
Abstract
Gastrodiscidae species are neglected but significant paramphistomes in small ruminants, which can lead to considerable economic losses to the breeding industry of livestock. However, knowledge about molecular ecology, population genetics, and phylogenetic analysis is still limited. In the present study, we firstly sequenced and analyzed the full mitochondrial (mt) genome of Homalogaster paloniae (14,490 bp). The gene contents and organization of the H. paloniae mt genome is the same as that of other digeneans, such as Fasciola hepatica and Paramphistomum cervi. It is interesting that unlike other paramphistomes, H. paloniae is flat in shape which is similar with Fasciola, such as F. hepatica. Phylogenetic analysis of H. paloniae and other 17 selected digeneans using concatenated amino acid sequences of the 12 protein-coding genes showed that Gastrodiscidae is closely related to Paramphistomidae and Gastrothylacidae. The availability of the mt genome sequence of H. paloniae should provide an important foundation for further molecular study of Gastrodiscidae and other digeneans.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastrodiscidae species are important paramphistomes parasiting in the large intestine of small ruminants such as goats, sheep and cattles, and Homalogaster paloniae (Poirier, 1883) is one of the most common species (Li 2011; Taylor et al. 2007). As a neglected pathogen, H. paloniae have been reported in Burma, China, Formosa, India, Indonesia, Japan, Philippines, and Thailand (Yamaguti 1971). H. paloniae (Poirier, 1883) usually inhabits in the caecum of animals. Mature eggs are expelled with feces from the intestine into the environment and will develop into miracidium after several days under favor conditions. Subsequently, miracidium will invade into freshwater snails (intermediate host). Miracidium will develop into cercaria, and escape from the freshwater snails, then develop into metacercaria on the water plants. Ruminants infect H. paloniae by intaking water plants polluted by metacercaria. Although animals infected with H. paloniae usually do not show obvious symptoms, it can cause considerable losses to the breeding of sheep and cattle under heavy burden (Guoqing 2006).
Since no effective vaccine is available, application of chemical drugs is the main methods for the prevention and control of H. paloniae and other paramphistomes. Accurate diagnosis of H. paloniae infection is essential for the prevention and control of this species. Traditional morphological methods have been widely used for a long time; however, these methods are time-consuming and inaccurate (Bott et al. 2009). Based on these restricted factors, molecular methods based on PCR were developed for species identification (Itagaki et al. 2003; Morgan and Blair 1995). Recently, the mitochondrial genome has been used for species identification, population diversity, phylogenetic analysis, and so on (Cheng et al. 2016; Choi et al. 2012; Liu et al. 2012, 2015). Besides, advances in PCR-coupled sequencing together with bioinformatics analysis have been proved to be useful for the molecular study of species (Jex et al. 2010). In the present study, we aimed (i) to characterize the H. paloniae mt genome, and this is the first report about full mt genome of Gastrodiscidae; (ii) to assess the phylogenetic relationship between Gastrodiscidae and other family; and (iii) to provide useful information for further study of species identification, biology, population genetics and phylogenetic analysis.
Materials and methods
Parasites and DNA extraction
Adult flukes were collected from the caecum of a naturally infected black goat post-mortem in Macheng, Hubei province, PR China. After sufficiently washed with physiological saline, the specimens were fixed in 75 % (v/v) ethanol and preserved at −20 °C until use. These flukes were identified to be H. paloniae based on key morphological characteristics and parasitic positions (Guoqing 2006; Li 2011; Taylor et al. 2007).
Subsequently, total genomic DNA of H. paloniae was isolated from single worm by a kit (E.Z.N.A.® Tissue DNA Kit, D3396-01). The ITS-2 (the second internal transcribed spacer) region of H. paloniae was amplified and sequenced for further molecular identification (Itagaki et al. 2003); the sequence was 100 % similarity with a sequence available for H. paloniae (GenBank accession no. KM281535.1).
