
Abstract
Purpose
The molecular pathogenesis of solid tumour was first assessed in colorectal cancer (CRC). To date, ≤ 100 genes with somatic alterations have been found to inter-connectively promote neoplastic transformation through specific pathways. The process of colorectal carcinogenesis via genome landscape is reviewed on the basis of an adenoma-to-carcinoma sequence, as shown by serial histological and epidemiological observations.
Methods
The relevant literatures from PubMed (1980–2021) have been reviewed for this article.
Results
The major routes of CRC development, chromosomal instability (CIN), microsatellite instability (MSI), and the serrated route either via CIN or MSI, proceed through the respective molecular pathway of colorectal carcinogenesis. Particular aspects of CRC carcinogenesis can also be determined by evaluating familial CRCs (FCRC) and genotype–phenotype correlations. Specific causative gene alterations still leave to be identified in several FCRCs. Otherwise, recently verified FCRC can be particularly notable, for example, EPCAM-associated Lynch syndrome, polymerase proofreading-associated polyposis, RNF43-associated polyposis syndrome or NTHL1 tumour syndrome, and hereditary mixed polyposis syndrome. The oncogenic landscape is described, including representative pathway genes, the three routes of carcinogenesis, familial CRCs, genotype–phenotype correlations, the identification of causative genes, and consensus molecular subtypes (CMS).
Conclusion
Whole genome research using multi-gene panels (MGPs) has facilitated high through-put detection of previously unidentified genes involved in colorectal carcinogenesis. New approaches designed to identify rare variants are recommended to consider their alterations implicated in the molecular pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tumour initiation is traditionally regarded as a stepwise accumulation of alterations, leading to an expansion of clones with selective growth advantages, with the fittest clone eventually becoming dominant (Fearon and Vogelstein 1990). This sequential model of colorectal carcinogenesis has been confirmed by serial histological and epidemiological observations (Luebeck and Moolgavkar 2002). The most prevalent molecular alterations, including chromosomal instability (CIN), microsatellite instability (MSI), and somatic mutations involving the TGF-β and Wnt pathways, are synergistically involved in early carcinogenesis, cancer stem cell (CSC) maintenance, and tumour progression (Angius et al. 2021). By contrast, the “Big Bang” model suggests that tumours grow predominantly as a single expanding clone producing numerous mixtures of sub-clones that are not subject to stringent selection, with both public (clonal) and most detectable private (sub-clonal) alterations arising soon after the initial step of tumour progression (Sottoriva et al. 2015).
The gene causing familial adenomatous polyposis (FAP) was initially identified by a deletion in chromosome 5, as determined by linkage analysis of DNA markers from 124 subjects in 13 different FAP families (Bodmer et al. 1987), with further analysis showing that this locus was located at chromosome 5q22.2. The classical pathway of colorectal cancer (CRC) progression was hypothesized to involve an “adenoma–carcinoma” sequence, in which the initial molecular alterations lead to the formation of benign tumours, such as adenomas and serrated polyps, followed by additional molecular steps that give rise to more histologically invasive tumours (Fearon and Vogelstein 1990).
This review summarizes the oncogenic pathways associated with colorectal carcinogenesis, including identified genes and their interactions. The oncogenic landscape is described, including representative pathway genes, the three routes of carcinogenesis, familial CRCs, genotype–phenotype correlations, the identification of causative genes, and consensus molecular subtypes (CMS).
Oncogenomic landscape and colorectal cancer
To date, several hundred driver genes have been identified and shown to be altered by intragenic mutations, with other mutations being passengers not associated with selective growth advantages (Martínez-Jiménez et al. 2020). Alternatively, the differentiation between driver and passenger genes may depend simply on the strength of selections associated with given genetic mutations or epigenetic changes (Crouch and Bodmer 2020). Thus, the stronger drivers may be those genes that are more frequently mutated, whereas it may be difficult to determine whether particular genes with relatively few mutations are really “neutral” or “passengers”.
DNA methylation accompanied by transcriptional repression facilitates the oncogenic process but does not exclude the possibility of subsequent mutations. DNA methylation has been associated with a wide range of molecular changes, including changes in growth rate, inflammation, and metaplasia, not only genetic changes. Aberrant methylation in a subset of MLH1, MGMT, and HIC1 genes can be pathogenic in cancer (Grady and Carethers 2008). Nucleosomal core histones possess specific tails, which can be subjected to different modifications, including acetylation, methylation, ubiquitination, phosphorylation, and sumoylation (https://www.genecards.org/). Histone modifications resulting in gene dysregulation have been identified in various malignancies. These modifications can result in aberrant expression of histone deacetylases, such as HDAC1 and HDAC2; mutations in genes encoding histone acetyltransferases, such as P300, CBP, and PCAF; and/or overexpression of histone methyltransferases such as EZH2 in CRCs (Esteller 2007).
Enteric microbes can also participate in colorectal carcinogenesis and progression through their metabolites and toxins. Approximately 2% of CRCs have been linked to colitis, including colitis induced by Bacteroides fragilis toxins and the adherent-invasive Escherichia coli strain NC101 (Mizutani et al. 2020). Another periodontal pathogen, Fusobacterium nucleatum, was reported to induce non-colitis-associated CRCs. Secondary bile acids may also be involved in CRC cell proliferation, through β-catenin activation, ERK1/2 activation, the c-Myc target pathway, and the NF-κB signalling pathway (Pai et al. 2004).
Representative pathways and related genes
Molecular signalling of colorectal carcinogenesis can be divided into nine major pathways, called the Wnt/β-catenin, JAK/STAT, RAS/ERK, PI3K/AKT, TGF/SMAD, Notch, Hedgehog, TNF-related apoptosis-inducing ligand (TRAIL), and p53 pathways (Farooqi et al. 2019; Kim et al. 2008). These nine pathways interact closely to form intra- and inter-cellular networks that induce colorectal carcinogenesis (Fig. 1).

The nine major pathways associated with colorectal carcinogenesis. The nine representative pathways, drawn as circles, were the tumour suppressor (green), oncogenic (red), and facultative (blue) pathways. Molecular interactions between two pathways were either synergistic (green lines) or suppressive (red lines), as well as being strong (thick lines) or modest (thin lines)
Wnt/β-catenin
The canonical Wnt/β-catenin (or Wnt) pathway involves the accumulation of β-catenin in the cytoplasm and its translocation into the nucleus, where it acts as a transcriptional coactivator of transcription factors belonging to the TCF/LEF family (Deitrick and Pruitt 2016). Subsequently, β-catenin is targeted for ubiquitination and subsequent proteasomal degradation by a degradation complex consisting of axin, adenomatous polyposis coli (APC), casein kinase 1 (CK1), and glycogen synthase kinase-3β (GSK-3β). More than half of CRCs harbour oncogenic mutations in regulatory components of the Wnt pathway, with their deregulation being primary drivers of colorectal carcinogenesis (Farooqi et al. 2019). In particular, APC mutations, found in approximately 80% of all CRCs, result in the unrestrained activation of the Wnt pathway (van Neerven et al. 2021). Tumours with microsatellite instability (MSI) frequently harbour mutations in regulatory components of the Wnt pathway, including truncating, but mutually exclusive, mutations in AXIN2 and TCF7L2 (Kim et al. 2009). The E3 ubiquitin ligase RNF43, which is associated with serrated polyposis syndrome, was found to regulate Wnt signalling by inducing the degradation of the Wnt receptor Frizzled (Tsukiyama et al. 2020). Additionally, the Wnt-dependent endogenous Rspo2 and Rspo3 chromosomal rearrangements can initiate and maintain colorectal carcinogenesis (Han et al. 2017).
