
Abstract
Metagonimus Katsurada, 1912 is a genus of small intestinal parasites. The genus comprises eight species, primarily from far-eastern Asia, with two exceptions reported from Europe. Metagonimus yokogawai, the most widespread species, is the main agent responsible for the intestinal disease, metagonimiasis, in Japan and some other East Asian countries. On the basis of the ratio of the size of the ventral and oral suckers, Metagonimus has traditionally been morphologically divided into two groups; however, the genus has not been extensively studied using molecular data. To reveal phylogenetic relationships within Metagonimus based on molecular data, we analyzed six of the seven species present in Asia using samples collected in central Japan. Maximum likelihood and Bayesian analyses of a combined 28S ribosomal DNA (rDNA), internal transcribed spacer 2 (ITS2), and mitochondrial cox1 gene sequence dataset separated the six species into two well-supported clades. One clade comprised M. yokogawai, M. takahashii, M. miyatai, and M. hakubaensis, whereas the other consisted of M. otsurui and M. katsuradai. Genetic distances calculated from 28S rDNA and ITS2 nucleotide sequences and a comparison of the predicted amino acid sequences of cox1 gene suggested that M. otsurui and M. katsuradai may have diverged recently. None of the four main morphological characters used to delimit species of Metagonimus (i.e., sucker ratio, positions of the uterus and testes, and distribution of vitelline follicles) was consistent with the distribution of species in the molecular tree.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Trematodes of the genus Metagonimus Katsurada, 1912 (Digenea: Heterophyidae) are small intestinal parasites of fish-eating mammals and birds. Some of these species have been reported to be potential human parasites (Chai et al. 2005). Infection in the definitive hosts (humans and various fish-eating mammals and birds) occurs through consumption of raw flesh of freshwater and brackish-water fish infected with metacercariae (the infective stage). Infection in humans is generally asymptomatic, but heavy infections may cause symptoms of metagonimiasis: severe diarrhea, abdominal pain, and weight loss (Chai et al. 1991).
The Metagonimus life cycle includes two intermediate hosts, with the metacercariae encysted in the musculature and other organs of teleost fishes (Sohn 2009; Yu and Chai 2012). Many fish, including members of the genera Plecoglossus, Tribolodon, and Carassius, have been reported as second intermediate hosts (Kino et al. 2006; Sohn 2009; Yu and Chai 2012). In some cases, metacercariae of more than one Metagonimus species can be found in a single fish. For example, metacercariae of M. yokogawai Katsurada, 1912, M. takahashii Suzuki, 1930, and M. miyatai Saito et al., 1997 have been found in Plecoglossus altivelis from Korea and Japan (Kino et al. 2006; Yu and Chai 2012).
Based on an adult worm of M. yokogawai, the genus was originally described from Taiwan and Japan (Katsurada 1912; Kobayashi 1912). Seven additional species have been subsequently described. Two of these species, M. romanicus Ciurea, 1915 and M. minutus Katsuta, 1932, have been recorded in Europe and Taiwan, respectively; these species are of uncertain validity (Shimazu and Urabe 2002; Yu and Chai 2012). The most widely distributed species, M. yokogawai, has been found in far eastern Asia (Korea, China, Taiwan, and Japan), the Russian Far East, southeastern Europe (the Balkan Peninsula and Romania), and southwestern Europe (Spain) (Ito 1964; Yu and Mott 1994; Chai et al. 2009). M. minutus is known only from Taiwan (Katsuta 1932), whereas M. takahashii and M. miyatai have been found in Japan and Korea. The remaining three species, M. hakubaensis Shimazu, 1999, M. katsuradai Izumi, 1935, and M. otsurui Saito & Shimizu, 1968, have been recorded only from Japan (Shimazu 1999, 2003; Shimazu and Urabe 2002; Kino et al. 2006). Three of the six species known to occur in Japan, M. yokogawai, M. takahashii, and M. miyatai, have been reported as human pathogens causing metagonimiasis, with M. yokogawai being the main species causing the disease in Japan and other Asian countries (Chai et al. 2000; Uppal and Wadhwa 2005; Yamada et al. 2008).