PCR-coupled sequencing of H. paloniae mitochondrial genome
Firstly, seven pairs of primers (Table 1) were designed and synthesized to amplify short fragments from nad1, nad4, cox1, cox2, nad5, rrnS, and cytb based on the conserved regions of the mt genomes of Fasciola hepatica (Le et al. 2001a) and Paramphistomum cervi (Yan et al. 2013). PCR reactions (25 μl) were as follow: 1 × Taq polymerase buffer, 0.2 mM each of dNTP, 0.5 μM of each primer, 2 U Taq polymerase (Takara), and 2.5 μl genomic DNA in a thermocycler (Biometra) under the following conditions: 94 °C for 5 min, followed by 35 cycles of 94 °C/30 s, 50 °C/30 s, 72 °C/1 min, and a final extension of 72 °C/7 min. Subsequently, PCR products were identified by agarose gel electrophoresis, and positive amplicons were sent for sequencing in both directions. Later, seven pairs of primers were designed based on the obtained seven short fragments to amplify the remaining sequences of the complete mt genome in seven long-PCR reactions (Table 1). The long-PCR reactions (25 μl) were performed in 0.8 mM of each dNTPs, 2.5 μl 10× LA Taq buffer, 0.5 μM of each primer, 2.5 U LA Taq polymerase (Takara), and 2.5 μl genomic DNA sample. PCR reactions were carried out under the following condition: 94 °C for 5 min, then 35 cycles at 94 °C/30 s, and annealed at 50 °C/30 s, followed by extension at 72 °C/8 min, and a final extension of 72 °C/7 min. Then, positive PCR amplicons were purified and cloned into pGEM-T vector (Promega, USA) for sequencing using a primer-walking strategy.
Assembly, annotation, and bioinformatics analyses
The complete H. paloniae mt sequences were assembled by bioinformatics analysis and manually adjustment. Gene boundaries of the mt genome sequence of H. paloniae were predicted by aligning against that of F. hepatica (Le et al. 2001a) and P. cervi (Yan et al. 2013) using the software Clustal X 1.83 (Thompson et al. 1997). The protein-coding genes, transfer RNA, ribosomal RNA, and non-coding regions were annotated as reported (Yan et al. 2013).
Nucleotide comparison of the complete mt genome of H. paloniae and other 17 selected digeneans was conducted. Meanwhile, the AT-skew and GC-skew were calculated as previous study (Baek et al. 2014; Yan et al. 2014).
Sliding window analysis of nucleotide variation
Pairwise alignment of the complete mt genome of H. paloniae, Fischoederius elongates, P. cervi, Gastrothylax crumenifer, and Ogmocotyle sikae was accomplished by MEGA v6.0 to predict variable nucleotide sites (Tamura et al. 2013). Subsequently, a sliding window analysis of H. paloniae, F. elongates, P. cervi, G. crumenifer, and O. sikae was accomplished using DnaSP v.5.0 to assess the nucleotide variation diversity for the 12 protein-coding genes among these five paramphistomes (Librado and Rozas 2009).
Phylogenetic analysis
All 12 protein-coding genes of H. paloniae mt genome and other 17 selected digeneans were translated, concatenated, and aligned for phylogenetic analysis, including Clonorchis sinensis (NC_012147) (Shekhovtsov et al. 2010), Fasciola gigantica (NC_024025) (Liu et al. 2014), F. hepatica (NC_002546) (Le et al. 2001a), Fischoederius elongatus (KM_397348) (Yang et al. 2015b), G. crumenifer (KM_400624), Haplorchis taichui (NC_022433.1) (Lee et al. 2013), Hypoderaeum conoideum (KM111525) (Yang et al. 2015a), Metagonimus yokogawai (KC330755.1), O. sikae (KR006934) (Ma et al. 2015), Opisthorchis felineus (EU_921260) (Shekhovtsov et al. 2010), Opisthorchis viverrini (JF729304.1) (Cai et al. 2012), P. cervi (NC_023095.1) (Yan et al. 2013), Schistosoma haematobium (NC_008074) (Littlewood et al. 2006), Schistosoma japonicum (AF215860) (Le et al. 2001b), Schistosoma mekongi (NC_002529) (Le et al. 2000), Schistosoma spindale (NC_008067) (Littlewood et al. 2006), and Trichobilharzia regent (NC_010976) (Webster et al. 2007). And Taenia solium (NC_004022.1) (Nakao and Sako 2003) was included as an outgroup control.
