
Abstract
Objectives
Postoperative early relapse of early-stage lung adenocarcinoma is implicated in poor prognosis. The purpose of our study was to develop an integrated mRNA and non-coding RNA (ncRNA) signature to identify patients at high risk of early relapse in stage I–II lung adenocarcinoma who underwent complete resection.
Methods
Early-stage lung adenocarcinoma data from Gene Expression Omnibus database were divided into training set and testing set. Propensity score matching analysis was performed between patients in early relapse group and long-term nonrelapse group from training set. Transcriptome analysis, random survival forest and LASSO Cox regression model were used to build an early relapse-related multigene signature. The robustness of the signature was evaluated in testing set and RNA-Seq dataset from The Cancer Genome Atlas (TCGA). The chemotherapy sensitivity, tumor microenvironment and mutation landscape related to the signature were explored using bioinformatics analysis.
Results
Twelve mRNAs and one ncRNA were selected. The multigene signature achieved a strong power for early relapse prediction in training set (HR 3.19, 95% CI 2.16–4.72, P < 0.001) and testing set (HR 2.91, 95% CI 1.63–5.20, P = 0.002). Decision curve analyses revealed that the signature had a good clinical usefulness. Groups divided by the signature exhibited different chemotherapy sensitivity, tumor microenvironment characteristics and mutation landscapes.
Conclusions
Our results indicated that the integrated mRNA–ncRNA signature may be an innovative biomarker to predict early relapse of early-stage lung adenocarcinoma, and may provide more effective treatment strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lung cancer is the leading cause of cancer‐related death worldwide, with 2.1 million new cases and 1.8 million deaths in the year 2018 (Bray et al. 2018). Lung adenocarcinoma is becoming the most common histological type of lung cancer, and its morbidity and mortality are also increasing (Travis et al. 2015). Even for patients with early-stage (stage I and stage II) lung adenocarcinoma, more than 30% of patients who underwent radical surgical resection will relapse and die of tumor recurrence (Padda et al. 2014). Patients underwent early relapse in lung adenocarcinoma tend to have poorer survival rates and is attributed mainly to poor clinicopathological features such as advanced tumor stage, poorly differentiated tumors, visceral pleural involvement, insufficient resection, incomplete nodal sampling and resistance to adjuvant chemotherapy (Kozu et al. 2013; Kiankhooy et al. 2014). In addition, the relapse of early-stage lung adenocarcinoma can be characterized as a process with time sequence. The first 2 years after surgery accounted for the most relapse cases. Currently, the tumor–node–metastasis (TNM) staging system carried out by American Joint Commission on Cancer/International Union against Cancer (AJCC/UICC) has been widely used for treatment selection and relapse prediction in early-stage lung adenocarcinoma (Amin et al. 2017). Though multiple clinicopathological features can be incorporated with TNM staging system to improve relapse prediction accuracy for lung adenocarcinoma, prognosis often varies in patients even with comparable stage and clinicopathological features. These problems reflect the potential tumor heterogeneity of lung adenocarcinoma. Consequently, more accurate strategies are warranted for early relapse detection. Nowadays, some studies have focused on transcriptional profiles in lung adenocarcinoma and several transcriptional multigene signatures have been developed for overall survival prediction in lung adenocarcinoma patients (Raponi et al. 2006; Der et al. 2014). However, very few molecular classifiers have been developed for early relapse prediction. More importantly, although ncRNAs have been confirmed to have important roles in multiple cancers (Kornienko et al. 2013; Kung et al. 2013; Khorkova et al. 2015), no previous study has combined mRNAs and ncRNAs to construct an integrated signature for early relapse prediction in early-stage lung adenocarcinoma. Thus, with multiple public transcriptome data and novel bioinformatic methods, identifying a robust and practical mRNA–ncRNA signature to predict early relapse of early-stage lung adenocarcinoma is feasible and of great clinical significance.
In the present study, we performed an integrative analysis of mRNAs and ncRNAs expression profiles for the prediction of early relapse in early-stage lung adenocarcinoma. We believed that the integrated signature with more transcript information would improve risk stratification, reveal the biological behavior of different risk groups and provide a more accurate individualized treatment strategy in early-stage lung adenocarcinoma.