Species of Metagonimus are differentiated from one another by the oral and ventral sucker size ratio, position of testes and uterus, and distribution of vitelline follicles (Saito et al. 1997; Shimazu 1999, 2003; Kim et al. 2006). Among these morphological characters, the sucker size ratio is the major diagnostic character dividing the genus into two major groups: M. yokogawai, M. miyatai, M. takahashii, and M. minutus, having an oral sucker smaller than the ventral one, and M. katsuradai and M. otsurui, of which the oral sucker is larger (Saito et al. 1997; Shimazu and Urabe 2002; Shimazu 2003; Shimazu and Kino 2015). In M. hakubaensis, this ratio is variable, but the oral sucker is typically larger than the ventral one (Shimazu 1999; Shimazu and Kino 2015). A preliminary molecular study on species of Metagonimus was performed by Lee et al. (2004), who used partial 28S ribosomal DNA (rDNA) and mitochondrial cytochrome c oxidase subunit I (cox1) sequences to analyze relationships among M. yokogawai, M. takahashii, and M. miyatai from Korea. Their results appear to support the monophyly of Metagonimus and to confirm the close relationship among the three species inferred by morphology. To further clarify relationships within Metagonimus, we analyzed six of the seven nominal species of Metagonimus distributed in the Manchurian subregion. For this purpose, samples were collected from central Japan, and phylogenetic relationships were reconstructed based on three DNA markers likely to have different levels of resolution: 28S rDNA (D1–D3 regions), the rDNA internal transcribed spacer 2 (ITS2) region, and the mitochondrial cox1 gene. We then compared these molecular phylogenetic relationships with the original published morphological descriptions of the species.
Materials and methods
Sample collection
Metacercariae of Metagonimus were collected from fishes from different localities in Japan (Fig. 1) by digestion with artificial gastric juice (Table 1). Doses of 36–100 metacercariae from each locality were orally administered to Syrian golden hamsters (Mesocricetus auratus Waterhouse, 1839). Adult worms were collected from the small intestines of hamsters 14–15 days post-infection. The hamsters were handled and maintained according to the guidelines provided in the Guide for the Care and Use of Laboratory Animals of Hamamatsu University School of Medicine, Japan (approval nos. 2007085 and 2012012). The worms were identified under a light microscope according to described morphological characteristics (Hong and Seo 1969; Chai et al. 1991; Saito et al. 1997; Shimazu 1999; Shimazu and Urabe 2002; Shimazu and Kino 2015). Adult worms of four species, M. yokogawai, M. takahashii, M. miyatai, and M. hakubaensis, were collected. Metacercariae of M. otsurui and M. katsuradai were collected and identified by one of the authors (Takeshi Shimazu) according to published morphological criteria and host specificity of these two parasites (Shimazu and Urabe 2002; Shimazu 2003; Shimazu and Kino 2015). These two species were not obtained from an infected experimental host. All samples were preserved in absolute alcohol and stored at −20 °C until use.

Collection localities of six Metagonimus spp. in central Japan
DNA extraction, PCR, and DNA sequencing
Genomic DNA from three individuals of each species was extracted using a Genomic DNA mini kit (Tissue) (Geneaid, Taipei, Taiwan) according to the manufacturer’s protocol. A portion of the 28S rRNA gene (D1–D3 regions), the ITS2 region of rDNA, and the mitochondrial cox1 gene were amplified by polymerase chain reaction (PCR) using the primers listed in Table 2. Amplification conditions for the 28S rDNA, modified from Olson et al. (2003), consisted of one cycle of denaturation at 95 °C for 3 min, followed by 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 5 min. PCR amplification for the ITS2 region followed Skov et al. (2009), whereas the PCR conditions used for the cox1 gene were according to Bowles and McManus (1994). PCR amplicons were size-checked by 1.0 % agarose gel electrophoresis and visualized under a UV transilluminator. All amplicons were sequenced in both directions with the same primers used for the PCRs on an ABI Prism 3730XL DNA sequencer (Biobasic, Toronto, Canada).