JAK/STAT
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signalling pathway is involved in colorectal carcinogenesis via immune function and cell growth (Spano et al. 2006). Activated JAKs phosphorylate receptors at specific tyrosine residues, resulting in the recruitment of cytoplasmic monomeric STAT proteins via their SH2 domains (Spano et al. 2006). The SOCS protein belongs to a group of cytokine-inducible genes that have been shown to inhibit STAT signalling by binding to JAKs. STATs participate in oncogenesis by upregulating the expression of genes encoding apoptosis inhibitors (Bcl-x1, Mcl-1), cell cycle regulators (cyclins D1/D2, c-Myc), and inducers of angiogenesis (VEGF) (Buettner et al. 2002).
Ras/ERK
The Ras/ERK signalling pathway is a well-characterized series of kinase cascades involved in cell growth and proliferation. Several feedback loops can promote MEKi resistance, and there is considerable cross-talk between components of the Ras/ERK signalling pathway involving nearly all major signalling cascades (Neuzillet et al. 2014). ERK activation can inhibit apoptosis induced by the death receptors of the ligands FAS1, TRAIL, and TNF. In addition, aberrant activation of the Ras/ERK pathway contributes to evasion of cell senescence by upregulating the expression of telomerase. The Ras/ERK pathway is also required for epithelial–mesenchymal transition (EMT) and contributes to the maintenance of an undifferentiated or mesenchymal state in the tumour microenvironment (TME). Specifically, the Ras/ERK pathway was shown to cooperate with the TGF-β/SMAD pathway in upregulating EMT-related genes (Maurer et al. 2011).
PI3K/AKT
PI3K/AKT(/mTOR) signalling is an important event in colorectal carcinogenesis, with mutations in PIK3CA, the catalytic subunit of PI3K, found in > 30% of solid malignancies (Samuels and Ericson 2006). Constitutively, PTEN directly inhibits PI3K/AKT signalling by dephosphorylating a key second messenger, thereby blocking cell cycle progression and inducing apoptosis. PTEN inactivation frequently involves promoter hyper-methylation resulting from genomic instability, explaining the correlation between MSI and PTEN loss (Goel et al. 2004). Functional analyses of PIK3CA mutations revealed that its enzymatic activity was increased, stimulating AKT/GSK-3β signalling and resulting in growth factor-independent growth, as well as invasion and metastasis (Samuels et al. 2004). PIK3CA mutations have been found to occur when benign colorectal tumour cells acquire the ability to invade, i.e., at the adenoma–carcinoma transition (Ijichi et al. 2001).
TGF-β/SMAD
The TGF-β/SMAD signalling pathway plays an important role in colorectal carcinogenesis, although the role of TGF-β in carcinogenesis depends on its stage (Ijichi et al. 2001). TGF-β, a prototype of a family of secreted polypeptides, regulates a wide variety of biological activities, including cell growth, differentiation, and apoptosis; extracellular matrix (ECM) production; and immune function. Members of the TGF-β superfamily form a heteromerically organized receptor complex with types I and II surface receptors, transducing intracellular signals via the SMAD complex (Pai et al. 2004).
Notch
Overexpression or constitutive activation of the Notch signalling pathway may be mechanistically involved in colorectal carcinogenesis (Qiao and Wong 2009). Alternatively, STRAP disassembles the PRC2, resulting in the activation of Notch signalling via epigenetic modification (Jin et al. 2017). A novel STRAP–Notch1–HES1 molecular axis has been shown to act as a CSC regulator in CRC, whereas DAB1 under-expression was found to suppress tumour invasion and metastasis in Notch signalling-activated mice (Jin et al. 2017; Sonoshita et al. 2015). Activation of the Notch pathway may be associated with a loss of FBXW7, resulting in the development of adenomas in mice aged 9–10 months (Babaei-Jadidi et al. 2011).
Hedgehog
The biological activity of the hedgehog (HH) signalling pathway involves signalling that terminates at glioma-associated oncogene (GLI) transcription factors, alternating between activator and repressor forms. The important components of this pathway include the hedgehog ligands (SHH), patched (PTCH) receptor, smoothened (SMO), suppressor of fused (SuFu), and GLI transcription factors (Niyaz et al. 2019). Activation of the SHH–GLI1 pathway correlates positively with colorectal tumour development, suggesting that this pathway is activated during colorectal carcinogenesis. The expression of components of the HH pathway varies during the progression from colon adenoma to carcinoma, with the expression of SMOH and GLI1 similarly showing gradual increases from normal colon to adenoma to CRC (Xu et al. 2016). The levels of expression of SHH, PTCH, and GLI1 were found to be higher in patients with Peutz–Jeghers syndrome than in normal tissue, showing gradual increases during the adenoma-to-carcinoma sequence.
TRAIL
The TRAIL pathway, which includes death inducing signalling complex (DISC), FAS-associated protein with death domain (FADD) and pro-caspase-8 interactions, involves signalling through death receptors and the induction of apoptosis (Farooqi et al. 2019). TRAIL pathway practically indicates TRAIL-resistant one caused by deficient TRAIL death receptor transport to the cell surface. Although loss of function of TRAIL-receptor genes by mutations or methylation is not frequently found in CRC, expression of members of the IAP families, including survivin and XIAP, can contribute to TRAIL resistance in CRC (van Geelen et al. 2004). TRAIL sensitivity during colorectal carcinogenesis was previously attributed to changes in the balance between the TRAIL receptors TRAIL-R1/R2 and the decoy receptors TRAIL-R3/R4 during the progression of malignancy (Hague et al. 2005).
TP53
TP53 encoding p53 protein is a critical transcription factor that suppresses colorectal carcinogenesis through major pathways, acting as a “guardian of the genome” to maintain the integrity of DNA (Lane 1992). The p53 protein has been shown to further facilitate components of the DNA repair machinery and to directly trans-activate apoptosis-associated genes, including Bax, Puma, and Noxa, while suppressing tumourigenic and anti-apoptotic genes, such as survivin and Pdk2 (Toledo and Wahl 2006). In addition, p53 was shown to activate caspase-8 pathways through the activation of cell death receptors, such as FAS, DR5, and PIDD. TP53 is located on the short arm of chromosome 17p13.1, and its central sequence-specific DNA-binding domain (codons 101–306) allows binding of p53 to DNA. Mutations in TP53, which are frequent in colorectal tumours, impede the physiological function of p53 (Toledo and Wahl 2006). The E3 ubiquitin–protein ligase MDM2 is one of the central enzymes that labels p53 with ubiquitin, maintaining p53 under-expression under normal physiological conditions (Cheok and Lane 2017). p53 protein activates p21 (WAF1), a cyclin-dependent kinase (CDK) inhibitor, which is involved in inhibiting cell transition from G1 to S phase. Mutations in TP53 appear to occur during later stages of colon adenoma-to-carcinoma progression (Pino and Chung 2010).
