. Moreover, a database can be constructed and then used to evaluate the relations between paramphistomum species, their hosts, and its larvae on genomic bases. This will help in the control of such parasites in animals and humans. For this reason, the present work aimed to demonstrate the sequencing of 18S ribosomal RNA gene (18S rRNA) of P. cervi and C. gregarius Sadat isolates and to determine the phylogenetic relationship as well as the level of similarity and diversity between them and those recorded in the GenBank.
Materials and methods
Parasite and DNA extraction
Adult P. cervi and adult C. gregarius were collected from the rumen of naturally infected cattle at an abattoir. The flukes were washed extensively in physiological saline and identified to species based on morphological characters in the key of Toledo and Fried (2014) and Jones (2005). Total genomic DNA was extracted from about 10 flukes of each paramphistome species using a Qiagen Tissue Kit (Qiagen, Germany) according to the manufacturer’s instructions and eluted into 100 ml H2O, followed by RNase treatment step. Extracted DNA was distance spectrophotometrically quantified at 260/280 nm and was used for polymerase chain reaction (PCR). The treated DNA sample was stored at (−20 °C) until use.
Amplification, sequencing, and assembling of 18S rRNA fragments
This was performed according to methods previously described by Littlewood et al. (2006). Primers were designed based on the conserved regions from published sequences of Paramphistomoidea sp. S4 isolate P5 (GenBank Accession No. GU735643) which used PCR amplification with C. gregarius adults and Paramphistomum cervi isolate PCD (GenBank Accession No. KJ459938). Primers used for P. cervi F 5′- AGAACATCGACATCTTGAAC -3 and R 5′- TATGCTTAAATTCAG CGGGT -3′ and primers used for C. gregarius F 5′- TTGCGCTGATTACGTCCCTG -3′ and R 5′- TTGGCTGCGCTCTTCATCGAC-3′. These sets of primers amplified overlapping fragments to facilitate eventual assembly using Taq polymerase—KOD FX Neo (TOYOBO, Japan). The cycling conditions used were 94 °C for 5 min (initial denaturation), then 94 °C for 1 min (denaturation), 50 °C for 35 s (annealing), 72 °C for 1–3 min (extension) for 30 cycles, and a final extension at 72 °C for 10 min. Each PCR reaction yielded a single band detected in a 1.0% (w/v) agarose gel stained with ethidium bromide (Jia et al. 2010). PCR products were directly sequenced on an ABI 3370 DNA sequencer at a molecular biology unit (Assiut University, Egypt) using a primer walking strategy. The complete rRNA sequence of P. cervi and C. gregarius was assembled using DNAStar software as a sequence editor (Burland 2000).
Prediction of coding genes for small subunit ribosomal RNA genes
The GORF finder tool at NCBI (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) was used to find coding gene sequences, which were subsequently used to search for homologous digenean sequences deposited in the GenBank by using tBLASTn. The rhabditophoran platyhelminth genetic code was specified (Telford et al. 2000). Gene boundaries were confirmed based on comparison and alignment with other published genomes of other species in Fasciolidae, Opisthorchiidae, and Paragonimidae (Le et al. 2000; Cai et al. 2012). Genes for small (rrnS) subunit ribosomal RNA genes were identified by comparison with the 18S rRNA genes of P. cervi isolate PCD (KJ459938), Paramphistomoidea sp. S4 isolate P5 (GU735643), P. cervi isolate PCE (KJ459934), Carmyerius spatiosus (HM159381.1), Gastrodiscoides hominis (EF027096), Cotylophoron cotylophorum (KC503917), Calicophoron microbothrioides isolate V2 (KF791030), Paramphistomum leydeni isolate 3.2 (KJ995529), Cotylophoron cotylophorum isolate Dharmanagar (JX678257), and Paramphistomum epiclitum (KF564870).