The amino acid sequences were aligned and subjected to phylogenetic analysis by maximum likelihood methods using MEGA v.6.0 with default settings (Tamura et al. 2013).
Results
Genome content and organization
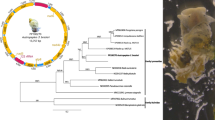
The complete mitochondrial (mt) genome of H. paloniae (GenBank accession no. KT266674) is 14,490 bp in length (Fig. 1) and contains 12 protein-coding genes, 22 transfer ribonucleic acid (tRNA) genes, two ribosomal ribonucleic acid (rRNA) genes (rrnS and rrnL), and two non-coding regions (Table 2). All the genes are transcribed in the same direction, which is in accordance with other digeneans (Le et al. 2001a; Yan et al. 2013). The gene arrangement is similar with other digeneans except for S. haematobium and S. spindale (Littlewood et al. 2006).

Arrangement of the mitochondrial genome of Homalogaster paloniae. Genes organization of 12 protein-coding genes, 22 tRNA gene, two RNA genes, and two non-coding regions in the mitochondrial genome of Homalogaster paloniae
As for the nucleotide composition, H. paloniae mt genome is obviously favor in T (Table 3). The nucleotide contents in the complete mt genome are 21.92 % (A), 9.28 % (C), 43.21 % (T), and 25.6 % (G). And the A + T content of mt genes range from 63.23 to 71.04 %, total A + T content is 65.12 %.
Annotation of H. paloniae mt genome
The H. paloniae mt genome has 12 protein-coding genes (Fig. 1). For these genes, the most commonly used start codon is ATG (nine of 12 protein genes), and GTG is used by the remaining genes (three of 12 protein genes) (Table 2), which is in agreement with other digeneans (Cai et al. 2012; Le et al. 2001a; Littlewood et al. 2006; Yan et al. 2013; Yang et al. 2015a). The termination codon is TAA for nad5, and TAG for the rest genes. No incomplete codons are used in the mt genome of H. paloniae.
The 12 protein-coding genes encode 3359 amino acids excluding the termination codons (Table 4). Among all the amino acids, Phe (TTT 10.00 %) is the most used, followed by Leu (TTG 7.09 %), and Leu (TTA 6.91 %). The least used codon is Arg (CGC 0.12 %), followed by Leu (CTC 0.15 %), and Arg (CGG 0.15 %).
As for the tRNA genes and rRNA genes, the length of the 22 tRNA genes ranged from 59 to 72 bp (Table 2). The size of rrnS and rrnL were 741 and 984 bp, respectively (Table 3). The location of rrnS is between trnC and cox2 and rrnL is between trnT and trnC, and their A + T content was 63.70 and 65.14 %, respectively (Table 3).
In the H. paloniae mt genome, two non-coding regions were recognized based on their AT-rich features and locations (Yan et al. 2013), one short non-coding region (SNR 71 bp) and one long non-coding region (LNR 1022 bp) (Table 2). The location of SNR is between cytb and nad4L, and LNR is located between trnE and cox3.
Comparative analyses of the mt genomes of H. paloniae and other digeneans
Nucleotide composition, AT skews and GC skews of the mt genome of H. paloniae and other digeneans were presented in Table 5. All the 18 digeneans mt genomes are rich in A + T. The nucleotide composition of H. paloniae is biased to T compared with A (AT skew = −0.327), and biased to G compared with C (GC skew = 0.468), which is in accordance with that of other digeneans.
Nucleotide variability
The sliding window analysis was showed in Fig. 2; the highest level of nucleotide variability was within cox1, and the lowest was within cox3. In our study, cox1 and nad6 are the most conserved genes, and cox3 and cytb are the least conserved.

