
Abstract
To assess the long-term efficacy of burosumab for pediatric patients with X-linked hypophosphatemia, focusing on linear growth. This multi-center retrospective study included 35 pediatric patients who began treatment with burosumab between January 2018 and January 2021. We collected clinical data, anthropometric measurements, laboratory results, and Rickets Severity Score (RSS), from 2 years prior to treatment initiation and up to 4 years after. Burosumab was initiated at a mean age of 7.5 ± 4.4 years (range 0.6–15.9), with a mean initial dose of 0.8 ± 0.3 mg/kg, which was subsequently increased to 1.1 ± 0.4 mg/kg. The patients were followed for 2.9 ± 1.4 years (range 1–4) after initiating burosumab. Serum phosphorus levels increased from 2.7 ± 0.8 mg/dl at burosumab initiation to 3.4 ± 0.6 mg/dl after 3 months and remained stable (p < 0.001). Total reabsorption of phosphorus increased from 82.0 ± 6.8 to 90.1 ± 5.3% after 12 months of treatment (p = 0.041). The RSS improved from 1.7 ± 1.0 at burosumab initiation to 0.5 ± 0.6 and 0.3 ± 0.6 after 12 and 24 months, respectively (p < 0.001). Both height z-score and weight z-score improved from burosumab initiation to the end of the study: from − 2.07 ± 1.05 to − 1.72 ± 1.04 (p < 0.001) and from − 0.51 ± 1.12 to − 0.11 ± 1.29 (p < 0.001), respectively. Eight children received growth hormone combined with burosumab treatment. Height z-score improved among those who received growth hormone (from − 2.33 ± 1.12 to − 1.94 ± 1.24, p = 0.042) and among those who did not (from − 2.01 ± 1.01 to − 1.66 ± 1.01, p = 0.001).
Conclusion: Burosumab treatment in a real-life setting improved phosphate homeostasis and rickets severity and enhanced linear growth.
What is Known: • Compared to conventional therapy, burosumab treatment has been shown to increase serum phosphate levels and reduce the severity of rickets. • The effect of burosumab on growth is still being study. | |
What is New: • Height z-score improved between the start of burosumab treatment and the end of the study (-2.07 ± 1.05 vs. -1.72 ± 1.04, p < 0.001). • Eight children received burosumab combined with growth hormone treatment without side effects during the concomitant treatments. |
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
X-linked hypophosphatemia (XLH) is the most common heritable form of rickets, with an incidence of approximately 1:25,000 live births [1, 2]. The etiology of this X-linked dominant condition is loss-of-function mutations in the phosphate-regulating endopeptidase homolog X-linked (PHEX) gene, which results in increased fibroblast growth factor 23 (FGF23) levels. Increased FGF23 levels lead to renal phosphate wasting, hypophosphatemia, and decreased levels of calcitriol, due to the inhibitory effect on 25-hydroxyvitamin D 1-α-hydroxylase [3]. In children and adolescents, this results in rickets, impaired growth, deformations of the weight-bearing joints, dental complications, muscle weakness, and pain [4].
Individuals with XLH typically have normal birth lengths. However, their height may be impacted during childhood and adolescence. This ultimately results in an adult height that is often two standard deviation scores below the population normative values [5, 6]. Moreover, impaired limb growth, in parallel with relatively preserved truncal growth, results in disproportionate short stature [7].
Conventional therapy for XLH typically involves multiple daily doses of oral phosphate salts and active vitamin D metabolites (calcitriol or alfacalcidol) [8]. Despite its demonstrated benefits, this therapy may not be adequate for severely affected persons, and adherence can be difficult. Additionally, it is not effective in improving growth, as evidenced by decreases in height z-scores in some treated children [9]. Moreover, side effects and complications may present, such as secondary hyperparathyroidism and nephrocalcinosis [10].