Materials and methods
Data collection and preprocessing
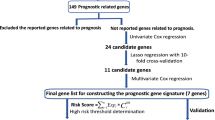
Raw gene microarray expression profiles were downloaded from the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/). All datasets fulfilling the following criteria were included: (1) detected gene expression profiles of primary lung cancer; (2) used the chip platform of Affymetrix Human Genome U133 Plus 2.0 (GPL570); (3) availability of basic clinicopathological information, follow‐up and relapse status; (4) a sample size of more than 50. Finally, datasets of GSE31210, GSE50081, GSE30219 and GES37745 were recruited. Among the four datasets, only patients with stage I–II lung adenocarcinoma. In addition, considering the potential for uncured resection, patients who relapse within 1 month after surgery are deleted. Finally, clinical data and raw CEL files of the remaining 476 patients from 4 datasets were merged as a meta-dataset for further analysis. Using the robust multichip average (RMA) algorithm (Irizarry et al. 2003), raw CEL files of the four microarray datasets of lung adenocarcinoma were processed for background correction, normalization, and log2 transformation. To get mRNA and ncRNA expression profiles separately, we performed a probe reannotation pipeline as proposed in previous studies (Du et al. 2013). First, the probe sequences of Affymetrix HG‐U133 Plus 2.0 array were remapped to the latest version of the NetAffx Annotation File. When multiple gene probes were mapped to the same EntrezGeneID, the mean value was used as average expression level. Second, the chromosomal coordinates of the retained probes were matched to the chromosomal coordinates of ncRNAs derived from the GENCODE project (https://www.gencodegenes.org/, release 28). The probes that mapped to both ncRNAs and protein‐coding genes were discarded. The ComBat method was used to remove the potential internal and external batch effects among different datasets.
Identification of mRNA and ncRNA related to early relapse
Early relapse is defined as relapse within 2 years after radical resection. The cases in the meta-dataset were randomly allocated to generate a training set and a test set according to the ratio of 7:3 (training set, n = 334; testing set, n = 142), and the training set was further divided into an early relapse group and long-term nonrelapse group (at least 5 years of follow-up without relapse). To eliminate the interference of confounding factors between the groups, propensity score matching (PSM) analysis was performed between the two groups based on clinicopathological information such as gender, age, smoking history, and tumor staging. The matching ratio was set to 1:1. Finally, 56 paired patients were selected for transcriptome analysis in the training set. Linear Models for Microarray data (LIMMA) method was used to screen differentially expressed mRNAs (fold change > 1.5, adjusted P < 0.05) and ncRNAs (fold change > 1.25, adjusted P < 0.05) between samples from paired patients of early relapse and long-term nonrelapse groups. Random survival forest (RSF) analysis was used to perform dimensionality reduction and importance ranking of differentially expressed genes (DEGs). LASSO Cox regression model was then used to construct the final prognostic model using the DEGs related to early relapse after dimensionality reduction by RSF.
Establishment and clinical application of risk score and prognostic model
The risk score of each patient is calculated combining the expression levels of RNAs and the LASSO Cox regression coefficient. Patients from training and testing sets were divided into high-risk and low-risk groups using the median risk score of the training set as the cutoff value. Kaplan–Meier estimator was used to compare the relapse differences between high-risk and low-risk groups in training and testing sets. Univariate and multivariate Cox regression analysis and stratified survival analysis were used to test the independent role of risk score in predicting relapse. The time-dependent receiver operating characteristic curve (ROC) was used to evaluate the predictive accuracy of each feature and signature at different times. Integrated mRNA–ncRNA signature and clinicopathological characteristics were combined to construct a nomogram for early relapse prediction. Survival decision curve analysis (DCA) was used to evaluate the net benefits derived from the integrated mRNA–ncRNA signature or nomogram. We predicted the chemotherapeutic response for each sample based on the Genomics of Drug Sensitivity in Cancer (GDSC) database (https://www.cancerrxgene.org/) using R package “pRRophetic” (Geeleher et al. 2014). Seven commonly used chemotherapy drugs including paclitaxel, fluorouracil, cisplatin, etoposide, vinorelbine, gemcitabine, and docetaxel were used for analysis, the samples’ half-maximal inhibitory concentration (IC50) for each drug was estimated by ridge regression and the prediction accuracy was evaluated by tenfold cross-validation.