A sliding window analysis of complete mt genome sequences of Homalogaster paloniae, Fischoederius elongates, Paramphistomum cervi, Gastrothylax crumenifer, and Ogmocotyle sikae. The black line in the picture showed nucleotide diversity in the sliding window analysis (windows = 300 bp; steps = 10 bp). There are two overlapping genes in the protein-coding genes, one is between Nad4L and nad4, and the other is between cox2 and nad6. All the 12 protein-coding genes are indicated using grey boxes
Phylogenetic analyses
Waeschenbach and colleagues reported that the complete mt sequences are more reliable for phylogenetic analyses (Waeschenbach et al. 2012). Based on previous study, the concatenated amino acid sequence data of the 12 protein-coding genes of H. paloniae and other 17 digeneans (C. sinensis, F. gigantica, F. hepatica, F. elongatus, G. crumenifer, H. taichui, H. paloniae, H. conoideum, M. yokogawai, O. sikae, O. felineus, O. viverrini, P. cervi, S. haematobium, S. japonicum, S. mekongi, S. spindale, and T. regent) and one tapeworm (T. solium, as an outgroup) were used for the phylogenetic study. The relationship of H. paloniae with selected digeneans was showed in Fig. 3. The phylogenetic tree contains two clades with significantly strong support (100 %), one contains 13 members from eight families (Opisthorchiidae, Heterophyidae, Echinostomatidae, Fasciolidae, Notocotylidae, Gastrodiscidae, Paramphistomidae, and Gastrothylacidae), and the other contains five members from the Schistosomatidae family. The tree indicated that H. paloniae was together with other paramphistomes including F. elongatus, G. crumenifer, O. sikae, and P. cervi in one sub-clade, but separated from F. gigantica and F. hepatica from Fasciolidae, and H. paloniae has the closest relationship with members from Paramphistomidae and Gastrothylacidae that inhabiting in small ruminants. Nevertheless, more mt genomes from digeneans are needed for further phylogenetic analyses in the future.

The phylogenetic relationships of Homalogaster paloniae and other digeneans based on concatenated amino acid sequence data representing 12 protein-coding genes using Taenia solium as an outgroup
Discussion
As an important paramphistome, H. paloniae can lead to considerable economic losses to the breeding industry of small ruminants under heavy burden. Although the development of advances in technology, knowledge about epidemiology, biology, and genetics is still limited.
The present study firstly characterized the mt genome of H. paloniae. The gene content and organization are the same as other digeneans. Knowledge of the H. paloniae mt genome should provide useful for comparative study of this species and other digeneans.
As for the complete mt of H. paloniae, the gene arrangement is the same as other digeneans except for S. haematobium and S. spindale (Littlewood et al. 2006). All the protein-coding genes use complete codons, which is in accordance with other selected digeneans. Among all the 18 digeneans, all species show strand asymmetry (AT skew = −0.513 ∼ −0.159; GC skew = 0.359 ∼ 0.512). With the accomplishment of sliding window analysis, cox1 gene is the most conserved region among these four paramphistomes; this is in accordance with previous studies, which indicated the conserved characteristics of cox1 gene (Chibwana et al. 2013; Pérez-del-Olmo et al. 2014; Rollinson et al. 2009). Phylogenetic analyses can provide a basic understanding of the relationship of H. paloniae with other digeneans.
Although H. paloniae is closer to Fasciolidae in shape, phylogenetic analysis based on the complete mt genome of H. paloniae and other digeneans indicated that H. paloniae is closely related to paramphistomes, this is in accordance with their relationship in taxonomy.
Now, the H. paloniae mt genome is available; this should provide useful information for the study of epidemiology, biology, species identification, population genetic, and phylogenetic analyses.
Conclusions
In conclusion, our study firstly reported the complete mt genome sequence of H. paloniae and compared the mt genome of H. paloniae with other selected digeneans. The H. paloniae mt genome is the first mt genome available for Gastrodiscidae. Knowledge of mt genome of H. paloniae should enrich the mt genome databases of digeneans and also provide useful information for the study of epidemiology, biology, population genetics, as well as phylogenetic analyses.