The major routes of colorectal carcinogenesis
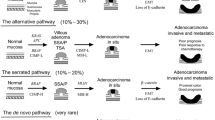
Current knowledge of the genome suggests that colorectal carcinogenesis proceeds along well-known canonical routes, constituting the adenoma-to-carcinoma sequence. The major routes of CRC evolution, namely the CIN and MSI routes, can be distinguished by their site-specificity, being predominant in the left and right colon, respectively, whereas the third route of CRC evolution, the serrated route, is located on both sides of the colon (Fig. 2).

The three major routes of colorectal carcinogenesis. A The chromosomal instability route (CIN). B The microsatellite instability route (MSI). C The serrated route, involving either traditional serrated adenomas (TSA) or sessile serrated adenomas (SSAs). aMSI polyps included all polyps, whether adenomatous, sessile, or hamartomatous
Chromosomal instability route
More than 70% of CRCs develop through CIN routes, involving somatic mutations in APC, accompanied by chromosomal changes, including somatic copy number alterations (SCNAs) caused by aneuploidy, insertion–deletion mutants (in-dels), amplifications, and/or loss of heterozygosity (LOH) (Fig. 2A) (Pino and Chung 2010; Nguyen et al. 2020). However, next-generation sequencing (NGS) recently showed that the mutation profiles of CIN and microsatellite-and-chromosomal-stable (MACS) CRCs were similar, although the causes of aneuploidy or retention of diploidy could not be determined (Ham-Karim et al. 2019). The areas of genomic alterations frequently harbour mutations, leading to the activation of KRAS, the loss of p53, and LOH of genes on the long arm of chromosome 18 (Markowitz and Bertagnolli 2009). Increased structural defects associated with CIN presumably expand the repertoire of driver mutations promoting carcinogenesis. Few MYC mutations have been observed in CRCs, but MYC amplifications have been reported in approximately 10% of CRCs, resulting in a feature associated with poorer outcomes (Kozma et al. 1994). CRCs that develop through the CIN route are positively associated with the risk of developing metastases, whereas those that develop through the MSI route are not (Angius et al. 2021). In some patients with polyposis, the hamartoma–carcinoma sequence could be explained by three potential steps associated with cellular and molecular pathogenesis, namely dysplastic transformation, altered turnover rate of stem cell lineage, and hamartoma–adenoma transition (Bosman 1999; Korsse et al. 2013; Jansen et al. 2009). Hamartoma–dysplasia transformation was associated with stepwise alterations of SMAD4/STK11, along with loss of TGF/β-catenin signalling, whereas hamartoma–adenoma transition included mixed hamartomatous and adenomatous components in a polyp, or mixed clusters of hamartoma and adenoma in an individual.
Deficient mismatch repair route
Microsatellite instability (MSI) is a feature observed in approximately 10–20% of CRCs (Fig. 2B). MSI in colorectal tumours primarily arises from dysfunction of the mismatch repair (MMR) genes, MLH1, MSH2, MSH6, and PMS2, and leads to numerous mutations, in particular within highly repetitive microsatellite regions (Farooqi et al. 2019). Alternatively, deletions in the 3′ end of EPCAM, a gene located 17 Kb upstream of MSH2, eliminate termination signals, resulting in MSH2 promoter hyper-methylation with epigenetic silencing (Tutlewska et al. 2013). Although EPCAM mutations are rare, enhanced EPCAM expression, which correlates with the downregulation of E-cadherin, has oncogenic potential linked to CSCs and EMT, resulting in poor differentiation, vascular and marginal invasion, and lymph node metastasis (Kempers et al. 2011). Germline mutations in MMR genes are rare in CRCs, except in patients with Lynch syndrome (LS). MSI-containing CRCs have been associated with the BRAF V600E mutation, which has been observed in around 80–90% of sporadic MSI-H CRCs but rarely in CRCs due to LS (Müller et al. 2016). Another deficient MMR caused by MLH1 promoter hypermethylation may also be useful in distinguishing sporadic from LS-associated CRCs. Up to 90% of MSI CRCs carry TGFBR2 mutations, which cannot prevent cellular proliferation (Pinheiro et al. 2015). Other genes disrupted by MSI included genes encoding proteins that regulate proliferation (e.g., GRB1, TCF4, WISP3, ACVR2, IGF2R, AXIN2, and CDX), cell cycle arrest or apoptosis (e.g., CASP5, PRDM2, BCL10, PTEN, PA2G4, and FAS), and DNA repair (e.g., MBD4, BLM, CHK1, RAD50, MSH3, and MSH6), although most of these gene alterations were not associated with major functional consequences (Guinney et al. 2015). APC mutations have been found in 35–50% of MSI tumours, suggesting that the initiating event in adenoma formation may be shared by MSI and CIN tumours without showing any correlations, distinguishing these tumours from MSI tumours initiated by BRAF mutations (Müller et al. 2016). Other CRCs may be distinguished by genome stability lacking hypermutation and aneuploidy, but enriched in DNA hypermethylation and mutations in KRAS, SOX9, and PCBP1 (Liu et al. 2018).
Serrated route
Approximately 10–20% of CRCs may develop through a different sequence of morphological changes, known as the serrated route (Fig. 2C). Although the majority of serrated polyps are hyperplastic, without malignant transformation, a subset of serrated lesions can progress to CRCs (Nguyen et al. 2020). Based on genetic and morphological pathogenesis, the serrated route involves dual traits from either the CIN or deficient MMR pathways, producing two types of premalignant precursor lesions, traditional serrated adenomas (TSAs) and sessile serrated adenomas (SSAs), respectively (Müller et al. 2016). Approximately 80% of TSAs carry KRAS mutations, whereas approximately 80% of SSAs carry BRAF and MMR-associated mutations or gene silencing caused by promoter hypermethylation (Ham-Karim et al. 2019). The latter phenotype frequently includes MGMT promoter methylation as a consequence of the low-level MSI (MSI-L) phenotype. The characteristic morphological features of TSAs include architectural dysplasia with ectopic crypt formation and serration, likely associated with molecular alterations that result in hyper-proliferation and inhibition of apoptosis (Nguyen et al. 2020). TSAs, which are diagnosed based on their characteristic cytology (eosinophilic cytoplasm and central, elongated hyperchromatic nuclei) and slit-like epithelial serrations with ectopic crypt formation, may progress to adenocarcinoma (Davies et al. 2002). By contrast, SSAs have BRAF mutations and MSI, which correlate with mucinous or poorly differentiated tumours, and may progress to serrated or mucinous adenocarcinomas (Advani et al. 2018). These tumours are frequently found in the right colon, and predominantly in older and female patients.