Burosumab is a recombinant human monoclonal antibody that targets FGF23. Based on several clinical trials, in 2018, both the European Medicines Agency and the US Food and Drug Administration (FDA) approved burosumab as a monotherapy for individuals with XLH of age 1 year or older [11,12,13,14]. The FDA subsequently extended its approval to children as young as 6 months. Compared to conventional therapy, burosumab treatment has been shown to improve renal tubular phosphate reabsorption, increase serum phosphorus to normal levels, increase 1,25-dihydroxyvitamin D levels, and reduce the severity of rickets [15]. Burosumab treatment has also improved patient-reported outcomes, such as pain interference, physical function, mobility, and fatigue scores [16]. Among children and adolescents with XLH, burosumab treatment was shown to decrease adiposity while increasing muscle mass [17].
Burosumab is a novel, etiology-based treatment for patients with XLH. However, long-term studies in a real-life, pediatric setting are limited. Therefore, our study aimed to evaluate the effectiveness and safety of burosumab for children with XLH in real-life clinical practice. We focused on the impact of the treatment on linear growth.
Materials and methods
Study population
This retrospective multi-center real-life study included 35 pediatric patients with XLH, who received care at seven medical centers throughout Israel: Edmond and Lily Safra Children’s Hospital in Sheba Medical Center, Dana-Dwek Children’s Hospital in Tel Aviv Sourasky Medical Center, Schneider Children’s Medical Center, Kaplan Medical Center, Rambam Health Care Campus, Hadassah Hebrew University Medical Center, and Shaare Zedek Medical Center. The study protocol was approved by the institutional review board of each individual medical center, with Sheba Medical Center as the primary site.
Burosumab treatment
The study included all the patients who began treatment with burosumab between January 1, 2018, and January 1, 2021. The criteria for starting burosumab treatment were clinical or genetic diagnosis of XLH and radiographic evidence of bone disease. Treatment prior to the study period, with oral phosphate supplement and alfacalcidol, was according to the XLH conventional treatment guidelines [2]. Such therapy was discontinued 7 days before starting burosumab. Burosumab was initiated at a dose of 0.4 to 0.8 mg/kg of body weight, rounded to the nearest 10 mg. Subcutaneous injections were to be given every 2 weeks by trained nurses or parents. The dosage was increased stepwise according to laboratory results, consistent with clinical practice guidelines, up to a maximum dose of 2 mg/kg body weight or 90 mg [2, 18]. For this study, data on side effects were obtained from information provided by the patients and their parents during clinic visits.
Clinical data, anthropometric measurements, and laboratory results were collected from the medical records, starting 2 years prior to burosumab initiation. The end of the follow-up period was defined as the earliest of the following time points: 4 years after burosumab initiation or January 31, 2023, the study census date.
Anthropometric measurements
The weight and height of the participants were measured at clinic visits approximately every 3 months, both during the conventional and the burosumab treatment. Body mass index (BMI) was calculated by dividing body weight in kilograms by the square of height in meters. Height, weight, and BMI z-scores were then calculated using growth charts that were specific to sex and age, based on the Center for Disease Control and Prevention 2000 Growth Charts.
Laboratory tests results
Laboratory studies were obtained according to the practice guidelines [2, 18] and the clinical discretion of the physicians. The samples were obtained just before the burosumab injection and tested for serum phosphorus, calcium, creatinine, alkaline phosphatase (ALP), 25-hydroxy vitamin D, 1,25-dihydroxyvitamin D (1,25vit D), intact parathyroid hormone (PTH), intact FGF23, and insulin-like growth factor 1 (IGF-1). Spot urine studies included phosphorus, calcium, and creatinine. Total reabsorption of phosphate (TRP) was calculated as 1- [Phos (u) X Crea (s) / Phos (s) X Crea (u)] and presented as percentages. Tubular maximum transport of phosphate to glomerular filtration rate (TmP/GFR) was calculated from values in serum and spot urine as previously described [19], and later updated [20]. The normal reference limits of intact FGF-23 were those that were recently determined [21].