References
Baek SY, Choi EH, Jang KH, Ryu SH, Park SM, Suk HY, Chang CY, Hwang UW (2014) Complete mitochondrial genomes of Carcinoscorpius rotundicauda and Tachypleus tridentatus (Xiphosura, Arthropoda) and implications for chelicerate phylogenetic studies. Int J Biol Sci 10:479–489
Bott NJ, Campbell BE, Beveridge I, Chilton NB, Rees D, Hunt PW, Gasser RB (2009) A combined microscopic-molecular method for the diagnosis of strongylid infections in sheep. Int J Parasitol 39:1277–1287
Cai XQ, Liu GH, Song HQ, Wu CY, Zou FC, Yan HK, Yuan ZG, Lin RQ, Zhu XQ (2012) Sequences and gene organization of the mitochondrial genomes of the liver flukes Opisthorchis viverrini and Clonorchis sinensis (Trematoda). Parasitol Res 110:235–243
Cheng T, Liu GH, Song HQ, Lin RQ, Zhu XQ (2016) The complete mitochondrial genome of the dwarf tapeworm Hymenolepis nana—a neglected zoonotic helminth. Parasitol Res 115:1253–1262
Chibwana FD, Blasco Costa S, Georgieva S, Hosea KM, Nkwengulila G, Scholz T, Kostadinova A (2013) A first insight into the barcodes for African diplostomids (Digenea: Diplostomidae): brain parasites in Clarias gariepinus (siluriformes: Clariidae). Infect Genet Evol 17:62–70
Choi KS, Koekemoer LL, Coetzee M (2012) Population genetic structure of the major malaria vector Anopheles funestus s.s. and allied species in southern Africa. Parasit Vectors 5:283
Guoqing L (2006) Veterinary parasitology. China Agricultural Science and Technology Press
Itagaki T, Tsumagari N, Tsutsumi K, Chinone S (2003) Discrimination of three amphistome species by PCR-RFLP based on rDNA ITS2 markers. J Vet Med Sci 65:931–933
Jex AR, Hall RS, Littlewood DTJ, Gasser RB (2010) An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res 38:522–533
Le TH, Blair D, Agatsuma T, Humair PF, Campbell NJ, Iwagami M, Littlewood DT, Peacock B, Johnston DA, Bartley J, Rollinson D, Herniou EA, Zarlenga DS, McManus DP (2000) Phylogenies inferred from mitochondrial gene orders-a cautionary tale from the parasitic flatworms. Mol Biol Evol 17:1123–1125
Le TH, Blair D, McManus DP (2001a) Complete DNA sequence and gene organization of the mitochondrial genome of the liverfluke, Fasciola hepatica L. (Platyhelminthes; Trematoda). Parasitology 123:609–621
Le TH, Humair PF, Blair D, Agatsuma T, Littlewood DT, McManus DP (2001b) Mitochondrial gene content, arrangement and composition compared in African and Asian schistosomes. Mol Biochem Parasitol 117:61–71
Lee D, Choe S, Park H, Jeon HK, Chai JY, Sohn WM, Yong TS, Min DY, Rim HJ, Eom KS (2013) Complete mitochondrial genome of Haplorchis taichui and comparative analysis with other trematodes. Korean J Parasitol 51:719–726
Li XR (2011) Color atlas of animal parasitosis, 2nd edn. China Agriculture Press, Beijing
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Littlewood DT, Lockyer AE, Webster BL, Johnston DA, Le TH (2006) The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Mol Phylogenet Evol 39:452–467
Liu GH, Wang SY, Huang WY, Zhao GH, Wei SJ, Song HQ, Xu MJ, Lin RQ, Zhou DH, Zhu XQ (2012) The complete mitochondrial genome of Galba pervia (Gastropoda: Mollusca), an intermediate host snail of Fasciola spp. Plos One 7:e42172
Liu GH, Gasser RB, Young ND, Song HQ, Ai L, Zhu XQ (2014) Complete mitochondrial genomes of the ‘intermediate form’ of Fasciola and Fasciola gigantica, and their comparison with F. hepatica. Parasit Vectors 7:150