Phylogenetic analysis
The obtained sequences were analyzed using the BioEdit software version 7 computer program. Also, nucleotide sequences were aligned using pair-wise comparison and multiple alignments of nucleotide sequences by Clustal W 12.1 V (http://blast.ncbi.nlm.nih.gov/Blast.cgi ) (Larkin et al. 2007). Phylogenetic tree was constructed using Bayesian analyses in MrBayes v3.1 (Ronquist and Huelsenbeck 2003) of the nucleotides sequences of the coding genes in the ribosomal genomes of P. cervi and C. gregarius; the tree was rooted with the Cotylophoron cotylophorum isolate Dharmanagar (JX678257) and was used as an out group. In every case, two runs, each of four chains, were specified. For the nucleotide alignment, the GTR + I + G model was as described previously (Wang et al. 2009), partitioned by codon position. Bayesian analysis was run for 5,000,000 generations and sampled every 1000 generations. The first 25% of trees were omitted as burn in and the remaining trees were used to calculate Bayesian posterior probabilities (Ronquist and Huelsenbeck 2003). The conversion of the alignment format to PHYLIP or NEXUS was done by the program Seqret of EMBOSS (Olson et al. 2003). The percentage divergence values of nucleotide sequences were computed by PAUP4.0b10 (Shalaby and Amer 2012). PAUP: phylogenetic analysis using parsimony 4.0 beta, (Sinauer Associates, Sunderland, Massachusetts, USA.) using Kimura’s two parameter model (Tamura et al. 2013) with a c-shaped parameter (a = 1). PAUP4.0b10 was employed for the neighbor-joining method analysis using the DNA data set. The tree is drawn to scale, with branch lengths (next to the branches) in the same units as those of the evolutionary genetic distances (0.007%) used to infer the phylogenetic tree.
Results
Morphologically, both flukes have conical body. Only the characteristic difference between them is the presence of ventral pouch in C. gregarius. Hence, P. cervi belongs to the family Paramphistomidae while C. gregarius belongs to the family Gastrothylacidae.
The full-length sequences of 18S rRNA gene of P. cervi Sadat isolate was (456) nucleotides. It was AT rich, with (A + T) content of (47.75%) to (C + G) content of (52.25%) in this isolate. There are nine single nucleotide polymorphisms (SNPs); the first ten nucleotides are missed or deleted. Also displayed is a variable number of thymidine that has two deletions of two thymidines; it has an insertion of five bases.
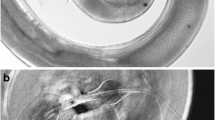
But for C. gregarius Sadat isolate, the full-length sequences of 18S rRNA gene was 401 nucleotides; it was AT rich, with A + T content of 41.75% to C + G content of 58.25% in this isolate. There are nine SNPs in the form of deletion, as in Fig. 1a, b. The multiple sequence alignment of 18S rRNA gene between C. gregarius Sadat isolate and P. cervi Sadat isolate indicates that the identity was 79.33% and alignment score of 75.56 with genetic distance of 0.12219 between them after clustering together by the neighbor-joining method analysis using the DNA data set.