DNA analysis
The generated DNA sequences were checked using BioEdit version 7.1.3.0 (Hall 2011) and aligned using CLUSTAL X2 (Larkin et al. 2007). For inclusion in the phylogenetic analyses, out-groups were selected on the basis of known relationships among heterophyid intestinal flukes (Thaenkham et al. 2012); their corresponding sequences were downloaded from GenBank and incorporated along with two additional published Metagonimus ITS2 sequences into the aligned sequence datasets (Table 1). Genetic distances among 28S rDNA, ITS2, and cox1 sequences were estimated by MEGA 6.06 (Tamura et al. 2013). To examine genetic differentiation within Metagonimus, the cox1 sequences were translated into amino acid sequences using MEGA.
Phylogenetic analysis
Before conducting the phylogenetic analyses, nucleotide substitution saturation was measured to evaluate the degree of saturation of each marker using the DAMBE (data analysis in molecular biology and evolution) program (Xia 2013). Phylogenetic analyses were conducted on individual 28S rDNA, ITS2, and cox1 sequence datasets. Phylogenetic trees were reconstructed using maximum likelihood (ML) as implemented in MEGA 6.06 and MrBayes v.3.2 (Ronquist et al. 2012). For the ML analyses, best-fit models of nucleotide substitution were selected based on the Akaike information criterion as determined in MEGA. The ML trees were constructed with and without out-groups, and support for each node in the resulting trees was estimated using 1000 bootstrap replicates. Haplorchis taichui Nishigori, 1924 (Trematoda: Heterophyidae) was used as a potential out-group because of indicating close phylogenetic position to genus Metagonimus (Thaenkham et al. 2012).
Phylogenetic analyses were also conducted on a combined dataset consisting of concatenated sequences of all three DNA regions. Before generating the combined dataset, partition homogeneity testing in PAUP* 4.0b10 (Swofford 1998) was carried out to confirm congruence between the three partitions at a significance level of P < 0.02 (Cunningham 1997). Maximum likelihood and Bayesian inference were used to generate trees from the combined dataset using H. taichui as an out-group. The ML tree was constructed following the method described above. For the Bayesian analysis, Bayesian posterior probabilities were calculated from the combined sequence dataset under the same best-fit model of evolution used in the ML analysis (Ronquist et al. 2012) using the default priors in MrBayes. Two simultaneous Markov chain Monte Carlo (MCMC) runs, each comprising four chains (one cold and three heated), were conducted. Each MCMC run was performed for 1,000,000 generations, with sampling every 100 generations. The convergence between runs was visually estimated by plotting the number of generations vs. log-likelihood values and using the potential scale reduction factor provided in the “sump” output of MrBayes (Ronquist et al. 2012). After discarding results from the first 25 % of generations as “burn in,” majority-rule consensus trees were generated from the remaining trees. Node support was assessed using the Bayesian posterior probabilities.
Results
Genetic distances among species of Metagonimus
Pairwise genetic distances were estimated from the 28S rDNA (1140 bp), ITS2 (406 bp), and cox1 (355 bp) aligned sequences (Table 3). The average genetic distance among six Metagonimus species based on the 28S rDNA sequences was approximately 0.012, although the genetic distance between M. katsuradai and M. otsurui was 0.002. Using the ITS2 sequence dataset, the average distance among six Metagonimus was approximately 0.03, with a distance of 0.008 calculated between M. katsuradai and M. otsurui. Based on the cox1 sequences, the genetic distance among six Metagonimus species was about 0.15; the genetic distance between M. katsuradai and M. otsurui was 0.115, close to the average.
Amino acid differences based on the cox1 gene
Because of the relatively large genetic distances between cox1 sequences compared with distances estimated from 28S rDNA and ITS2 sequences, especially those between M. katsuradai and M. otsurui, we examined differences in amino acid sequences predicted from Metagonimus cox1 nucleotide sequences. A comparison of 115 predicted amino acids within this region uncovered no differences between M. katsuradai and M. otsurui (Table 4). In contrast, amino acid sequence differences were found among the other four Metagonimus species, i.e., M. takahashii, M. yokogawai, M. miyatai, and M. hakubaensis.
