
Abstract
Congenital central hypoventilation syndrome (CCHS) is a rare condition characterized by central hypoventilation, leading to the majority of patients being dependent on ventilatory support during sleep. This condition is often accompanied by various associated symptoms, due to a PHOX2B gene variant involved in neuronal crest cell migration. This study is the first to review the characteristics and outcomes in children with CCHS on long-term mechanical ventilation in the Netherlands. We performed a retrospective study of all CCHS patients treated in the 4 Centers of Home Mechanical Ventilation of the University Medical Centers in the Netherlands from 2000 till 2022 by collecting information from the electronic medical records, documented during follow-up. We included 31 patients, out of which 27 exhibited a known genetic profile associated with CCHS, while no PHOX2B variant was identified in the remaining patients. Among the 27 patients with known genetic profiles, 10 patients had a non-polyalanine repeat expansion mutation (NPARM), followed by 20/27, 20/25, and 20/26 polyalanine repeat expansion mutations (PARMs) in descending order. The most common presentation involved respiratory failure or apneas during the neonatal period with an inability to wean off ventilation. The majority of patients required ventilatory support during sleep, with four patients experiencing life-threatening events related to this dependency. Daily use of ventilatory support varied among different genetic profiles. All genotypes reported comorbidities, with Hirschsprung’s disease and cardiac arrhythmias being the most reported comorbidities. Notably, Hirschprung’s disease was exclusively observed in patients with a 20/27 PHOX2B variant.
Conclusion: Our study results suggest that in our cohort, the genotype is not easily associated to the phenotype in CCHS. Consistent with these findings and international literature, we recommend a thorough annual evaluation for all patients with CCHS to ensure optimal management and follow-up.
What is Known: • The majority of CCHS patients are dependent on ventilatory support. • Variants in the PHOX2B gene are responsible for the characteristics of CCHS. | |
What is New: • This study provides insight into the clinical course and long-term outcomes of CCHS patients in the Netherlands. • In CCHS, the genotype is not easily associated with the phenotype, requiring a thorough life-long follow-up for all patients. |
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Congenital central hypoventilation syndrome (CCHS) is a lifelong disease, with approximately 1000–1200 cases diagnosed until 2010 [1], occurring in approximately 1:200,000 newborns [2]. It is characterized by abnormal control of breathing and presents with symptoms of alveolar hypoventilation resulting in ventilator-dependency during sleep in most patients, as well as symptoms of autonomic dysregulation. In 2003, variants in the PHOX2B gene were discovered to be responsible for the characteristics of CCHS [3, 4]. This gene plays a role in neuronal crest cell migration, elucidating the relationship between CCHS, Hirschsprung’s disease, cardiac arrhythmias, and neuroblastoma. In 2010, the American Thoracic Society published a statement on CCHS, supporting the clinician in optimizing care based on genetic information, through annual in-hospital evaluations [1]. Since then, care for CCHS patients in the Netherlands has been guided by this statement. Recently, a new multidisciplinary guideline for the management and follow-up for patients with CCHS and a review with recommendations for a comprehensive multidisciplinary approach were published [5, 6].
Over the past decade, we have been able to perform genetic testing to identify specific variants of the PHOX2B gene. Specific variants are thought to be associated with a different disease course, different comorbidities and varying severity of the phenotype, known as the genotype-phenotype correlation. Due to the rarity of this disease, only a few studies have been conducted to provide insight into the clinical course and long-term outcomes of CCHS patients. No studies have been conducted in the Dutch CCHS population until now, implying that we are currently not informed about the clinical course and outcome of our Dutch population, as well as the way care is established for children and adolescents as they transition into adulthood.
The aim of this study is to review the characteristics and outcomes of Dutch children and adults with CCHS and to provide a recommendation for follow-up for this specific patient group, focusing on ventilatory management and chronic care.