Hereditary colorectal cancer
Approximately 5% and 20% of all CRCs have been found to be familial and hereditary CRCs (FCRC and HCRC), respectively. The former category includes a variety of genetically verified syndromes with high penetrance, whereas the latter can be applied to any familial occurrences of CRC due to multi-genic variants, each with low-level effects as in the analysis of polygenic risk scores (Crouch and Bodmer 2020). Two dominant routes of colorectal carcinogenesis were revealed through analysis of patients with LS and FAP. LS has been erroneously called hereditary nonpolyposis CRC, but “nonpolyposis” is a misnomer as almost all colorectal polyps can be LS precursor lesions, which typically present with villous growth and high-grade dysplasia (Burt et al. 2004). Recently, the classical perspectives of LS as an “accelerating” disease have been challenged to provide many alternatives to classical adenoma–carcinoma sequence, importantly by the discovery of MMR-deficient crypt foci (MMR-DCF) (Ahadova et al. 2018). Ahadova et al. suggested LS CRCs to be possibly developed through three distinct pathways, i.e., MMR-proficient adenomas after secondary inactivation of the MMR system (pathway 1), the other larger parts from precursor lesions in which MMR deficiency is an early event, likely to include MMR-DCF, either through an adenomatous phase (pathway 2) or as non-polypus lesions with immediate invasive growth (pathway 3). Although there are some genotypic and phenotypic overlaps, FCRC represents 14 syndromes, including six LS and LS-associated spectra and eight inherited polyposis syndromes. The former category includes traditional LS, Muir–Torre syndrome, Turcot syndrome, constitutional MMR deficiency (CMMRD) syndrome, EPCAM-associated LS (EALS), and familial CRC type X (FCCTX). The latter category includes a broad spectrum of inherited polyposis syndromes, including FAP, MUTYH-associated polyposis (MAP), polymerase proofreading-associated polyposis (PPAP), serrated polyposis syndrome (SPS), hereditary hamartomatous polyposis including Peutz–Jeghers syndrome (PJS), juvenile polyposis syndrome (JPS), PTEN hamartoma tumour syndrome, and hereditary mixed polyposis syndrome. The lifetime risks of associated neoplasms in patients with FCRC are summarized in Table 1.
Lynch syndrome and associated spectra
Men and women with LS have estimated lifetime risks for CRC of 70% and 40%, respectively (Wells and Wise 2017). Endometrial adenocarcinoma is the most common extra-colonic cancer, with a lifetime risk of 32–45%, followed by ovarian, small bowel, gastric, urinary tract, pancreas, and brain cancers, in that order. Variants in MLH1, MSH2, MSH6, and PMS2 were found in 40%, 34%, 18%, and 8%, respectively, of patients with LS (Peltomäki et al. 2020). EPCAM deletions are also considered a cause of LS, being present in up to 30% of patients with MSH2-mutation (−) tumours and 20% of those without MMR mutations (Tutlewska et al. 2013). EPCAM-associated LS carries a risk of CRC, with the phenotype of these tumours being similar to those of tumours with MLH1 and MSH2 mutations, whereas the cumulative risk of endometrial cancer in patients with EPCAM-associated LS is much lower (Tutlewska et al. 2013; Kempers et al. 2011). Muir–Torre syndrome and Turcot syndrome are particular LS subtypes caused by MMR alterations, selectively displaying sebaceous neoplasms of the skin and brain tumours, respectively. Muir–Torre syndrome carries MLH1, MSH2, and MSH6 mutations, along with several recently described autosomal recessive traits, including MUTYH alterations in the absence of MSI (Gadish et al. 2005). LS and FAP can co-segregate with Turcot syndrome, mostly inherited through autosomal recessive transmission with bi-allelic MLH1/PMS2 and APC mutations, respectively (Gadish et al. 2005). Glioblastomas occur in patients with MMR gene mutations, specifically those with MLH1 mutations, whereas medulloblastomas have been associated with APC mutations. Another LS subset, a specific phenotype designated constitutional mismatch repair deficiency (CMMRD), is a highly penetrating cancer-predisposition syndrome caused by bi-allelic MMR alterations, more frequently in PMS2 and MSH6 than in other MMR genes (Nejadtaghi et al. 2017). Tumours frequently observed in patients with CMMRD include brain (48%), gastrointestinal (32%), and haematological (15%) tumours. As many as 40% of CRCs fulfilling the clinical criteria for LS exhibit MSS, transiently called familial colorectal cancer type X (FCCTX) (Nejadtaghi et al. 2017). Causative genes for FCCTX remain to be verified, although several candidate genes have been proposed by multi-gene panels (MGPs), including AXIN2, BCR, BLM, BMPR1A, BRCA1, BRCA2, BRF1, CHEK2, FAN1, GABBR2, GALNT12, HABP4, KIF24, MSH3, MUTYH, OGG1, POLD1, RPS20, SEMA4A, and ZNF367 (Hansen et al. 2017; Yurgelun et al. 2015a, b; Gupta et al. 2019).
Inherited polyposis syndrome
Inherited polyposis syndrome can be stratified by histological morphology as adenomatous, hamartomatous, and mixed polyposis. Germline APC mutations around codon 1300 (codons 1286–1513) have been associated with severe colorectal polyposis of FAP (Fearnhead et al. 2001). Otherwise, somatic APC mutations of codons 1400–1580 have been observed in upper gastrointestinal polyps, including severe duodenal polyposis clusters, with these APC gene products retaining only one of the 20-amino acid β-catenin-binding degradation repeats (Groves et al. 2002). MAP tends to present later in life (age > 25 years) than FAP, and develops predominantly in the proximal colon. Histologically, these lesions are mucin-rich, with abundant lymphocyte infiltration, and patients with MAP have a better prognosis than patients with sporadic CRCs (Kanth et al. 2017). The two most common MUTYH founder mutations, Y179C and G396D (previously called Y165C and G382D, respectively), are present in 70–80% of individuals of Northwestern European ancestry with MAP, with these mutations inherited in an autosomal recessive manner (Stoffel and Boland 2015). However, approximately one-third of individuals with biallelic MUTYH mutations develop CRC in the absence of polyposis, suggesting incomplete penetrance (Sereno et al. 2014).
Germline pathogenic variants in the exonuclease domain (ED) of polymerases POLE1 and POLD predispose to PPAP (Church et al. 2013). Most POLE variant heterozygotes carry a colorectal tumour phenotype, with their median ages at diagnosis of polyps and CRCs being 36 and 44 years, respectively (Palles et al. 2021). Endometrial and ovarian cancers are the most common malignancies found in women with POLD1 variant heterozygotes under age 50 years (Church et al. 2013; Palles et al. 2021). The updated diagnostic criteria for SPS (WHO, 2019) include ≥ 5 serrated polyps above the rectum that are ≥ 5 mm in diameter with at least two ≥ 10 mm polyps, or ≥ 20 serrated polyps of any size throughout the colon with ≥ 5 above the rectum, along with increased risk of CRC (Stanich and Pearlman 2019). Non-synonymous mutations in RNF43, encoding E3 ubiquitin-protein ligase, have been observed only in the affected siblings of patients with SPS, suggesting that these mutations are causative genetic variants (Quintana et al. 2018). NTHL1 tumour syndrome, a novel type of familial CRC predisposing to adenomatous polyposis and CRC, is caused by germline bi-allelic pathogenic variants in NTHL1 (Weren et al. 2015). This condition is also accompanied by increased lifetime risks for breast cancer and variable extra-colonic tumours of both the LS- and FAP-associated spectra.
The Peutz–Jeghers syndrome (PJS) phenotype manifests when germline STK11 (LKB1) mutations, inherited in an autosomal-dominant manner, are accompanied by acquired defects in the other allele in somatic cells (Tacheci et al. 2021). JPS is caused by SMAD4 and BMPR1A (ALK3) mutations, inherited in an autosomal-dominant manner with incomplete penetrance, with collective rates of 23 and 21–38%, respectively (Woodford-Richens et al. 2000a, b; Zhou et al. 2001). BMPR1A is located upstream of SMAD4 in the TGF-β pathway, and mutations encoding BMP receptors result in loss of intracellular BMP signalling via SMAD 4 (Chow and Macrae 2005). In contrast to the initial landscaper-defect hypothesis of JPS (Kinzler and Vogelstein 1998), fluorescence in situ hybridization showed that epithelial malignancies in JPS are likely to develop through direct progression in the epithelial components of hamartomas, suggesting that SMAD4 acts as a gatekeeper in both JPS and sporadic cancers (Woodford-Richens et al. 2000). Patients with PTEN hamartoma tumour syndrome (PHTS), including those with Bannayan–Riley–Ruvalcaba syndrome, Cowden syndrome, Gorlin syndrome, and Proteus-like syndrome, are at increased risk of developing cancer (Hendricks et al. 2021). Mutations in PTEN (10q23.3) and PTCH (9q31) can rule out almost all JPS patients that are exclusively considered in the context of PHTS (Zhou et al. 2001). Patients with Cowden syndrome and similar phenotypes may also have associated hypermethylation of the KLLN promoter, deregulating p53-induced apoptosis (Nizialek et al. 2015).