Molecular characteristics and tumor microenvironment analysis of different risk groups
The gene set enrichment analysis (GSEA) was performed on the expression profile data to investigate the potential mechanisms in the MSigDB database of h.all.v7.2.symbols, c2.cp.kegg.v7.2.symbols and c2.cp.reactome.v7.2.symbols using the JAVA program (https://www.gsea-msigdb.org/gsea/index.jsp) and R package “clusterProfiler” (Subramanian et al. 2005; Yu et al. 2012). The random number is set to 1000, the significance threshold is set to adjusted P < 0.05, and false discovery rate (FDR) < 0.25. The CIBERSORT algorithm was used to calculate the composition of 22 immune cells of each sample. The gene expression data with standard annotation were uploaded to the CIBERSORT web portal (https://cibersort.stanford.edu/), 22 immune cell feature matrix (LM22) was used to perform 1000 random times for deconvolution. For accuracy evaluation, samples with a CIBERSORT output of P < 0.05 were selected.
RNA-seq and mutation landscape analysis
TCGA RNA-Seq raw read counts data of 226 stage I–II lung adenocarcinoma patients with complete follow-up information was downloaded from GDC database (https://portal.gdc.cancer.gov/). Ensembl ID for genes was annotated in GENCODE 28 to generate Gene Symbol names. To be consistent with the distribution of microarray data, raw read counts data were normalized across samples using voom algorithm. Mutation data that stored in Mutation Annotation Format (MAF) contained somatic variants were also downloaded from GDC. Nonsynonymous mutations were used for mutation load investigations.
Statistical analysis
All statistical tests were executed by R/3.6.1 and SPSS/23.0 using a χ2 or Fisher’s exact test for categorical data when appropriate, a two-sample Wilcoxon test (Mann–Whitney test) for continuous data. Pearson’s correlation test was used for correlation analysis. Survival analysis were depicted using the Kaplan–Meier method and compared using the log-rank tests. Univariable and multivariable Cox regression were performed to investigate whether the gene signature was independent of other. All statistical tests were two sided, and a P value < 0.05 was considered statistically significant.
Results
Preparation of lung adenocarcinoma dataset
Four hundred and seventy-six patients with stage I–II lung adenocarcinoma from GEO database were selected and comprehensively studied, including 226 patients from GSE31210 cohort; 125 patients from GSE50081 cohort; 82 patients from GSE30219 cohort; and 43 patients from GSE37745 cohort. The clinical information of all patients can be found in Supplementary Table 1. Plots of the first and second principal components before and after removing batch effects among the four cohorts are shown in Fig. S1.
Establishment of early relapse-related mRNA–ncRNA signature from the training set
Patients in the training set were divided into early relapse and long-term nonrelapse group. The baseline clinicopathologic characteristics before and after PSM analysis are shown in Table 1. Before PSM analysis, there were more patients with stage II disease in the early relapse group. After PSM analysis, there were no significant differences between the two groups among age, gender, T stage, N stage, and TNM stage. Through gene annotation, a total of 3419 ncRNAs and 17,561 mRNAs were identified. Transcriptome change profiling was then performed between the matched two groups. The results are shown in Supplementary Table 2 and 3. A total of 193 mRNAs and 49 ncRNAs that were differentially expressed were included. After dimensionality reduction using RSF analysis, 41 RNAs including 39 mRNA and 2 ncRNA are retained (Fig. 1A, B). The differentially expressed genes and their distributions on chromosomes is shown in Fig. 2. LASSO coefficient profiles of the 41 RNAs are shown in Fig. 1C, D. A coefficient profile plot was produced against the log (λ) sequence. Vertical line was drawn at the value selected using tenfold cross-validation, and the minimize λ method resulted in 12 mRNAs and 1 ncRNA. Finally, Risk score was calculated for each patient in training set based on the expression levels of the 13 RNAs and LASSO Cox regression coefficients: Risk score = (0.006 × CCL20) + (0.069 × ANLN) + (0.061 × ARNTL2) + (− 0.088 × CYP4B1) + (0.036 × FAM83A) + (0.005 × GREM1) + (0.005 × IL1R2) + (0.001 × SPOCK1) + (0.034 × DLGAP5) + (0.022 × COL11A1) + (0.063 × TPX2) + (0.095 × TK1) + (0.171 × LINC01116).