Methods
We performed a multi-center retrospective study that included all CCHS patients who received care at one of the four Centers for Home Mechanical Ventilation (HMV) in the Netherlands between 2000 and 2022. HMV in the Netherlands can only be provided by these four centers, folllowing the same guidelines. Consequently, all CCHS patients receive consistent treatment. According to national guidelines, all patients are screened annually for comorbidities and/or chronic respiratory failure requiring ventilatory support [7].
We collected the following data from electronic medical records: demographics, living condition, type and intensity of ventilatory support, (disease-related) comorbidities and complications, hospital admissions and results of genetic testing. After obtaining informed consent from the patient and/or their parents, the patient’s primary physician copied the data from (electronic) patient records and stored it in a provided database by completing an anonymized questionnaire in Redcap®. The database was then analysed by the principal researchers, using Redcap® and Excel. There were no exclusion criteria. The study protocol was approved by the Ethical Committee (Centrale Toetsingscommissie UMCG, UMCG Research Register number 202000420).
Results
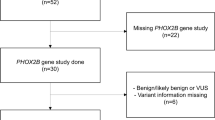
Thirty-one patients were included in this study, ten of whom were female. Patient demographics are presented in Table 1. The mean age of the patients is 20.9 years (SD 15.9). The majority of the patients lived at home, with one of them being hospitalized at the time of the review, and one residing in a residential home. The patients had varying levels of education, with 25.9% of them attending a special needs school for children with learning disabilities.
Presenting symptoms and genetic profile
Table 1 also presents the genetic diagnosis profile. In this study, 10 patients (32.2%) were diagnosed with a non-polyalanine repeat expansion mutation (NPARM) variant of the PHOX2B gene. A 20/27 PHOX2B variant was found in 7 patients (22.5%), and a 20/25 PHOX2B variant in 5 patients (16.1%). The remaining reported PHOX2B variants are 20/24 20/26 and 20/31. For one patient, a PHOX2B mutation was confirmed by genetic testing, but a specific variant was not recorded. In 4 patients, the genetic substrate was unknown. These diagnoses were based on a well-documented polysomnography showing central hypoventilation and hypercapnia, along with a clinical course consistent with the diagnosis CCHS. Additionally, one of these patients had two first-degree family members with a genetically confirmed diagnosis of CCHS.
Table 2 displays the clinical characteristics of the patients. Respiratory failure at birth or apneas in the neonatal period were the most frequently reported presenting symptoms. Among our patients, 51.6% received a diagnosis of CCHS after the age of 1 month, corresponding to a diagnosis of late-onset CCHS (LOCCHS). Regarding patients with a genetic diagnosis, most of those with a 20/26 and a 20/27 (n = 9) variant experienced respiratory failure in the early neonatal period. Six NPARM patients were diagnosed with a non-specific presentation characterized by fatigue (n = 4) or were detected due to comorbidities associated with CCHS (n = 1) or a close family member with CCHS (n = 3).
Mechanical ventilation
In this study, 29 out of 30 patients are dependent on ventilatory support (see Table 2). One patient is not on ventilatory support because this patient is in a palliative setting at the time of data collection. This patient is monitored through annual polysomnography. The main reasons for ventilatory support are central apneas (19 patients) and hypercapnia (20 patients). Additionally, hypoventilation during sleep and recurrent respiratory infections were considered indications to initiate ventilation. For one patient, in addition to hypercapnia, there was also a component of pulmonary hypertension, which led to the decision to start ventilatory support. The other patient had been respiratory insufficient and hypercapnic since the neonatal period and therefore continued ventilation. All patients with a 20/26 or 20/27 variant could not be weaned off the ventilator via endotracheal tube due to respiratory failure at birth. Nine out of ten patients with a NPARM variant initiated ventilatory support after the neonatal period, ranging from the age of 4 years to 49 years.