Phylogenetic relationships
To show phylogenetic relationships among six Metagonimus species studied, trees were constructed from 28S rDNA, ITS2, and cox1 sequences using H. taichui as an out-group. None of these genetic markers showed evidence of nucleotide substitution saturation; consequently, they could all be used for revealing phylogenic relationships. As shown in the Supplement Data 1, the topologies recovered using the three different markers were incongruent with one another. The same was true for the phylogenetic trees constructed without an out-group. As indicated by the high ML bootstrap proportions and Bayesian posterior probabilities, most nodes in each tree were well supported.
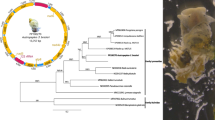
To more fully resolve phylogenetic relationships using all available evolutionary evidence, the three sequence datasets were combined and analyzed using H. taichui as the out-group. Although the trees generated from the individual datasets were incongruent, a partition homogeneity test indicated that the 28S rDNA, ITS2, and cox1 sequence partitions could be combined (nonsignificant for the homogeneity test; P = 0.5). ML and Bayesian analyses were run on the combined dataset under the HKY + G + I model of nucleotide substitution. As shown in Fig. 2, the topologies recovered by these analyses were congruent, with the inferred phylogenetic relationships among Metagonimus species generally well supported—as indicated by ML bootstrap proportions and most Bayesian posterior probabilities. In the tree in Fig. 2, species of Metagonimus are separated into two clades. Within clade I, M. yokogawai and M. takahashii constitute a sister clade to M. hakubaensis, with these three species in turn forming a sister clade to M. miyatai. Clade II comprises the remaining two species, M. katsuradai and M. otsurui.

Phylogenetic tree of Metagonimus spp. based on a combined 28S rDNA, ITS2, and cox1 sequence dataset. H. taichui was used as an out-group. Maximum likelihood bootstrap support values/Bayesian posterior probabilities are shown above branches. Branch lengths are drawn to scale, with the scale bar indicating the number of nucleotide substitutions. Morphological characters traditionally used to delimit Metagonimus spp. are indicated to the right of the tree as follows: OSV, oral sucker smaller than ventral sucker; OLV, oral sucker larger than ventral sucker; RTP, right testis located at the posterior end of the body; RTB, right testis located at a distance from the posterior extremity, in the intercecal field of the body; UAT, uterus occupies the space from the ventral sucker to the anterior border of the testes; UPT, uterus extends between the testes near the posterior end of the body; VAT, vitelline follicles distributed along the lateral side of body and mostly terminating at the front of the anterior two-thirds of the right testis; and VPT, vitelline follicles distributed along the lateral side of the body and extending beyond the posterior end of the right testis
We compared the inferred molecular phylogenetic relationships of the six Metagonimus species with their previously described morphological characteristics (Hong and Seo 1969; Chai et al. 1991; Saito et al. 1997; Shimazu 1999, 2002, 2003; Shimazu and Urabe 2002; Shimazu and Kino 2015). Mapping of morphological characteristics used to distinguish individual species of Metagonimus, namely position of testes and uterus position and distribution of vitelline follicle (see Materials and Methods for references), onto the phylogenetic tree has revealed incongruence of molecular and morphological data (Fig. 2). No morphological character was consistent with respect to the two main lineages of Metagonimus. Another taxonomically important characteristic, sucker ratio, was also inconsistent because of the variable size of the suckers of M. hakubaensis.
Discussion
Pairwise comparison of genetic distances based on three different DNA markers—28S rDNA, ITS2, and cox1—revealed the evolutionary divergence of the six Metagonimus species collected from central Japan. According to genetic distances estimated from 28S rDNA and ITS2 sequences, M. yokogawai, M. takahashii, M. miyatai, and M. hakubaensis were clearly highly genetically differentiated from one another at the species level, while M. katsuradai and M. otsurui were closely related to each other. Interestingly, the divergence between cox1 sequences of the latter two species was similar to the average genetic distance among all six Metagonimus species. No changes in amino acids were evident between M. otsurui and M. katsuradai, whereas the predicted amino acid sequences of the other four Metagonimus species were different from one another (Table 4). This similarity in amino acids supports the evidence obtained from 28S rDNA and ITS2 sequences that M. otsurui and M. katsuradai are closely related.