Integrated mRNA–ncRNA signature selection using RSF and LASSO Cox regression. A, B Dimensionality reduction using RSF analysis. C, D LASSO coefficient profiles of the 41 candidate mRNAs and ncRNAs

The differentially expressed genes and their distributions on chromosomes in the PSM paired groups after dimensionality reduction using RSF
The prognostic value of risk score in different datasets
By applying the median risk score as cutoff value, patients in the training set were divided into a low-risk group and high-risk group (n = 167, respectively). The distribution of risk scores and relapse status shows that patients with low risk scores have better RFS than patients with high risk scores (Fig. 3A, left panel). Time-dependent ROC analysis evaluated the prognostic accuracy of the integrated mRNA–ncRNA risk score, and the AUC at 1, 3 and 5 year were 0.747, 0.736, and 0.764, respectively (Fig. 3A, middle panel). The RFS rates for patients in low-risk group were 94.6% at 1 year, 80.8% at 3 year, and 54.5% at 5 year, compared with 80.2%, 47.3%, and 31.7% in high-risk group, respectively (HR 3.19, 95% CI 2.16–4.72, P < 0.001, Fig. 3A, right panel). Then, the same analysis was performed in the testing set. In the testing set, the time-dependent ROC AUC at 1, 3 and 5 year were 0.749, 0.711, and 0.728, respectively. The 1, 3 and 5-year RFS rates for the low-risk group were 96.4%, 78.2%, and 52.7%, respectively, while for the high-risk group were 79.3%, 51.7%, and 32.2%, respectively (HR 2.91, 95% CI 1.63–5.20, P = 0.002) (Fig. 3B). In the entire dataset, the classification based on the risk score yield similar results (Fig. 3C). Subgroup analysis based on T stage and mutation background suggested that in T1N0, T2N0, EGFR mutant and EGFR wild-type subgroups, the high-risk patients tend to have significantly unfavorable RFS (Fig. 4A–D).

Distribution of risk score, time-dependent ROC curves at 1, 3 and 5 years and Kaplan–Meier analysis between patients at low and high risk of relapse in training set (A), testing set (B) and entire dataset (C)

Kaplan–Meier analysis for the entire dataset with stages I–II lung adenocarcinoma based on the integrated mRNA–ncRNA signature stratified by T stage and EGFR mutation status
Clinical application of risk score established based on mRNA–ncRNA
After multivariate analysis adjusted by clinicopathological factors, the integrated mRNA–ncRNA classifier remained a powerful and independent factor in the training set and testing set (Fig. 5). To provide a quantitative method for predicting the likelihood of relapse, a nomogram that integrates the mRNA–ncRNA classifier and clinicopathological factors was constructed (Fig. 6A). The calibration curve (Fig. 6B–D) showed that the nomogram model performed well agreement in predicting the RFS rate at 1, 3 and 5 years. Similarly, the decision curve (Fig. 6E) showed that both the mRNA–ncRNA classifier and the classifier based nomogram have a higher net income and better prediction accuracy than the TNM staging system. The drug sensitivity analysis of seven chemotherapeutics including paclitaxel, fluorouracil, cisplatin, etoposide, vinorelbine, gemcitabine, and docetaxel (Fig. 7) showed that patients in the high-risk group have lower IC50 values, which indicated higher sensitivity to these seven chemotherapeutics (P < 0.001), suggesting that the mRNA–ncRNA classifier can be used as a potential indicator for postoperative adjuvant chemotherapy.

Univariable and multivariable Cox regression analysis in training and testing datasets with stages I–II lung adenocarcinoma

Construction of nomogram based on mRNA–ncRNA signature and its clinical utility. A Nomograms integrated with the mRNA–ncRNA signature to predict 1-, 3- and 5-year RFS probability in the entire dataset. C, D Calibration curve in predicting the RFS rate at 1, 3 and 5 years. E Decision curve analysis of the nomogram

Drug sensitivity analysis of paclitaxel, fluorouracil, cisplatin, etoposide, vinorelbine, gemcitabine, and docetaxel in the low- and high-risk groups using IC50 values
Pathway enrichment analysis and immunophenotyping analysis related to mRNA–ncRNA signature
GSEA using HALLMARK, KEGG and REACTOME gene sets (Fig. 8) showed that the high-risk group has high enrichment levels in cell cycle regulation, DNA replication, mismatch repair, glucose metabolism, and immune pathways related to antigen presentation. The immune lineage analysis using CIBERSORT algorithm (Fig. 9) showed that all the selected 13 RNAs had significant correlations with immune cell composition in the tumor microenvironment, meanwhile, a higher risk score was positively correlated with the composition of suppressive immune cells such as M2 macrophage and Treg, as well as a variety of antigen-presenting cells in resting state, while negatively correlated with the antigen-presenting cells in activated state. The risk score is also positively correlated with expression levels of multiple inhibitory immune checkpoint coding genes including CD274, PDCD1, HAVCR2, LAG3, PDCD1LG2, IDO1, TIGIT, CTLA4, and LAIR1 (Fig. 10).