Most patients (73%) require ventilatory support only during the night, while 7 patients (23%) also need ventilatory support during daytime rest periods or activities. Both patients who are ventilator-dependent for more than 16 h a day were diagnosed with a 20/27 PHOX2B variant. One patient (3%) requires ventilatory support all day long, although this patient was only a few months old during data collection. A NPARM variant was most often diagnosed in patients with less than 8 h of ventilation-dependency. Mean transcutaneous CO2 (TcCO2) before the initiation of ventilation was measured in 24 patients, twenty-two of them had a TcCO2 above 6 kPa (45 mmHg).
Ten patients transitioned from invasive ventilatory support (via a tracheostomy) to non-invasive positive pressure ventilatory support between the ages 5 and 22 years. However, in two of them, this change was not well tolerated, leading to a return to invasive ventilation via a tracheostomy. In this study, none of the patients were supported by diaphragm pacing.
Three out of five patients without a specific genetic diagnosis are on non-invasive positive pressure ventilation, the remaining two are ventilated via tracheostomy.
Complications of mechanical ventilation
Three patients reported a complication caused by the tracheostomy, which included tracheomalacia, granulation tissue, and anxiety disorders due to difficult recannulations. Due to the continuous use of the nasal mask, three patients developed midface hypoplasia. Four patients experienced life-threatening events: one patient with aPHOX2B 20/25 variant developed a hypercapnic coma during a respiratory tract infection, one patient with a NPARM variant suffered from acute respiratory failure, and one had an unexplained collapse. The fourth patient died while sleeping without ventilatory support; the other three events were not fatal.
Comorbidities
In Table 3, comorbidities are listed. Only 13 (42%) patients are diagnosed with comorbidities related to CCHS, of which Hirschsprung’s disease (n = 5, 39%) and cardiac arrhythmia (n = 4, 31%) — for which in two patients required a pacemaker — were the most common. All five patients with Hirschsprung’s disease are diagnosed with a 20/27 PHOX2B variant. In two patients (15%), both with a NPARM variant, a neural crest tumour was diagnosed. Neuropsychological examination was performed in 3 patients, although the reported results were not complete. In one of these patients, overall neurocognitive development was average, while in one of them it was below average. One patient had impaired language development.
Discussion
This is the first study reviewing the Dutch cohort of CCHS patients. In this cohort, the most common presentation of CCHS is respiratory failure at birth, especially in patients with a 20/25, 20/26 and 20/27 variant. Almost all patients are dependent on ventilatory support, mainly during the night. This dependency makes patients more vulnerable to complications, shown by 4 reported life-threatening events. In this study, none of the patients were supported by diaphragm pacing. A recent review article by Kasi suggested diaphragm pacing as an option for stable patients requiring full-time ventilatory support, as it may provide more freedom during the day [6].
A recently reported cohort study comparing ventilatory support and long-term outcomes showed that most CCHS patients were able to convert from all-day assisted ventilation to sleep-only assisted ventilation in their first year [8]. This is in line with our findings that most patients (73%) only require assisted ventilation during night-time sleep, taking into account the mean age of 16 in our cohort. The cohort study by Fain et al. also shows a comparable percentage of patients changing from invasive-ventilatory support via a tracheostomy to non-invasive positive pressure ventilatory support. In contrast, in our cohort, no patients changed to diaphragm pacing.
Because of our limited study population, we are not able to make clear statements about the relationship between genotype and phenotype. Nevertheless, it is noticeable that all 5 patients with Hirschsprung’s disease in this cohort were diagnosed with a 20/27 variant. This high incidence of Hirschsprung’s disease in patients with a 20/27 variant was previously seen in literature [9]. Furthermore, only patients with NPARM variants in our cohort had a neural crest tumour.
We observed that patients with shorter PARMs (20/25, 20/26) also needed ventilatory support during some hours of the day. This is not in line with previous findings suggesting that these genotypes have a milder phenotype with regard to the respiratory profile [1, 10, 11]. We observed that most NPARM genotypes had a relatively mild and atypical presentation regarding respiration, with ventilatory support only required during the night. These conflicting results with the literature in this national, although small, cohort underline the statements made by Trang [5] and Kasi [6] that the relationship between genotype and phenotype is variable and includes many exceptions.