Hereditary mixed polyposis syndrome (HMPS) is characterized by a small number of polyps with mixed phenotypes, most commonly adenoma and non-dysplastic mixed serrated/inflammatory polyps. The causative genetic alteration in HMPS was found to be a 40 Kb duplication at the 3’ end of the SCG5 (Ashkenazi Jewish founder mutation), resulting in aberrant epithelial expression of the mesenchymal BMP antagonist, Gremlin, transmitted via autosomal-dominant inheritance (Davis et al. 2015).
Genotype–phenotype correlations and causative gene discovery
Particular traits of CRC carcinogenesis have been investigated by evaluating genotype–phenotype correlations. Approximately 16% of CRC patients aged < 50 years (early-onset CRC, EOC) were found to carry at least one pathogenic cancer-susceptible gene mutation (Pearlman et al. 2017). One NGS study according to the revised Bethesda guidelines found that FAT4 mutation rates were lower in patients with EOC than in those with later-onset CRCs, potentially defining an early-onset MSS subtype (Kim et al. 2021). Another EOC study by the polygenic risk score demonstrated that the cumulative burden of common genetic variants associated with CRCs was higher in patients with EOCs, particularly in the absence of a family history (Archambault et al. 2020). Furthermore, EOCs tend to present with higher histological grade and higher rates of recurrence and metastasis. The incidence of CRC is modestly higher in patients with Li–Fraumeni syndrome, characterized by germline mutations in TP53, than in the overall population. One registry-based analysis found that germline alterations in TP53were associated with EOCs (Yurgelun et al. 2015a, b). Taken together, these findings indicated that EOCs are not only associated with rare monogenic or high-penetrance genetic syndromes in high-risk families, but also with low-penetrant multi-gene variants. The morphology and histology of left-sided (LCRC) and right-sided (RCRC) CRCs also differ markedly. Epidemiologically, younger individuals and men are more likely to have LCRC, whereas elderly persons and women are more likely to develop RCRC. Nevertheless, LCRC remains the most commonly diagnosed form of CRC across all age groups, constituting approximately 70% of all CRCs (Jess et al. 2013).
Most comprehensive MGPs have utilized NGS to identify pathogenic single or multiple gene variants. However, gene discovery via MGPs identifies considerable numbers of variants of unknown significance (VUS), as well as clinically questionable or non-actionable variants (Hall et al. 2014). Actionable CRC gene variants identified by MGPs include mutations/alterations in APC (I1307K polymorphism), AXIN2, CHEK2, GREM1, GALANT12, MSH3, MUTYH (monoallelic), NTHL1, POLD1, and POLE (Gupta et al. 2019) A genome-wide association study (GWAS) of a Finnish cohort of patients with CRC found an association between the intronic SNP rs992157 at 2q35 with PNKD/TMBIM1, a finding independently replicated in a meta-analysis of CRC patients of European ancestry (Tanskanen et al. 2018). Another study demonstrated that TRIM4 and PYGL, which encode proteins that influence redox homeostasis and cellular metabolic reprogramming, respectively, may be implicated in a novel CRC pathway linked to cell growth and proliferation (Bien et al. 2019). Another custom-made MGP (HaloPlex®) targeting 112 genes, covering previously identified and candidate CRC susceptibility genes, identified 17 pathogenic variants as potential gene alterations associated with CRC susceptibility; these included variants of MUTYH, ATM, AXIN1, AXIN2, BRCA1, CHEK2, BMP4, CCDC18, NUDT7, PICALM, PTPRJ, SLC5A9, TLR2, TWSG1, UBAP2, USP6NL, and ZFP14 (Hansen et al. 2017). New approaches designed to identify rare variants should herein consider the two main criteria suggested by the historical overview, i.e., genes in which obviously severe disruption of function gives rise to a severe, usually familial, version of the disease being studied and genes known to be involved in the biology of the disease based on biochemical and physiological analyses (Bodmer and Bonilla 2008).
Consensus molecular subtype
Classification of CRCs by consensus molecular subtype (CMS) may assist in the development of personalized medicine to treat these patients. These classifications, however, have limitations, mainly confined to MSI, mesenchymal cells, and specific driver gene mutations. Additionally, regional tumour heterogeneity in molecular classifiers, particularly concerning CMS4 with a stromal mixture, can result in tumour misclassification (Dunne et al. 2016). CMS nevertheless appears to increase understanding of the molecular and immune signatures that predict clinical behaviour and responses to different therapeutic agents (Farooqi et al. 2019). CMS1 tumours are characterized by widespread hypermethylation, whereas CMS2-4 tumours show development via the CIN route, as measured by SCNA counts (Guinney et al. 2015). CMS1 CRCs are hyper-mutated tumours with MSI due to defective MMR, usually caused by MLH1 promoter hypermethylation. Nearly 80–90% of sporadic hyper-mutated CRCs also carry BRAF V600E mutations. POLE or POLD1 ED (proofreading) mutations insert incorrect nucleotides during DNA replication, resulting in an ultra-mutated phenotype. Additionally, gene expression profiling has shown evidence of strong immune activation in CMS1, with high tumour infiltration by CD8+ cytotoxic T lymphocytes. CMS2 CRCs display epithelial differentiation and strong upregulation of Wnt and MYC downstream targets, classically implicated in colorectal carcinogenesis. By contrast, multiple metabolic signatures are enriched in CMS3 CRCs, in agreement with the occurrence of KRAS activating mutations that induce metabolic adaptations. Finally, CMS4 CRCs show clear upregulation of genes implicated in EMT and signatures associated with the activation of the TGF-β signalling, angiogenesis, and matrix remodelling pathways, and activation of serum complement. Finally, mixed CMS subtypes, probably resulting from multiple clones in an individual tumour, have been detected in as many as 13% of CRCs, although these are considered to be outliers. Irrespective of patient cohort, patients with CMS4 tumours have poorer survival outcomes, whereas the percentage of long-term survivors is higher in patients with CMS2 tumours than in those with other subtypes. Interestingly, survival in patients with CMS1 tumours is very poor after relapse, in agreement with studies showing a poorer prognosis in patients with MSI and BRAF-mutated CRCs following tumour recurrence (Guinney et al. 2015).