Liu GH, Jia YQ, Wang YN, Zhao GH, Zhu XQ (2015) The complete mitochondrial genome of the gullet worm Gongylonema pulchrum: gene content, arrangement, composition and phylogenetic implications. Parasit Vectors 8:100
Ma J, He JJ, Liu GH, Blair D, Liu LZ, Liu Y, Zhu XQ (2015) Mitochondrial genome of Ogmocotyle sikae and implications for phylogenetic studies of the Notocotylidae trematodes. Infect Genet Evol 37:208–214
Morgan JAT, Blair D (1995) Nuclear rDNA ITS sequence variation in the trematode genus Echinostoma: an aid to establishing relationships within the 37-collar-spine group. Parasitology 111:609–615
Nakao M, Sako YA (2003) The mitochondrial genome of the tapeworm Taenia solium: a finding of the abbreviated stop codon U. J Parasitol 89:633–635
Pérez-del-Olmo A, Georgieva S, Pula HJ, Kostadinova A (2014) Molecular and morphological evidence for three species of Diplostomum (Digenea: Diplostomidae), parasites of fishes and fish-eating birds in Spain. Parasit Vectors 7:502
Rollinson D, Webster JP, Webster B, Nyakaana S, Jørgensen A, Stothard JR (2009) Genetic diversity of schistosomes and snails: implications for control. Parasitology 136:1801–1811
Shekhovtsov SV, Katokhin AV, Kolchanov NA, Mordvinov VA (2010) The complete mitochondrial genomes of the liver flukes Opisthorchis felineus and Clonorchis sinensis (Trematoda). Parasitol Int 59:100–103
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Taylor MA, Coop RL, Wall RL (2007) Veterinary parasitology, 3rd edn. Blackwell Publishing Ltd, London
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Waeschenbach A, Webster BL, Littlewood DT (2012) Adding resolution to ordinal level relationships of tapeworms (Platyhelminthes: Cestoda) with large fragments of mtDNA. Mol Phylogenet Evol 63:834–847
Webster BL, Rudolfová J, Horák P, Littlewood DTJ (2007) The complete mitochondrial genome of the bird schistosome Trichobilharzia regenti (Platyhelminthes: Digenea), causative agent of cercarial dermatitis. J Parasitol 93:553–561
Yamaguti S (1971) Synopsis of digenetic trematodes of vertebrates, vol I. Keigaku Publishing Ltd, Tokyo
Yan HB, Wang XY, Lou ZZ, Li L, Blair D, Yin H, Cai JZ, Dai XL, Lei MT, Zhu XQ, Cai XP, Jia WZ (2013) The mitochondrial genome of Paramphistomum cervi (Digenea), the first representative for the family Paramphistomidae. Plos One 8:e71300
Yan Y, Wang Y, Liu X, Winterton SL, Yang D (2014) The first mitochondrial genomes of antlion (Neuroptera: Myrmeleontidae) and split-footed lacewing (Neuroptera: Nymphidae), with phylogenetic implications of Myrmeleontiformia. Int J Biol Sci 10:895–908
Yang X, Gasser RB, Koehler AV, Wang L, Zhu K, Chen L, Feng H, Hu M, Fang R (2015a) Mitochondrial genome of Hypoderaeum conoideum—comparison with selected trematodes. Parasit Vectors 8:97
Yang X, Zhao Y, Wang L, Feng H, Tan L, Lei W, Zhao P, Hu M, Fang R (2015b) Analysis of the complete Fischoederius elongatus (Paramphistomidae, Trematoda) mitochondrial genome. Parasit Vectors 8:279
Acknowledgments
This work was supported by the “Special Fund for Agro-scientific Research in the Public Interest” (Grant No. 201303037), “National Key Basic Research Program (973Program) of China” (Grant No. 2015CB150300).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yang, X., Wang, L., Feng, H. et al. Characterization of the complete mitochondrial genome sequence of Homalogaster paloniae (Gastrodiscidae, Trematoda) and comparative analyses with selected digeneans. Parasitol Res 115, 3941–3949 (2016). https://doi.org/10.1007/s00436-016-5160-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-016-5160-9