This study also showed that not only CCHS patients with longer PARMs but also those with other PHOX2B variants, in particular NPARMs, are at risk for serious comorbidities and adverse events due to their need for ventilatory support and even live-threatening events. This finding is in line with the findings of Bachetti and Ceccherini regarding PHOX2B phenotypic heterogeneity [12].
Unfortunately, neuropsychological examinations were performed in only 4 patients, whereas this should be part of the regular follow-up for CCHS patients [5, 6]. In our cohort, 26% of the patients attended special-needs schools, which is comparable to the literature that reports learning disabilities in 30% of the CCHS patients and additional educational needs or attending special schools in 50% of the patients [5]. Recently, an association was found between oxygen saturation and heart rate during challenges and cognitive outcomes in patients with CCHS [13], underlining the importance of adequate ventilatory management for optimal functioning. Furthermore, ocular disorders were only found in one patient in this group (decreased pupillary reaction), while Goldberg and Ludwig found ocular disorders in 46–92% of patients with CCHS [14]. We hypothesize that not all patients were regularly screened for ocular diseases.
In this study, we were not able to focus on the quality of life. A small French cohort study with 12 CCHS patients found that this disease is associated with a moderate impairment of health-related quality of life in young adults [15]. We observed that the majority of patients with CCHS were able to live at home and could pursue normal education despite their life-affecting disease. While this is a very reassuring outcome, further research should definitely include an assessment of the quality of life.
The recent guideline by Trang and the review Kasi can be used to formalize an adequate follow-up [5, 6]. A recent expert consensus by Slattery and colleagues [15] can serve as a guide for transitional care for adolescents and young adults. While it is still not possible to precisely predict a clinical profile based on the PHOX2B variant, we recommend regularly evaluating the respiratory status and giving specific attention to expected comorbidities in all patients, regardless of their genetic variant. We suggest an annual multidisciplinary evaluation in a University Medical Center where an HMV center is located. Experts in HMV should be part of this multidisciplinary team. Considering the high risk of life-threatening complications, clinicians should monitor patients’ clinical profile and implement individualized interventions focused on respiratory and circulatory disruptions.
In the Appendix, we formulate a proposal for the evaluation of CCHS patients in the Dutch setting. Guided by a patient’s clinical profile, follow-up in adolescents and adults should be performed in an individualized way. The paediatric evaluation should consist of an evaluation of the respiratory situation with titration of the ventilatory support through polysomnographic investigation, and in case of a tracheostomy, management by an otolaryngologist. Besides respiratory management, cardiac monitoring for arrhythmias should be performed annually, as well as surveillance for neural crest tumours, gastrointestinal motility disorders and ocular diseases (strabismus, pupillary abnormalities, convergence insufficiency and ptosis). Neurodevelopmental testing should be conducted annually, at least until school age. Thereafter, it can be conducted in coordination with the school. Recently, Welbel et al. found that the NIH Toolbox® Cognition Battery (NTCB) is a valid tool for measuring cognitive outcomes in patients with CCHS [16]. This can be used as an accessible tool in neurodevelopmental follow-up. Throughout the year, there should be close contact between the patient (or parents) and preferably a specialized nurse for HMV. In the transition to adult healthcare, a multidisciplinary team should be responsible for the follow-up of these patients.
Based on the incidence of CCHS, we expected 40 to 45 living patients in the Netherlands. With a sample size of 31 patients, we believe that this study covers the main part of this population. However, some CCHS patients with a milder phenotype might not have been included if they were not treated in an HMV center or if their diagnosis was not confirmed through genetic testing. Considering this possibility, this study could be biased as it then included only patients with a more severe profile of CCHS. For this retrospective cohort study, data was collected by patients’ caregivers. This implies a vulnerability to omissions in medical documentation. We are planning to follow the Dutch CCHS patients prospectively to minimalize this effect.