A study correlating CMS subtypes with tumour site suggested that the mutational profiles of tumours in the transverse colon differed from the profiles of right-sided, and especially left-sided, tumours (Loree et al. 2018). Concerning EOC, possibly related to heredity, CMS1 was the most common subtype, whereas CMS3 and CMS4 were uncommon, whereas CMS2 was relatively stable across age groups (Willauer et al. 2019). The CMS classification holds clear potential for clinical use in predicting both prognosis and response to systemic therapy, which seems to be independent of the classifier used (Ten Hoorn et al. 2021). Although fluoropyrimidine-monotherapy lacked benefits for dMMR CMS1 tumours, whereas additional bevacizumab seemed beneficial and immune checkpoint inhibitors received a new agnostic indication for unresectable or metastatic CRCs with high tumour mutational burden in CMS1. Adjuvant chemotherapy in stage II and III CRC increased overall survival in CMS2 and CMS3, although not effective in CMS4 tumors. However, CMS4 metastatic CRC predicted benefits from irinotecan combined with cetuximab (in KRAS wt tumours) and bevacizumab (in KRAS mutant tumours), recommended as preferred first-line options in CMS4 metastatic CRC patients. On the other hand, CRC microbiomes might be associated CMS, for example, elevated abundance of Fusobacterium, Peptostreptococcus, or Parvimonas, to be associated with CMS1, underscored the potential role of oral poly-microbial communities in the development of a subset of CRC pathogenesis (Purcell et al. 2017).
A recent global transcriptomic immune classification proposed six immune subtypes (ISs), along with their interplay with CMS types (Soldevilla et al. 2019). Only five ISs, all but C5, were identified in CRCs, with C1 (wound healing subtype, 77%) and C2 (IFN-γ dominant subtype, 17%) being the most frequent. CMS1 showed the highest proportion of C2 (53%), whereas C1 was particularly frequent in CMS2 (91%). CMS3 had the highest representation of C3 (inflammatory subtype, 7%) and C4 (lymphocyte depleted subtype, 4%), whereas all C6 TGF-β dominant subsets belonged to CMS4 (2.3%). The immunologically quiescent C5 subtype showed the lowest lymphocyte and highest macrophage responses, dominated by M2 macrophages. Another study found that 54 (87%) of 62 colorectal adenomas could be classified according to the CMS (Komor et al. 2018). The metabolic type of CMS3, which was least common among CRCs, was the most prevalent among adenomas (73%), followed by CMS2 (13%) and CMS1 (2%). None of the adenomas presented with the mesenchymal subtype CMS4, consistent with the lack of invasion-associated stroma in adenomas.
Concluding remarks
Current knowledge of genomic colorectal carcinogenesis is generally based on the adenoma-to-carcinoma sequence, and to some extent on the hamartoma–carcinoma sequence, serrated neoplasia, and LS pathway. The classical routes of carcinogenesis, via CIN, MSI, and serrated transition have been identified, each exhibiting site-specificity and distinctive tumour biology. Although the events initiating adenoma formation may be common to MSI and CIN tumours, TSAs and SSAs are confined to the serrated route, with most carrying KRAS mutations and MSI, respectively. Post-transcriptional modifications also participate in oncogenic transformation throughout all the steps of carcinogenesis, including DNA methylation and dysregulation of histone modifications. Environmental factors, including alterations of the enteric microbiome, participate in the genomic orchestration of colorectal carcinogenesis through gene–environment interactions. FCRCs have played important roles in determining aspects of colorectal carcinogenesis. Recent advances in the NGS platform may define the genes involved in FCRC by, for example, determining FCCTX using comprehensive MGPs.
References
Advani SM, Advani P, DeSantis SM, Brown D, VonVille HM, Lam M et al (2018) Clinical, pathological, and molecular characteristics of CpG island methylator phenotype in colorectal cancer: a systematic review and meta-analysis. Transl Oncol 11(5):1188–1201
Ahadova A, Gallon R, Gebert J, Ballhausen A, Endris V, Kirchner M et al (2018) Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int J Cancer 143(1):139–150
Angius A, Scanu AM, Arru C, Muroni MR, Rallo V, Deiana G et al (2021) Portrait of cancer stem cells on colorectal cancer: molecular biomarkers, signaling pathways and miRNAome. Int J Mol Sci 22(4):1603
Archambault AN, Su YR, Jeon J, Thomas M, Lin Y, Conti DV et al (2020) Cumulative burden of colorectal cancer-associated genetic variants is more strongly associated with early-onset vs late-onset cancer. Gastroenterology 158(5):1274–1286
Babaei-Jadidi R, Li N, Saadeddin A, Spencer-Dene B, Jandke A, Muhammad B et al (2011) FBXW7 influences murine intestinal homeostasis and cancer, targeting Notch, Jun, and DEK for Degradation. J Exp Med 208(2):295–312
Bien SA, Su YR, Conti DV, Harrison TA, Qu C, Guo X et al (2019) Genetic variant predictors of gene expression provide new insight into risk of colorectal cancer. Hum Genet 138(4):307–326
Bodmer W, Bonilla C (2008) Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 40(6):695–701
Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ, Ellis A, Gorman P et al (1987) Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature 328(6131):614–616
Bosman FT (1999) The hamartoma-adenoma-carcinoma sequence. J Pathol 188(1):1–2
Buettner R, Mora LB, Jove R (2002) Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res 8:945–954
Burt RW, Leppert MF, Slattery ML, Samowitz WS, Spirio LN, Kerber RA et al (2004) Genetic testing and phenotype in a large kindred with attenuated familial adenomatous polyposis. Gastroenterology 127(2):444–451
Cheok CF, Lane DP (2017) Exploiting the p53 pathway for therapy. Cold Spring Harb Perspect Med 7(3):ra026310
Chow E, Macrae F (2005) A review of juvenile polyposis syndrome. J Gastroenterol Hepatol 20(11):1634–1640
Church DN, Briggs SE, Palles C, Domingo E, Kearsey SJ, Grimes JM et al (2013) DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet 22(14):2820–2828
Crouch DJM, Bodmer WF (2020) Polygenic inheritance, GWAS, polygenic risk scores, and the search for functional variants. Proc Natl Acad Sci USA 117(32):18924–18933
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al (2002) Mutations of the BRAF gene in human cancer. Nature 417(6892):949–954
Davis H, Irshad S, Bansal M, Rafferty H, Boitsova T, Bardella C et al (2015) Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat Med 21(1):62–70
Deitrick J, Pruitt WM (2016) Wnt/β catenin-mediated signaling commonly altered in colorectal Cancer. Prog Mol Biol Transl Sci 144:49–68
Dunne PD, McArt DG, Bradley CA, O’Reilly PG, Barrett HL, Cummins R et al (2016) Challenging the cancer molecular stratification dogma: intratumoral heterogeneity undermines consensus molecular subtypes and potential diagnostic value in colorectal cancer. Clin Cancer Res 22(16):4095–4104