To confirm the genetic relationships suggested above, we reconstructed rooted and unrooted phylogenies of the six Metagonimus species using the three genetic markers (28S rDNA, ITS2, and cox1). Most nodes in the resulting trees had high statistical support, but the topologies were largely incongruent (Supplement Data 1). Such high levels of incongruence have not typically been observed in analyses of trematode genera such as Paragonimus Braun, 1899 and Schistosoma Weinland, 1858, where trees constructed from different genetic markers were generally congruent (Lockyer et al. 2003; Thaenkham and Waikagul 2008; Doanh et al. 2007). In the genus Haplorchis, some inconsistencies were uncovered between rDNA and ITS2 trees; in that case, however, the tree constructed from the ITS2 region was characterized by low bootstrap support (Thaenkham et al. 2010). After performing a partition homogeneity test (Cunningham 1997), which supported the merging of the three data partitions, we analyzed the combined dataset to better resolve phylogenetic relationships among the six Metagonimus species. The phylogenetic tree obtained from analysis of the combined DNA sequences (Fig. 2) contained two distinct Metagonimus clades. Clade I comprised M. yokogawai, M. takahashii, M. miyatai, and M. hakubaensis. M. yokogawai and M. takahashii, which were most closely related to one another, were in a sister relationship with M. hakubaensis. Within this clade, M. miyatai was distinct from the other three species. Clade II consisted of M. katsuradai and M. otsurui. The high similarity of their 28S rDNA and ITS2 sequences and their identical cox1 translated amino acid sequences suggest that these two species may have recently diverged from one another.
To look for correlations between the molecular phylogeny and the morphological characters used to delimit the six Metagonimus species (described in the legend in Fig. 2), we labeled the species in the tree in Fig. 2 according to their morphological characteristics. No clear correspondence was found between the two phylogenetic clades and sucker size ratio, which is considered to be the major defining morphological character within Metagonimus. In M. hakubaensis, this ratio has been reported to range from 0.94 to 1.33 (Shimazu 1999). The minor morphological diagnostic characters, including position of testes and uterus, and distribution of vitelline follicles, were also incongruent with phylogenetic relationships. No congruence between morphological characters and phylogenetic clades was thus observed in the six representatives of Metagonimus from Japan. This result contrasts with the situation observed in members of the subfamily Haplorchiinae (Heterophyidae), where the monophyletic clade of the genus Haplorchis is characterized by a single testis and is distinct from other heterophyid genera (Thaenkham et al. 2010).
Morphological characters of the Japanese representatives investigated in this study seem to be homoplastic, but a more robust phylogenetic hypothesis should be available. Morphological adaptations among species of Metagonimus, which may have diverged within a small region of central Japan, should be clarified by future studies on morphological polymorphisms and genetic variation of Metagonimus populations from other geographical localities, especially the mainland region, but also using other molecular markers such as microsatellites.
In conclusion, this study was the first to reveal phylogenetic relationships of six of eight species of Metagonimus, which have obviously diverged into two separate clades. The results of our phylogenetic analyses also suggest that the major morphological diagnostic character, sucker ratio, may have no correspondence with evolutionary divergence within Metagonimus. Because our sampling was limited to a small number of Japanese populations, more samples from other regions should be analyzed to provide more robust data on the evolutionary history of these sympatrically living parasites. Additionally, another Asian species, M. minutus endemic to Taiwan, should be collected to assess its relationships with the Japanese taxa. For future analyses, samples should be collected more broadly from various geographical localities to assess genetic variation within and among species. For phylogenetic analysis of Metagonimus, 28S rDNA, ITS2, and cox1 markers can be used in combination to reconstruct a reliable phylogenetic tree.