Gene set enrichment analysis using HALLMARK, KEGG and REACTOME gene sets

Correlation of selected RNAs, risk score and immune cell composition in the tumor microenvironment

Correlation of risk score and expression levels of inhibitory immune checkpoint coding genes including CD274, PDCD1, HAVCR2, LAG3, PDCD1LG2, IDO1, TIGIT, CTLA4, and LAIR1
Validation and mutation analysis in a database based on RNA sequencing
Validation using the RNA-Seq data of 226 stage I–II lung adenocarcinoma cases from TCGA database showed a similar result to the training set and testing set. The clinical information of all patients can be found in Supplementary Table 4. The high-risk group also tended to have unfavorable RFS (HR 1.70, 95% CI 1.14–2.53, P = 0.008). The 2-year ROC curve showed that the mRNA–ncRNA classifier has a higher AUC value (AUC = 0.680) than the TNM staging system (AUC = 0.615), and the combination of the mRNA–ncRNA classifier and the TNM staging system provided a stronger predictive power (AUC = 0.708). The RNA-seq-based external validation further showed that the integrated mRNA–ncRNA classifier can be used as an effective prognostic indicator for stage I–II lung adenocarcinoma patients (Fig. 11). Based on the mutation analysis of the above patients in the TCGA database, patients in the high-risk group tended to have higher mutation frequencies, and the mutation rate of driver genes such as TP53 (63%) and KRAS (33%) was also higher, while in the low-risk score group, EGFR mutation (33%) was more common (Fig. 12). In addition, patients in the high-risk group tended to have higher mutation burdens (P < 0.001) (Fig. 13).

Validation of the relapse prediction efficiency in the TCGA lung adenocarcinoma dataset. A Kaplan–Meier analysis for the TCGA dataset with stages I–II lung adenocarcinoma. B The 2-year ROC curve comparison of the mRNA–ncRNA signature and the TNM staging system

Mutation landscape of patients in the TCGA database. A Top ranked mutations in the high-risk group. B Top ranked mutations in the low-risk group

Mutation burdens of patients in low and high risk groups in the TCGA database
Discussion
Although early-stage lung adenocarcinoma can benefit from radical resection and postoperative adjuvant chemotherapy, early relapse is still the main cause of unfavorable prognosis (Birim et al. 2006). At present, prognostic systems such as the TNM staging system of AJCC have been widely used to assess the prognosis of lung adenocarcinoma patients (Woodard et al. 2016). However, they cannot always be sufficient to predict relapse and prognosis, especially for early relapse of early-stage patients, which may be due to insufficient understanding of the different genetic backgrounds of tumors in current prediction methods (Borczuk et al. 2009). Although some literatures have explored the association between molecular markers and early postoperative relapse, most of the works have focused on analyzing the function of only one or a class of biomarkers. Compared with a single biomarker, integrating multiple biomarkers of different types and functions into a single model will significantly improve the prognostic value and provide indications for adjuvant therapy. However, most of these previous studies only included protein-coding genes into analysis while thousands of non-coding RNAs were excluded (Farhat et al. 2012; Matthaios et al. 2013; Fang and Wang 2014; Zhu and Tsao 2014). Increasing evidences have proved that ncRNAs affect various aspects of homeostasis in cells, and play key roles in cell proliferation, migration and genomic stability (Niedzwiecki et al. 2016). Hence, an integrated mRNA–ncRNA signature could provide more diversified information for early relapse prediction and biological characteristics identification.