a, b The multiple sequence alignment between Carmyerius gregarius Sadat isolate (401 nucleotides) and Paramphistomum cervi Sadat isolate (456 nucleotides) indicating that the length of the identity percent was 79.33%
Results of the phylogenetic analysis in Fig. 2 indicated that P. cervi belong to a multispecies complex that divided into several strains all over the world; one species cluspurified the species together on their own branch with very short branch lengths between them. P. cervi Sadat isolate lying on a short branch from the main first group that includes various isolates of P. cervi as Paramphistomoidea sp. S4 isolate P5 on a genetic distance (0.00366) and homology of 99% lying on the short branch and with P. cervi PCD isolate and P. cervi PCE isolate on a genetic distance of 0.01292 with 95% identity that lying on the long branch on the tree. But the identity was 79.33% between C. gregarius and P. cervi Sadat isolate, with a genetic distance of 0.12219 lying faraway on the tree from each other and gaps between them were 4.5%, maximum score 2793 bits, and E value is 0.0 with considerable high indels and SNPs for Cotylophoron cotylophorum (KC503917). It was also lying far away from our isolate on the tree with genetic distance of 0.23366 with low identity of 94%. Belonging the C. gregarius Sadat isolate, pair-wise comparison and multiple alignments of the gene sequences show that C. gregarius Sadat isolate and Gastrodiscoides hominis (EF027096) isolate have revealed a high sequence homology because they shared a high number of identical nucleotides. There was similar sequences and gaps between them were 0%, maximum score 3219 bits, and E value is 0.0 with considerable one indel and SNPs and identity value 99.9%, but Carmyerius spatiosus (HM159381) isolate was dissimilar; gaps between them were 2%, maximum score 2793 bits, and E value is 0.0 with considerable high indels and SNPs and identity value 71.1%. Also, for Paramphistomum epiclitum, it was dissimilar when compared with our strain. Gaps between them were 0.409%, maximum score 3219 bits, and E value is 0.0 with considerable one indel and SNPs and identity value 96.3%. The homology was 95% between C. gregarius Sadat isolate and C. cotylophorum isolate (KC503917). Phylogenetic tree indicated that C. gregarius Sadat isolate belong to a multispecies complex (Paramphistomoidea spp.) and showing that C. gregarius Sadat isolate and Carmyerius spatious were lying on a far away long branch of genetic distance with 0.15364 and on the same short branch with Gastrodiscoides hominis of genetic distance with 0.0014, but it is lying faraway from C. cotylophorum isolate (JX678257) of genetic distance with 0.1467 and it is shown that Carmyerius spatious and C. gregarius Sadat isolate were not identical to each other (71.1%), but somewhat identical to Gastrodiscoides hominis of 99% identity and faraway on the tree from P. cervi Sadat isolate within Paramphistomoidea spp.

Phylogenetic tree and evolutionary relationship of Carmyerius gregarius and Paramphistomum cervi Sadat isolate based on the 18S rRNA gene locus) using the clustal program for pair-wise and multiple sequence alignment; the tree was constructed by the neighbor-joining analysis (NJ) with genetic distance of 0.007
Discussion
Paramphistomum worms are difficult to identify because most have thick robust conical bodies in which the internal organs are hard to be seen. In the present study, the identification of P. cervi and C. gregarius put them in separate families. This result is proved by sequence application. The investigator may have to rely on another specific procedure of identifying adult worms. Obtaining the specific gene sequence of Paramphistomoidea spp. gives an effective method for subspecific differentiation using a phylogenic tree of these parasites can be developed (Lotfy et al. 2010). The development of alternative approaches to better delineate species is developed. PCR-based techniques using rDNA ITS2 sequences have proven to be a reliable tool to identify digenean species and to recover their phylogenetic relationships (Van Van et al. 2009). Moreover, 18S rRNA gene locus has been selected to be a useful marker for species identification of paramphistomes (Goswami et al. 2009). Results of the present