Conclusion
CCHS is a multiorgan disease with life-threatening respiratory problems. In our cohort, the genotype is not easily associated with the phenotype in CCHS, although it is remarkable that all 5 patients with Hirschsprung’s disease as a comorbidity had a 20/27 PHOX2B variant. Our findings highlight the importance of a multi-disciplinary approach and thorough lifelong annual evaluation for all patients with CCHS in order to provide the best care according to the latest standards and prevent patients from life-threatening events.
Data availability
The data that support the findings of this study are available from the authors but restrictions apply to the availability of these data, due to the small sample size of this cohort.
References
Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H (2010) An official ATS clinical policy statement: congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med 181(6):626
Trang H, Dehan M, Beaufils F, Zaccaria I, Amiel J, Gaultier C, French CCHS Working Group (2005) The French Congenital Central Hypoventilation Syndrome Registry: general data, phenotype, and genotype. Chest 127(1):72–79. https://doi.org/10.1378/chest.127.1.72
Amiel J, Laudier B, Attié-Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, Vekemans M, Munnich A, Gaultier C, Lyonnet S (2003) Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet 33(4):459–461. https://doi.org/10.1038/ng1130
Sasaki A, Kanai M, Kijima K, Akaba K, Hashimoto M, Hasegawa H, Otaki S, Koizumi T, Kusuda S, Ogawa Y, Tuchiya K, Yamamoto W, Nakamura T, Hayasaka K (2003) Molecular analysis of congenital central hypoventilation syndrome. Hum Genet 114(1):22–26. https://doi.org/10.1007/s00439-003-1036-z
Trang H, Samuels M, Ceccherini I, Frerick M, Garcia-Teresa MA, Peters J, Schoeber J, Migdal M, Markstrom A, Ottonello G, Piumelli R, Estevao MH, Senecic-Cala I, Gnidovec-Strazisar B, Pfleger A, Porto-Abal R, Katz-Salamon M (2020) Guidelines for diagnosis and management of congenital central hypoventilation syndrome. Orphanet J Rare Dis 15(1):252. https://doi.org/10.1186/s13023-020-01460-2
Kasi AS, Li H, Harford KL, Lam HV, Mao C, Landry AM, Mitchell SG, Clifton MS, Leu RM (2022) Congenital central hypoventilation syndrome: optimizing care with a multidisciplinary approach. J Multidiscip Healthc 15:455–469. https://doi.org/10.2147/JMDH.S284782
Ruys I (ed) (2015) Veldnorm chronische beademing bij kinderen. VSCA. Retrieved 22 May 2023 from https://www.vsca.nl/wp-content/uploads/veldnorm_webversie.pdf
Fain ME, Westbrook AL, Kasi AS (2023) Congenital central hypoventilation syndrome: diagnosis and long-term ventilatory outcomes. Clin Med Insights Pediatr 26(17):11795565231169556. https://doi.org/10.1177/11795565231169556
Balakrishnan K, Perez IA, Keens TG, Sicolo A, Punati J, Danialifar T (2021) Hirschsprung disease and other gastrointestinal motility disorders in patients with CCHS. Eur J Pediatr 180(2):469–473. https://doi.org/10.1007/s00431-020-03848-5 (Epub 2020 Oct 28. Erratum in: Eur J Pediatr. 2021 Jan 5)
Berry-Kravis EM, Zhou L, Rand CM, Weese-Mayer DE (2006) Congenital central hypoventilation syndrome: PHOX2B mutations and phenotype. Am J Respir Crit Care Med 174(10):1139–1144. https://doi.org/10.1164/rccm.200602-305OC
Bishara J, Keens TG, Perez IA (2018) The genetics of congenital central hypoventilation syndrome: clinical implications. Appl Clin Genet 11:135–144. https://doi.org/10.2147/TACG.S140629
Bachetti T, Ceccherini I (2020) Causative and common PHOX2B variants define a broad phenotypic spectrum. Clin Genet 97(1):103–113. https://doi.org/10.1111/cge.13633