In this study, we used microarray probe reannotation and subsequently extracted mRNA and ncRNA transcriptional profiles from 476 early-stage (stage I–II) lung adenocarcinoma patients from the GEO database. PSM analysis was performed to exclude the interference of other clinicopathological factors between the early relapse group and the long-term nonrelapse group. The RSF algorithm and LASSO Cox regression were used to identify an early relapse-related signature including 12 mRNAs and 1 ncRNA. The survival analysis showed that the signature has accurate relapse prediction ability in both the training set and the testing set, and is further verified by the RNA-seq data in TCGA. When combined with other clinicopathologic information, multivariate Cox regression indicated that the signature can be used as an independent prognostic factor for relapse prediction. DCA confirmed that the nomogram which combined the mRNA–ncRNA signature and clinicopathological data are superior to the TNM staging system in relapse prediction. Chemotherapeutics sensitivity analysis showed that the risk score is positively correlated with the drug sensitivity of the seven commonly used chemotherapeutics, suggesting that the integrated mRNA–ncRNA signature could be used as a prediction of postoperative adjuvant chemotherapy. GSEA showed that the mRNA–ncRNA-based risk score is closely related to the tumor microenvironment, and the high-risk group showed active cell proliferation and glucose metabolism characteristics, as well as the enrichment of immune pathways related to antigen presentation. Combining mutation landscape analysis, immune cell composition, and immune checkpoint expression analysis, we hypothesize that the high-risk group has a heavier mutation load, which in turn produces more tumor neoantigens, promotes the antigen presentation process. But due to the inhibitory immune microenvironment, the antigen-presenting cells are more likely to stay in a resting or functionally inhibited state, and are unable to exert an effective immune surveillance effect, which suggests that early relapse may be related to the inhibitory tumor immune microenvironment in addition to the high proliferation characteristics of the tumor cells.
In addition, through literature search, we found that the genes included in the signature have been experimentally proved to be related to cancer. Among them, LINC01116 has been proved to be related to cell proliferation, G1/S transition, and apoptosis regulation in lung adenocarcinoma and has been proved to promote gefitinib resistance by affecting IFI44 expression, which is involved in the IFN/STAT1 pathway. (Wang et al. 2020). Blocade of CCL20 was confirmed a strong induction of circulating cancer-specific T cells in blood and can significantly reshape the tumor microenvironment (Da Silva et al. 2019). A single cell sequencing study confirmed that high expression of IL1R2 is related to activated tumor Tregs, and is correlated with poor prognosis in lung adenocarcinoma (Guo et al. 2018). In a study of early breast cancer, high COL11A1 expression was observed in tumor cells and surrounding stromal cells, and is associated with aggressive behavior, poor outcome and resistance to radiotherapy (Toss et al. 2019). SPOCK1 was proved to be a novel regulator of metastasis from the lung to the brain. It plays a crucial role in cancer stem cell self-renewal, and can modulate tumor initiation (Singh et al. 2017). GREM1 contributed to a tumor-associated mesenchymal stem cells (MSC) phenotype, enhanced the MSC’s ability to promote primary tumor cell dissemination, and contributed to an immunosuppressive tumor microenvironment (Fregni et al. 2018). FAM83A in lung cancer tissues was significantly increased and overexpression of FAM83A enhanced the proliferation, colony formation, and invasion of lung cancer cells, and was correlated with advanced TNM stage and poor prognosis. Meanwhile, overexpression of FAM83A increased the expression of active β-catenin and Wnt target genes and the activity of epithelial–mesenchymal transition (Zheng et al. 2020). Disc large homologue-associated protein 5 (DLGAP5), which required for AURKA-dependent, centrosome-independent mitotic spindle assembly is essential for the survival and proliferation of SMARCA4 mutant lung cancer cells (Tagal et al. 2017). Another gene related to AURKA in the signature is TPX2, which act as the coactivator of AURKA, can mitigate drug-induced lung cancer cell apoptosis, and hence emerges in response to chronic EGFR inhibition (Shah et al. 2019). Thymidine kinase 1 (TK1) overexpression is associated with significantly reduced RFS in lung adenocarcinoma patients. Transcriptional overexpression of TK1 in lung cancer cells is driven, in part, by MAP kinase pathway in a transcription factor MAZ-dependent manner (Malvi et al. 2019). High expression of the transcription factor ARNTL2 also predicts poor lung adenocarcinoma patient outcome. ARNTL2 initiated metastatic self-sufficiency by orchestrating the expression of complex pro-metastatic secreted factors (Brady et al. 2016). ANLN, a homologue of anillin, was transactivated in lung cancer cells and seemed to play a significant role in pulmonary carcinogenesis. Induction of small interfering RNAs against ANLN in NSCLC cells suppressed its expression and resulted in growth suppression; moreover, treatment with small interfering RNA yielded cells with larger morphology and multiple nuclei, which subsequently died. Interestingly, inhibition of phosphoinositide 3-kinase/AKT activity in NSCLC cells decreased the stability of ANLN and caused a reduction of the nuclear ANLN level. Immunohistochemical staining of nuclear ANLN on lung cancer tissue microarrays was associated with the poor survival of NSCLC patients, indicating that this molecule might serve as a prognostic indicator (Suzuki et al. 2005). CYP4B1 is one of the major xenobiotic-metabolizing enzymes (XME) coding genes, and plays a crucial role in maintaining normal bronchial epithelial cell structure and function. A decrease in CYP4B1 expression was observed in tumoral specimens. Furthermore, some of the XME coding genes are involved in the metabolism or transport of chemotherapeutics and may influence the response of tumors to chemotherapy (Leclerc et al. 2011).