Esteller M (2007) Cancer epigenomics: DNA methylomes and histone modification maps. Nature Rev Genet 8(4):286–298
Farooqi AA, de la Roche M, Djamgoz MBA, Siddik ZH (2019) Overview of the oncogenic signaling pathways in colorectal cancer: mechanistic insights. Semin Cancer Biol 58:65–79
Fearnhead NS, Britton MP, Bodmer WF (2001) The ABC of APC. Hum Mol Genet 10(7):721–733
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61(5):759–767
Gadish T, Tulchinsky H, Deutsch AA, Rabau M (2005) Pinealoblastoma in a patient with familial adenomatous polyposis: variant of Turcot syndrome type 2? Report of a case and review of the literature. Dis Colon Rectum 48(12):2343–2346
Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L et al (2004) Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res 64(9):3014–3021
Grady WM, Carethers JM (2008) Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 135(4):1079–1099
Groves C, Lamlum H, Crabtree M, Williamson J, Taylor C, Bass S et al (2002) Mutation cluster region, association between germline and somatic mutations and genotype-phenotype correlation in upper gastrointestinal familial adenomatous polyposis. Am J Pathol 160(6):2055–2061
Guillén-Ponce C, Castillejo A, Barberá VM, Pascual-Ramírez JC, Andrada E, Castillejo MI et al (2010) Biallelic MYH germline mutations as cause of Muir–Torre syndrome. Fam Cancer 9(2):151–154
Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicke A, Soneson C et al (2015) The consensus molecular subtypes of colorectal cancer. Nat Med 21(11):1350–1356
Gupta S, Provenzale D, Llor X, Halverson AL, Grady W, Chung DC et al (2019a) NCCN guidelines insights: genetic/familial high-risk assessment: colorectal, ver 2. 2019. J Natl Compr Canc Netw 17(9):1032–1041
Hague A, Hicks DJ, Hasan F, Smartt H, Cohen GM, Paraskeva C, MacFarlane M (2005) Increased sensitivity to TRAIL-induced apoptosis occurs during the adenoma to carcinoma transition of colorectal carcinogenesis. Br J Cancer 92(4):736–742
Hall MJ, Forman AD, Pilarski R, Wiesner G, Giri VN (2014) Gene panel testing for inherited cancer risk. J Natl Compr Canc Netw 12(9):1339–1346
Ham-Karim HA, Ebili HO, Bradshaw K, Richman SD, Fadhil W, Domingo E et al (2019b) Targeted next generation sequencing reveals a common genetic pathway for colorectal cancers with chromosomal instability and those with microsatellite and chromosome stability. Pathol Res Pract 215(7):152445
Han T, Schatoff EM, Murphy C, Zafra MP, Wilkinson JE, Elemento O et al (2017) R-Spondin chromosome rearrangements drive Wnt-dependent tumour initiation and maintenance in the intestine. Nat Commun 8:15945
Hansen MF, Johansen J, Sylvander AE, Bjørnevoll I, Talseth-Palmer BA, Lavik LAS et al (2017) Use of multigene-panel identifies pathogenic variants in several CRC-predisposing genes in patients previously tested for Lynch syndrome. Clin Genet 92(4):405–414
Hendricks LAJ, Hoogerbrugge N, Schuurs-Hoeijmakers JHM, Vos JR (2021) A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin Genet 99(2):219–225
Ijichi H, Ikenoue T, Kato N, Mitsuno Y, Togo G, Kato J et al (2001) Systematic analysis of the TGF-beta-Smad signaling pathway in gastrointestinal cancer cells. Biochem Biophys Res Commun 289(2):350–357
Jansen M, Ten Klooster JP, Offerhaus GJ, Clevers H (2009) LKB1 and AMPK family signaling: the intimate link between cell polarity and energy metabolism. Physiol Rev 89(3):777–798
Jess P, Hansen IO, Gamborg M, Jess T (2013) A nationwide Danish cohort study challenging the categorisation into right-sided and left sided colon cancer. BMJ Open 3(5):e002608
Jin L, Vu T, Yuan G, Datta PK (2017) STRAP Promotes stemness of human Colorectal cancer via epigenetic regulation of the NOTCH pathway. Cancer Res 77(20):5464–5478
Kanth P, Grimmett J, Champine M, Burt R, Samadder NJ (2017) Hereditary colorectal polyposis and cancer syndromes: a primer on diagnosis and management. Am J Gastroenterol 112(10):1509–1525
Kempers MJ, Kuiper RP, Ockeloen CW, Chappuis PO, Hutter P, Rahner N et al (2011) Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol 12(1):49–55
Kim JC, Cho YK, Roh SA, Yu CS, Gong G, Jang SJ et al (2008) Individual tumorigenesis pathways of sporadic colorectal adenocarcinomas are associated with the biological behavior of tumors. Cancer Sci 99(7):1348–1354
Kim MS, Kim SS, Ahn CH, Yoo NJ, Lee SH (2009) Frameshift mutations of Wnt pathway genes AXIN2 and TCF7L2 in gastric carcinomas with high microsatellite instability. Hum Pathol 40(1):58–64
Kim JC, Kim JH, Ha YJ, Kim CW, Tak KH, Yoon YS et al (2021) Analysis of genomic pathogenesis according to the revised Bethesda guidelines and additional criteria. J Cancer Res Clin Oncol 147(1):117–128
Kinzler KW, Vogelstein B (1998) Landscaping the cancer terrain. Science 280:1036–1037
Komor MA, Bosch LJ, Bounova G, Bolijn AS, Delis-van Diemen PM, Rausch C et al (2018) Consensus molecular subtype classification of colorectal adenomas. J Pathol 246(3):266–276
Korsse SE, Biermann K, Offerhaus GJ, Wagner A, Dekker E, Mathus-Vliegen EM et al (2013) Identification of molecular alterations in gastrointestinal carcinomas and dysplastic hamartomas in Peutz–Jeghers syndrome. Carcinogenesis 34(7):1611–1619
Kozma L, Kiss I, Szakáll S, Ember I (1994) Investigation of c-myc oncogene amplification in colorectal cancer. Cancer Lett 81(2):165–169
Lane DP (1992) Cancer. p53, guardian of the genome. Nature 358(6381):15–16
Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez-Vega F et al (2018) Comparative molecular analysis of gastrointestinal adenocarcinomas. Cell 33(4):721–735
Loree JM, Pereira AAL, Lam M, Willauer AN, Raghav K, Dasari A et al (2018) Classifying colorectal cancer by tumor location rather than sidedness highlights a continuum in mutation profiles and consensus molecular subtypes. Clin Cancer Res 24(5):1062–1072
Luebeck EG, Moolgavkar SH (2002) Multistage carcinogenesis and the incidence of colorectal cancer. PNAS 99(23):15095–15100
Markowitz SD, Bertagnolli MM (2009) Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 361(25):2449–2460
Martínez-Jiménez F, Muiños F, Sentís I, Deu-Pons J, Reyes-Salazar I, Arnedo-Pac C et al (2020) A compendium of mutational cancer driver genes. Nat Rev Cancer 20(10):555–572
Maurer G, Tarkowski B, Baccarini M (2011) Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 30(32):3477–3488
Mizutani S, Yamada T, Yachida S (2020) Significance of the gut microbiome in multistep colorectal carcinogenesis. Cancer Sci 111(3):766–773
Müller MF, Ibrahim AE, Arends MJ (2016) Molecular pathological classification of colorectal cancer. Virchows Arch 469(2):125–134
Nejadtaghi M, Jafari H, Farrokhi E, Samani KG (2017) Familial colorectal cancer type X (FCCTX) and the correlation with various genes-a systematic review. Curr Probl Cancer 41(6):388–937
Neuzillet C, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E (2014) MEK in cancer and cancer therapy. Pharmacol Ther 141(2):160–171
Nguyen LH, Goel A, Chung DC (2020) Pathways of colorectal carcinogenesis. Gastroenterology 158(2):291–302