Slattery SM, Zelko FA, Vu EL, Dunne EC, Rand CM, Bradley A, Zhou A, Carroll MS, Khaytin I, Brady KM, Stewart TM, Weese-Mayer DE (2023) Ventilatory and orthostatic challenges reveal biomarkers for neurocognition in children and young adults with congenital central hypoventilation syndrome. Chest 163(6):1555–1564. https://doi.org/10.1016/j.chest.2022.12.028
Goldberg DS, Ludwig IH (1996) Congenital central hypoventilation syndrome: ocular findings in 37 children. J Pediatr Ophthalmol Strabismus 33(3):175–180. https://doi.org/10.3928/0191-3913-19960501-11
Verkaeren E, Brion A, Hurbault A et al (2015) Health-related quality of life in young adults with congenital central hypoventilation syndrome due to PHOX2B mutations: a cross-sectional study. Respir Res 16(1):80. https://doi.org/10.1186/s12931-015-0241-3
Welbel RZ, Rand CM, Zhou A, Fadl-Alla A, Chen ML, Weese-Mayer DE, Zelko FA (2022) Neurocognitive monitoring in congenital central hypoventilation syndrome with the NIH Toolbox®. Pediatr Pulmonol 57(9):2040–2047. https://doi.org/10.1002/ppul.25973
Acknowledgements
We want to thank dr. E.S. Veldhoen and dr. E. Zonneveld-Huijssoon for their contributions to this study.
Author information
Authors and Affiliations
Contributions
This research was primarily conducted in the University Medical Center Groningen. Esther Evers-Bikker and Willemien de Weerd drafted the manuscript. All authors contributed to data collection and revising and final approval of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This research was carried out in line with the UMCG Research Code 2018. An ethical review was performed by the UMCG CTc (Central ethics Review Board). The authors confirm that legal and ethical requirements have been met with regards to the individuals described in the study. UMCG Research Register number 202000420.
Competing interests
The authors declare no competing interests.
Additional information
Communicated by Peter de Winter
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendix. Proposal for evaluation of CCHS patients in the Dutch setting
Appendix. Proposal for evaluation of CCHS patients in the Dutch setting
Clinical evaluation of respiratory situation (in a specialistic centre):
Annual (or at least bi-annual in age < 4 years)
-
Transcutaneous CO2 and if possible and accessible polysomnographic investigation
-
Evaluation of ventilatory support
-
If a tracheostomy is present, a clinical check by an otorhinolaryngologist should include airway endoscopy to evaluate the cannula position and size.
-
For non-invasive mechanical ventilation, consider an examination by a maxillofacial specialist or nurse for possible midface hypoplasia.
Outpatient Follow-up:
Annual (or bi-annual in age < 4 years)
-
Respiratory: Exercise test once during adolescence
-
Cardiovascular: 48–72 h ECG Holter, blood pressure, Echocardiogram
-
Gastro-intestinal: signs of constipation or Hirschsprung's disease
-
Nutrition
-
Ophthalmology: ocular testing
-
Neurology neurocognitive tests
-
Endocrinology: fasting glucose, and by indication 24 h glucose day curve and OGTT
-
Oncology: extensive physical examination, urine catecholamines, consider Chest X-ray and abdominal ultrasound
-
Social: Consultation of a social worker
We recommend organizing a multidisciplinary consultation for each patient, involving all caregivers and the patient’s care network, to raise awareness of CCHS and address potential risks and daily life challenges.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Evers-Bikker, E.E., de Weerd, W., Wijkstra, P.J. et al. Characteristics and outcomes in children with congenital central hypoventilation syndrome on long-term mechanical ventilation in the Netherlands. Eur J Pediatr 183, 791–797 (2024). https://doi.org/10.1007/s00431-023-05339-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-023-05339-9