References
Bowles J, McManus DP (1994) Genetic characterization of the Asian Taenia, a newly described taeniid cestode of humans. Am J Trop Med Hyg 50:33–44
Chai JY, Sohn WM, Kim MH, Hong ST, Lee SH (1991) Three morphological types of the genus Metagonimus encysted in the dace, Tribolodon taczanowskii, caught from the Sumjin River. Korean J Parasitol 29:217–225
Chai JY, Han ET, Park YK, Guk SM, Kim JL, Lee SH (2000) High endemicity of Metagonimus yokogawai infection among residents of Samchok-shi, Kangwon-do. Korean J Parasitol 38:33–36
Chai JY, Murrell KD, Lymbery AJ (2005) Fish-borne parasitic zoonoses: status and issues. Int J Parasitol 35:1233–1254
Chai JY, Shin EH, Lee SH, Rim HJ (2009) Foodborne intestinal flukes in Southeast Asia. Korean J Parasitol 47:S69–102
Cunningham CW (1997) Can tree incongruence tests predict when data should be combined? Mol Biol Evol 14:733–740
Doanh PN, Shinohara A, Horii Y, Habe S, Nawa Y, The DT, Le NT (2007) Morphological and molecular identification of two Paragonimus spp., of which metacercariae concurrently found in a land crab, Potamiscus tannanti, collected in Yenbai Province, Vietnam. Parasitol Res 100:1075–1082
Hall TA (2011) BioEdit: an important software for molecular biology. GERF Bull Biosci 2:60–61
Hong NT, Seo BS (1969) Study on Metagonimus yokogawai (Katsurada, 1912) in Korea. 1. On the metacercaria, its distribution in the second intermediate host and development in the final host. Korean J Parasitol 7:129–142
Ito J (1964) Metagonimus and other human heterophyid trematodes. Progr Med Parasitol Jpn 1:315–393
Katsurada F (1912) On a new trematode. Metagonimus yokogawai. Tokyo Iji Shinshi 1796:3483–3489 (in Japanese)
Katsuta I (1932) Studies on the metacercariae of Formosan brackish water fish, 2. On a new species, Metagonimus minutus n. sp. parasitic in Mugil cephalus. Taiwan Iggakai Zasshi 31:26 (in Japanese)
Kim DG, Kim TS, Cho SH, Song HJ, Sohn WM (2006) Heterophyid metacercarial infections in brackish water fishes from Jinju-man (Bay), Kyongsangnam-do, Korea. Korean J Parasitol 44:7–13
Kino K, Suzuki T, Oishi H, Suzuki S, Yamagiwa S, Ishiguro M (2006) Geographical distribution of Metagonimus yokogawai and M. miyatai in Shizuoka Prefecture, Japan, and their site preferences in the sweetfish, Plecoglossus altivelis, and hamsters. Parasitol Int 55:201–206
Kobayashi H (1912) On a new genus of the trematode (a preliminary report). Saikingaku Zasshi 204:780–786 (in Japanese)
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Lee SU, Huh S, Sohn WM, Chai JY (2004) Sequence comparisons of 28S ribosomal DNA and mitochondrial cytochrome c oxidase subunit I of Metagonimus yokogawai, M. takahashii and M. miyatai. Korean J Parasitol 42:129–135
Lockyer AE, Olson PD, Østergaard P, Rollinson D, Johnston DA, Attwood SW, Southgate VR, Horak P, Snyder SD, Le TH, Agatsuma T, McManus DP, Carmichael AC, Naem S, Littlewood DT (2003) The phylogeny of the Schistosomatidae based on three genes with emphasis on the interrelationships of Schistosoma Weinland, 1858. Parasitology 126:203–224
Olson PD, Cribb TH, Tkach VV, Bray RA, Littlewood DTJ (2003) Phylogeny and classification of the Digenea (Platyhelminthes: Trematoda). Int J Parasitol 33:733–755
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61:539–542
Saito S, Chai JY, Kim KH, Lee SH, Rim HJ (1997) Metagonimus miyatai sp. nov. (Digenea: Heterophyidae), a new intestinal trematode transmitted by freshwater fishes in Japan and Korea. Korean J Parasitol 35:223–232
Shimazu T (1999) Metagonimus hakubaensis sp. n. Digenea, Heterophyidae from Nagano, Japan: morphology and life cycle. Bull Natn Sci Mus Ser A 25:87–99