To our knowledge, this study was the first attempt to integrate mRNAs and ncRNAs to construct an early relapse predictive signature in early-stage lung adenocarcinoma. However, the limitations should be acknowledged. First, in addition to mRNA and ncRNA, the predictive value of methylation and single nucleotide polymorphisms in tumor prognosis has been verified. Multidimensional data analysis that integrates mRNA, ncRNA, CpG, single nucleotide polymorphisms and other multiomics information may further improve the prediction efficiency. Second, subject to the limitations of the clinical information available in the GEO database, some important clinicopathological characteristics, such as histological grade, histological subtypes, CT imaging information were not included in this study, which may affect the predictive value of the mRNA–ncRNA signature. Finally, the biological functions of the mRNAs and ncRNA incorporated in the integrated signature are still needed to be further explored.
Conclusions
In conclusion, we constructed a robust mRNA–ncRNA signature that can accurately identify patients at high risk of early relapse in stages I–II lung adenocarcinoma. Future clinical trials and confirmatory experiments are still essential to verify the clinical applicability and biological significance of the integrated signature in detecting postoperative early relapse in early-stage lung adenocarcinoma.
Data availability
Not applicable.
Code availability
Not applicable.
References
Amin MB, Greene FL, Edge SB et al (2017) The eighth edition AJCC cancer staging manual: continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin 67:93
Birim O, Kappetein AP, van Klaveren RJ, Bogers AJJC (2006) Prognostic factors in non-small cell lung cancer surgery. Eur J Surg Oncol 32:12
Borczuk AC, Toonkel RL, Powell CA (2009) Genomics of lung cancer. Proc Am Thorac Soc 6:152
Brady JJ, Chuang C-H, Greenside PG et al (2016) An arntl2-driven secretome enables lung adenocarcinoma metastatic self-sufficiency. Cancer Cell 29:697
Bray F, Ferlay J, Soerjomataram I et al (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394
Da Silva CG, Camps MGM, Li TMWY et al (2019) Effective chemoimmunotherapy by co-delivery of doxorubicin and immune adjuvants in biodegradable nanoparticles. Theranostics 9:6485
Der SD, Sykes J, Pintilie M et al (2014) Validation of a histology-independent prognostic gene signature for early-stage, non-small-cell lung cancer including stage IA patients. J Thorac Oncol 9:59
Du Z, Fei T, Verhaak RGW et al (2013) Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat Struct Mol Biol 20:908
Fang S, Wang Z (2014) EGFR mutations as a prognostic and predictive marker in non-small-cell lung cancer. Drug Des Devel Ther. https://doi.org/10.2147/DDDT.S69690
Farhat FS, Tfayli A, Fakhruddin N et al (2012) Expression, prognostic and predictive impact of VEGF and bFGF in non-small cell lung cancer. Crit Rev Oncol Hematol 84:149
Fregni G, Quinodoz M, Möller E et al (2018) Reciprocal modulation of mesenchymal stem cells and tumor cells promotes lung cancer metastasis. EBioMedicine 29:128
Geeleher P, Cox NJ, Huang RS (2014) Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol 15:1
Guo X, Zhang Y, Zheng L et al (2018) Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 24:1628
Irizarry RA, Hobbs B, Collin F et al (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249
Khorkova O, Hsiao J, Wahlestedt C (2015) Basic biology and therapeutic implications of lncRNA. Adv Drug Deliv Rev 87:15
Kiankhooy A, Taylor MD, LaPar DJ et al (2014) Predictors of early recurrence for node-negative t1 to t2b non-small cell lung cancer. Ann Thorac Surg 98:1175
Kornienko AE, Guenzl PM, Barlow DP, Pauler FM (2013) Gene regulation by the act of long non-coding RNA transcription. BMC Biol 11:59
Kozu Y, Maniwa T, Takahashi S et al (2013) Risk factors for both recurrence and survival in patients with pathological stage I non-small-cell lung cancer. Eur J Cardiothorac Surg 44:e53
Kung JTY, Colognori D, Lee JT (2013) Long noncoding RNAs: past, present, and future. Genetics 193:651
Leclerc J, Courcot-Ngoubo Ngangue E, Cauffiez C et al (2011) Xenobiotic metabolism and disposition in human lung: transcript profiling in non-tumoral and tumoral tissues. Biochimie 93:1012