study clarified the very high intraspecies homology between the isolated local strain (P. cervi) and Paramphistomoidea sp. S4 isolate P5 of high identity value; they were the closest interspecific pair. But it was low interspecies homology between P. cervi and C. gregarius Sadat isolate. On the other hand, homology was very high interspecies between the C. gregarius Sadat isolate and Gastrodiscoides hominis; they were the closest interspecific pair due to a high number of identical nucleotides. But intraspecies homology was low between it and both Carmyerius spatiosus (HM159381) and P. cervi Sadat isolate. This was due to several main variable regions which were identified in the 18S rRNA gene sequences. These regions were characterized by a considerable number of indels as SNPs in the form of deletion with low identity value. Homology was very high within P. cervi isolates, and the divergence was very low. The homology was much lower between P. cervi and Carmyerius spatious and the divergence was much higher. This was in agreement with the concept of Olson et al. (2003). And also, in agreement with Zheng et al. (2014) who reported that the intraspecific variation within P. cervi was between 0 and 0.2% for 18S rRNA. For the similarly of C. gregarius Sadat isolate with other species such as C. cotylophorum (KC503917) has revealed a low sequence homology, due to considerable high indels and SNPs, and the identity value was much low. Also, there was a complete homology between P. cervi Sadat isolate and Paramphistomoidea sp. S4 isolate P5 and share similar sequences, no gaps between them, with one indels and SNPs and complete identity. Phylogenetic results suggest that P. cervi and C. gregarius Sadat isolate belong to a multispecies complex (Paramphistomoidea spp.) that divided into several strains all over the world, one species cluspurified the species together on their own branch with very short branch lengths between them, which resemble results conducted by Shalaby and Amer (2012); our isolate in the present study lying on a short branch from the main first group, the tree including various isolates as the C. gregarius Sadat isolate and Carmyerius spatiosus were lying on the far away long branch and on the same short branch with Gastrodiscoides hominis, but it is lying faraway from C. cotylophorum isolate Dharmanagar (JX678257). From the previous data shown on the phylogenetic tree, it is shown that Gastrodiscoides hominis and C. gregarius Sadat isolate were completely identical, but somewhat differ from and of low identity with P. cervi of the present study.
Conclusion
The present study is constructed for the molecular characterization and phylogenetic analysis of P. cervi and C. gregarius from Sadat district, Menoufia province, Egypt. Depending on the complete sequence of 18S rRNA gene locus of the complete ribosomal genome sequence of selected species under the study, this is the first record about molecular characterization of these species in this province. Phylogenetic tree based on sequence alignment of genes of known species and two species under study revealed that P. cervi Sadat isolate lying on a short branch from the main first group including various isolates of P. cervi as Paramphistomoidea sp. S4 isolate P5 on genetic distance (0.00366) and homology of 99% with low identity with C. gregarius Sadat isolate (71.1%). On the other hand, C. gregarius Sadat isolate and Gastrodiscoides hominis were completely identical, but somewhat differ from and of low identity with Carmyerius spatiosus. In this study, the present tree will provide an important resource for the studies of inter- and intraspecific variation of the paramphistomes and a resource for comparative ribosomal genomics and systematic studies of digeneans in Egypt.
References
Burland TG (2000) DNASTAR’s Lasergene sequence analysis software. Methods Mol Biol 132:71–91
Cai XQ, Liu GH, Song HQ, Wu CY, Zou FC et al (2012) Sequences and gene organization of the mitochondrial genomes of the liver flukes Opisthorchis viverrini and Clonorchis sinensis (Trematoda). Parasitol Res 110:235–243
Goswami LM, Prasad PK, Tandon V, Chatterjee A (2009) Molecular characterization of Gastrodiscoides hominis (Platyhelminthes: Trematoda: Digenea) inferred from ITS rDNA sequence analysis. Parasitol Res 104:1485–1490