Shimazu T (2002) Life cycle and morphology of Metagonimus miyatai (Digenea: Heterophyidae) from Nagano, Japan. Parasitol Int 51:271–280
Shimazu T (2003) Morphology of metacercariae and adults of Metagonimus katsuradai Izumi (Digenea, Heterophyidae) from Shiga, Japan. Bull Natn Sci Mus Ser A 29:47–51
Shimazu T, Kino H (2015) Metagonimus yokogawai (Trematoda: Heterophyidae): from discovery to designation of a neotype. Korean J Parasitol 53:627–639
Shimazu T, Urabe M (2002) Morphology and life cycle of Metagonimus otsurui (Digenea, Heterophyidae) from Nara, Japan. Bull Natl Sci Mus Tokyo, Series A 28:21–28
Skov J, Kania PW, Dalsgaard A, Jorgensen TR, Buchmann K (2009) Life cycle stages of heterophyid trematodes in Vietnamese freshwater fishes traced by molecular and morphometric methods. Vet Parasitol 160:66–75
Sohn WM (2009) Fish-borne zoonotic trematode metacercariae in the Republic of Korea. Korean J Parasitol 47:S103–S113
Swofford DL (1998) PAUP: Phylogenetic analysis using parsimony (and other methods), version 4.0. Sinauer. Sunderland, Massachusetts
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA 6: molecular evolutionary genetic analysis version 6.0. Mol Biol Evol 30:2725–2729
Thaenkham U, Waikagul J (2008) Molecular phylogenetic relationships of Paragonimus pseudoheterotremus. SE Asian J Trop Med Publ health 39:217–221
Thaenkham U, Dekumyoy P, Komalamisra C, Sato M, Dung do T, Waikagul J (2010) Systematics of the subfamily Haplorchiinae (Trematoda: Heterophyidae), based on nuclear ribosomal DNA genes and ITS2 region. Parasitol Int 59:460–465
Thaenkham U, Blair D, Nawa Y, Waikagul J (2012) Families Opisthorchiidae and Heterophyidae: are they distinct? Parasitol Int 61:90–93
Uppal B, Wadhwa V (2005) Rare case of Metagonimus yokogawai. Ind J Med Microbiol 23:61–62
Xia X (2013) DAMBE5: a comprehensive software package for data analysis in molecular biology and evolution. Mol Biol Evol 30:1720–1728
Yamada SM, Yamada S, Takada H, Hoshiai YC, Yamada S (2008) A case of metagonimiasis complicated with multiple intracerebral hemorrhages and diabetes mellitus. J Nippon Med Sch 75:32–35
Yu YR, Chai JY (2012) Metagonimus. In: Liu D (ed) Molecular detection of human parasitic pathogens. CRC Press (Taylor and Francis Group), Boca Raton, pp 389–398
Yu S-H, Mott KE (1994) Epidemiology and morbidity of food-borne intestinal trematode infections. Trop Dis Bull 91:R125–R152
Acknowledgments
We thank the staff members of the Department of Helminthology, Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand for their technical support and permission to use its equipment and facilities. We express our thanks to David Blair, College of Marine and Environmental Science, James Cook University, for his valuable comments on this manuscript. We also thank Paul Adams and Glad Rotaru for language corrections of the manuscript. This research was supported by the Thailand Research Fund (TRF), Thailand (grant number MRG5380075) and the Faculty of Tropical Medicine, Mahidol University, Thailand.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The hamsters were handled and maintained according to the guidelines provided in the Guide for the Care and Use of Laboratory Animals of Hamamatsu University School of Medicine, Japan (approval nos. 2007085 and 2012012).
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 4281 kb)
Rights and permissions
About this article

Cite this article
Pornruseetairatn, S., Kino, H., Shimazu, T. et al. A molecular phylogeny of Asian species of the genus Metagonimus (Digenea)—small intestinal flukes—based on representative Japanese populations. Parasitol Res 115, 1123–1130 (2016). https://doi.org/10.1007/s00436-015-4843-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-015-4843-y