Niyaz M, Khan MS, Mudassar S (2019) Hedgehog signaling: an Achilles’ heel in cancer. Transl Oncol 12(10):1334–1344
Nizialek EA, Mester JL, Dhiman VK, Smiraglia DJ, Eng C (2015) KLLN epigenotype-phenotype associations in Cowden syndrome. Eur J Hum Genet 23(11):1538–1543
Pai R, Tarnawski AS, Tran T (2004) Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell 15(5):2156–2163
Palles C, Martin L, Domingo E, Chegwidden L, McGuire J, Cuthill V et al (2021) The clinical features of polymerase proof-reading associated polyposis (PPAP) and recommendations for patient management. Fam Cancer. https://doi.org/10.1007/s10689-021-00256-y
Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K et al (2017) Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol 3(4):464–471
Peltomäki P, Olkinuora A, Nieminen TT (2020) Expert Updates in the field of hereditary nonpolyposis colorectal cancer. Rev Gastroenterol Hepatol 14(8):707–720
Pinheiro M, Pinto C, Peixoto A, Veiga I, Lopes P, Henrique R et al (2015) Target gene mutational pattern in Lynch syndrome colorectal carcinomas according to tumour location and germline mutation. Br J Cancer 113(4):686–692
Pino MS, Chung DC (2010) The chromosomal instability pathway in colon cancer. Gastroenterology 138(6):2059–2072
Purcell RV, Visnovska M, Biggs PJ, Schmeier S, Frizelle FA (2017) Distinct gut microbiome patterns associate with consensus molecular subtypes of colorectal cancer. Sci Rep 7(1):11590
Qiao L, Wong BC (2009) Role of Notch signaling in colorectal cancer. Carcinogenesis 30(12):1979–1986
Quintana I, Mejías-Luque R, Terradas M, Navarro M, Piñol V, Mur P et al (2018) Evidence suggests that germline RNF43 mutations are a rare cause of serrated polyposis. Gut 67(12):2230–2232
Samuels Y, Ericson K (2006) Oncogenic PI3K and its role in cancer. Curr Opin Oncol 18(1):77–82
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S et al (2004) High frequency of mutations of the PIK3CA gene in human cancers. Science 304(5670):554
Sereno M, Merino M, López-Gómez M, Gómez-Raposo C, Zambrana Tébar F, Moreno Rubio J et al (2014) MUTYH polyposis syndrome: clinical findings, genetics issues and management. Clin Transl Oncol 16(8):675–679
Shuen AY, Lanni S, Panigrahi GB, Edwards M, Yu L, Campbell BB et al (2019) Functional repair assay for the diagnosis of constitutional mismatch repair deficiency from non-neoplastic tissue. J Clin Oncol 37(6):461–470
Soldevilla B, Carretero-Puche C, Gomez-Lopez G, Al-Shahrour F, Riesco MC, Gil-Calderon B et al (2019) The correlation between immune subtypes and consensus molecular subtypes in colorectal cancer identifies novel tumour microenvironment profiles, with prognostic and therapeutic implications. Eur J Cancer 123:118–129
Sonoshita M, Itatani Y, Kakizaki F, Sakimura K, Terashima T, Katsuyama Y et al (2015) Promotion of colorectal cancer invasion and metastasis through activation of NOTCH-DAB1-ABL-RHOGEF protein TRIO. Cancer Discov 5(2):198–211
Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J et al (2015) A Big Bang model of human colorectal tumor growth. Nat Genet 47(3):209–216
Spano JP, Milano G, Rixe C, Fagard R (2006) JAK/STAT signalling pathway in colorectal cancer: a new biological target with therapeutic implications. Eur J Cancer 42(16):2668–2670
Stanich PP, Pearlman R (2019) Hereditary or not? understanding serrated polyposis syndrome. Curr Treat Options Gastroenterol 17(4):692–701
Stoffel EM, Boland CR (2015) Genetics and genetic testing in hereditary colorectal cancer. Gastroenterology 149(5):1191–1203
Tacheci I, Kopacova M, Bures J (2021) Peutz–Jeghers syndrome. Curr Opin Gastroenterol 37(3):245–254
Tanskanen T, van den Berg L, Välimäki N, Aavikko M, Ness-Jensen E, Hveem K et al (2018) Genome-wide association study and meta-analysis in Northern European populations replicate multiple colorectal cancer risk loci. Int J Cancer 142(3):540–546
Ten Hoorn S, de Back TR, Sommeijer DW, Vermeulen L (2021) Clinical value of consensus molecular subtypes in colorectal cancer: a systematic review and meta-analysis. J Natl Cancer Inst. https://doi.org/10.1093/jnci/djab106
Toledo F, Wahl GM (2006) Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer 6(12):909–923
Tsukiyama T, Zou J, Kim J, Ogamino S, Shino Y, Masuda T et al (2020) A phospho-switch controls RNF43-mediated degradation of Wnt receptors to suppress tumorigenesis. Nat Commun 11(1):4586
Tutlewska K, Lubinski J, Kurzawski G (2013) Germline deletions in the EPCAM gene as a cause of Lynch syndrome—literature review. Hered Cancer Clin Pract 11(1):9
van Geelen CM, de Vries EG, de Jong S (2004) Lessons from TRAIL-resistance mechanisms in colorectal cancer cells: paving the road to patient-tailored therapy. Drug Resist Update 7(6):345–358
van Neerven SM, de Groot NE, Nijman LE, Scicluna BP, van Driel MS, Lecca MC et al (2021) Apc-mutant cells act as supercompetitors in intestinal tumour initiation. Nature 594(7863):436–441
Wells K, Wise PE (2017) Hereditary colorectal cancer syndromes. Surg Clin N Am 97(3):605–625
Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L et al (2015) A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 47(6):668–671
Willauer AN, Liu Y, Pereira AAL, Lam M, Morris JS, Raghav KPS et al (2019) Clinical and molecular characterization of early-onset colorectal cancer. Cancer 125(12):2002–2010
Woodford-Richens K, Bevan S, Churchman M, Dowling B, Jones D, Norbury CG et al (2000a) Analysis of genetic and phenotypic heterogeneity in juvenile polyposis. Gut 46(5):656–660
Woodford-Richens K, Williamson J, Bevan S, Young J, Leggett B, Frayling I et al (2000b) Allelic loss at SMAD4 in polyps from juvenile polyposis patients and use of fluorescence in situ hybridization to demonstrate clonal origin of the epithelium. Cancer Res 60(9):2477–2482
Xu X, Su J, Li R, Wang Y, Zeng D, Wu B (2016) Aberrant expression of Sonic hedgehog signaling in Peutz–Jeghers syndrome. Hum Pathol 50:153–161
Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P et al (2015a) Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology 149(3):604–613
Yurgelun MB, Masciari S, Joshi VA, Mercado RC, Lindor NM, Gallinger S et al (2015b) Germline TP53 mutations in patients with early-onset colorectal cancer in the colon cancer family registry. JAMA Oncol 1(2):214–221
Zhou XP, Woodford-Richens K, Lehtonen R, Kurose K, Aldred M, Hampel H et al (2001) Germline mutations in BMPR1A/ALK3 cause a subset of cases of juvenile polyposis syndrome and of Cowden and Bannayan–Riley–Ruvalcaba syndromes. Am J Hum Genet 69(4):704–711
Author information
Authors and Affiliations
Contributions
Conception and design: JCK; manuscript writing: JCK and WFB; final approval of manuscript: JCK and WFB. Accountable for all aspects of the work: JCK and WFB.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kim, J.C., Bodmer, W.F. Genomic landscape of colorectal carcinogenesis. J Cancer Res Clin Oncol 148, 533–545 (2022). https://doi.org/10.1007/s00432-021-03888-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-021-03888-w