Jia WZ, Yan HB, Guo AJ, Zhu XQ, Wang YC et al (2010) Complete mitochondrial genomes of Taenia multiceps, T. hydatigena and T. pisiformis: additional molecular markers for a tapeworm genus of human and animal health significance. BMC Genomics 11:447
Jones A (2005) Superfamily Paramphistomoidea Fischoeder, 1901. In: Jones A, Bray RA, Gibson DI (eds) Keys to the Trematoda. CABI Publishing and the Natural History Museum, New York Chapter 20& 27
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA et al (2007) Clustal W and clustal X version 2.0. Bioinformatics 23:2947–2948
Le TH, Blair D, Agatsuma T, Humair PF, Campbell NJ et al (2000) Phylogenies inferred from mitochondrial gene orders—a cautionary tale from the parasitic flatworms. Mol Biol Evol 17:1123–1125
Littlewood DT, Lockyer AE, Webster BL, Johnston DA, Le TH (2006) The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Mol Phylogenet Evol 39:452–467
Lotfy WM, Brant SV, Ashmawy KI, Devkota R, Mkoji GM, Loker ES (2010) A molecular approach for identification of paramphistomes from Africa and Asia. Vet Parasitol 174(2010):234–240
Olson PD, Cribb TH, Tkach VV, Bray RA, Littlewood DTJ (2003) Phylogeny and classification of the Digenea (Platyhelminthes: Trematoda). Int J Parasitol 33:733
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Shaheen HM, Bazh EK (2012) Morphological response of Paramphistomum cervi to treatment with oxyclozanide and niclosamide in vitro. J Egypt Vet med Assoc 72(4):561–574
Shaheen H, Sadek KM, Bazh EK (2013) Evaluation of oxyclozanide and niclosamide combination as alternative antiparamphistomal therapy in buffaloes. Afr J Pharm Pharmacol 7(30):2157–2166. doi:10.5897/AJPP2013.3493
Shalaby IMI, Amer SAM (2012) Preliminary molecular identification of two helminthes (Moniezia sp. and Paramphistomum sp.) in the province of Taif, Saudi Arabia. World Applied Sciences Journal 17(8):986–991
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Telford MJ, Herniou EA, Russell RB, Littlewood DT (2000) Changes in mitochondrial genetic codes as phylogenetic characters: two examples from the flatworms. Proc Natl Acad Sci U S A 97:11359–11364
Toledo R, Fried B (2014) (eds.) Digenetic Trematodes, Advances in Experimental Medicine and Biology, Volume 766 of the series pp 365–392 (Date: 21 May 2014) Chapter Amphistomes (Digenetic Trematodes) , © Springer Science & Business Media New York 2014. DOI: 10.1007/978-1-4939-0915-5_11
Van Van K, Dalsgaard A, Blair D, Le TH (2009) Haplorchis pumilio and H. taichui in Vietnam discriminated using ITS-2 DNA sequence data from adults and larvae. Exp Parasitol 123:146–151
Wang CR, Li L, Ni HB, Zhai YQ, Chen AH et al (2009) Orientobilharzia turkestanicum is a member of Schistosoma genus based on phylogenetic analysis using ribosomal DNA sequences. Exp Parasitol 121:193–197
Yang X, Wang L, Chen H, Feng H, Shen B, Hu M, Fang R (2016a) The complete mitochondrial genome of Gastrothylax crumenifer (Gastrothylacidae, Trematoda) and comparative analyses with selected trematodes. Parasitol Res 115(6):2489–2497. doi:10.1007/s00436-016-5019-0
Yang X, Wang L, Feng H, Qi M, Zhang Z, Gao C, Wang C, Hu M, Fang R, Li C (2016b) Characterization of the complete mitochondrial genome sequence of Homalogaster paloniae (Gastrodiscidae, Trematoda) and comparative analyses with selected digeneans. Parasitol Res 115(10):3941–3949. doi:10.1007/s00436-016-5160-9
Zheng X, Chang QC, Zhang Y, Tian SQ, Lou Y, Duan H, Guo DH, Wang CR, Zhu XQ (2014) Characterization of the complete nuclear ribosomal DNA sequences of Paramphistomum cervi. Sci World J 2014(2014):751907. doi:10.1155/2014/751907
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The cattle from which P. cervi and C. gregarius adults were collected were being processed at a local abattoir in Sadat district, Menoufia Province, Egypt, as part of the normal work of the abattoir.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
El-Bahy, N.M., Bazh, E.K., Abdel azizn, A.R. et al. New approach to molecular characterization of Paramphistomum cervi and Carmyerius gregarius and comparative analyses with selected trematodes. Parasitol Res 116, 1417–1422 (2017). https://doi.org/10.1007/s00436-016-5344-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-016-5344-3