Malvi P, Janostiak R, Nagarajan A et al (2019) Loss of thymidine kinase 1 inhibits lung cancer growth and metastatic attributes by reducing GDF15 expression. PLoS Genet. https://doi.org/10.1371/journal.pgen.1008439
Matthaios D, Hountis P, Karakitsos P et al (2013) H2AX a promising biomarker for lung cancer: a review. Cancer Invest 31:582
Niedzwiecki D, Frankel WL, Venook AP et al (2016) Association between results of a gene expression signature assay and recurrence-free interval in patients with stage II colon cancer in cancer and leukemia group B 9581 (alliance). J Clin Oncol 34:3047
Padda SK, Burt BM, Trakul N, Wakelee HA (2014) Early-stage non-small cell lung cancer: surgery, stereotactic radiosurgery, and individualized adjuvant therapy. Semin Oncol 41:40
Raponi M, Zhang Y, Yu J et al (2006) Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res 66:7466
Shah KN, Bhatt R, Rotow J et al (2019) Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat Med 25:111
Singh M, Venugopal C, Tokar T et al (2017) RNAi screen identifies essential regulators of human brain metastasis-initiating cells. Acta Neuropathol 134:923
Subramanian A, Tamayo P, Mootha VK et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102:15545
Suzuki C, Daigo Y, Ishikawa N et al (2005) ANLN plays a critical role in human lung carcinogenesis through the activation of RHOA and by involvement in the phosphoinositide 3-kinase/AKT pathway. Cancer Res 35:11314
Tagal V, Wei S, Zhang W et al (2017) SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nat Commun. https://doi.org/10.1038/ncomms14098
Toss MS, Miligy IM, Gorringe KL et al (2019) Collagen (XI) alpha-1 chain is an independent prognostic factor in breast ductal carcinoma in situ. Mod Pathol 32:1460
Travis WD, Brambilla E, Nicholson AG et al (2015) The 2015 world health organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol 10:1243
Wang H, Lu B, Ren S et al (2020) Long noncoding RNA LINC01116 contributes to gefitinib resistance in non-small cell lung cancer through regulating IFI44. Mol Ther Nucleic Acids 19:227
Woodard GA, Jones KD, Jablons DM (2016) Lung cancer staging and prognosis. Lung cancer. Springer, Cham, pp 47–75
Yu G, Wang L-G, Han Y, He Q-Y (2012) ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:287
Zheng Y-W, Li Z-H, Lei L et al (2020) FAM83A promotes lung cancer progression by regulating the wnt and hippo signaling pathways and indicates poor prognosis. Front Oncol. https://doi.org/10.3389/fonc.2020.00180
Zhu C-Q, Tsao M-S (2014) Prognostic markers in lung cancer: is it ready for prime time? Transl Lung Cancer Res 3:149
Funding
This work was supported by the National Natural Science Foundation of China (81930073), Shanghai Municipal Science and Technology Major Project (Grant No. 2017SHZDZX01, VBH1323001/026), Shanghai Municipal Key Clinical Specialty Project (SHSLCZDZK02104), and Pilot Project of Fudan University (IDF159034), Shanghai Sailing Program 19YF1408800.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Zhendong Gao declares that he has no conflict of interest. Han Han declares that he has no conflict of interest. Yue Zhao declares that he has no conflict of interest. Hui Yuan declares that he has no conflict of interest. Shanbo Zheng declares that he has no conflict of interest. Zhang Yang declares that he has no conflict of interest. Haiquan Chen declares that he has no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Consent of participant
Consent of participant was obtained from all individual participants included in the study.
Consent for publication
Consent for publication was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
432_2021_3718_MOESM2_ESM.tiff
Supplementary file2 (TIFF 1390 KB) Principal components plot of the first two principal components. (a) Before batch removing. (b) After batch removing.
Rights and permissions
About this article

Cite this article
Gao, Z., Han, H., Zhao, Y. et al. A tumor microenvironment-related mRNA–ncRNA signature for prediction early relapse and chemotherapeutic sensitivity in early-stage lung adenocarcinoma. J Cancer Res Clin Oncol 147, 3195–3209 (2021). https://doi.org/10.1007/s00432-021-03718-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00432-021-03718-z