Skeletal radiographs
Skeletal radiographs were obtained as needed for routine clinical care. For the purpose of this study, lower extremity radiographs were reviewed by two physician authors (LZ and RR) and scored using the Thacher Rickets Severity Score (RSS) [22, 23]. The images were scored from 0 to 6, with a higher score indicating more severe rickets. As wrist radiographs were not routinely obtained for all the patients, only the lower extremity radiographs were used for calculating the RSS.
Growth hormone treatment
During the study period, eight children with short stature (of whom two were GH deficient and six with GH sufficient levels) were treated with recombinant human growth hormone (rhGH). The two GH-deficient children had low GH levels in two stimulation tests, in accordance with consensus guidelines. The remaining six children had sufficient GH levels and were prescribed rhGH therapy due to severe short stature after careful consideration and discussion of the staff with the patients and their parents.
Statistical analysis
The data were analyzed using SPSS software version 28 (SPSS Inc., Chicago, IL). The results are presented as means and standard deviations, or as numbers and percentages, as appropriate. ANOVA with repeated measures was used to evaluate changes in variables over time (anthropometric measurements, laboratory results, TRP, TmP/GFR, and RSS). “End of study” values were calculated as the mean of values measured at 30 to 48 months after burosumab initiation. The paired t-test was used to compare between the measurements of 1,25vit D at burosumab initiation and 12 months later. The changes (delta) in values between these time points were calculated for the following parameters: height z-score, serum phosphorous, ALP, PTH, 1,25vit D, and TmP/GFR. The Pearson correlation coefficient was used to test for correlations between delta height z-score and the delta of the other parameters and between delta RSS and the delta of the other parameters. All the statistical tests were two sided and p-value < 0.05 was considered significant.
Results
The study included 35 patients with XLH who were treated with burosumab. The age range at study entry was 0.5–15.9 years. At presentation, all the patients had hypophosphatemia with hyperphosphaturia. XLH was diagnosed by the identification of a pathogenic variant in the PHEX gene in 31 (89%) patients. For the remaining four patients (11%), a clinical diagnosis was based on the presence of hypophosphatemia with hyperphosphaturia, elevated plasma levels of intact FGF23, and a positive family history of XLH [2, 18]. A family history of XLH was reported for 24 patients (69%). At the time of burosumab initiation, 27 (77%) patients were pre-pubertal. At the end of the study, 20 (57%) children were pre-pubertal, and 15 (43%) were at puberty. The clinical characteristics, anthropometric measurements, and laboratory results at burosumab initiation are presented in Table 1.
Treatment
Prior to the study period, 33 (94%) patients received conventional therapy, namely oral phosphate supplement and alfacalcidol. Such therapy was started at 2.1 ± 1.4 years (range 0.4–6.6) and continued for a mean duration of 5.4 ± 4.2 years (range 0.2–14.8). Two patients started burosumab treatment immediately after being diagnosed with XLH, without prior conventional therapy, at ages 2.6 and 4.3 years.
The mean age at the start of burosumab treatment was 7.5 ± 4.4 years (range 0.6–15.9). Burosumab was initiated at a mean dose of 0.8 ± 0.3 mg/kg, which yielded a total dose of 10–50 mg. Dosages were adjusted to target a low normal range serum phosphorus level. The mean maximal burosumab dose was 1.1 ± 0.4 mg/kg, and the maximal total dose was 10–80 mg. Patients were followed for a mean 2.9 ± 1.4 years (range 1–4) after beginning burosumab.
No significant adverse effects were identified during the course of burosumab treatment. Three patients reported experiencing pain, redness, or swelling at the injection site, while one patient reported a single episode of abdominal pain 1 day post injection. No other adverse side effects were reported.
Phosphate and calcium homeostasis
The results of blood tests taken during the study period are presented in Table 2. During the first 3 months of treatment with burosumab, the mean serum phosphorus level increased, from 2.7 ± 0.8 to 3.4 ± 0.6 mg/dl, p = 0.045. Twenty-five (71%) patients had serum phosphorus levels within the normal range for their age by the end of the third month and 28 (80%) by the end of 6 months. The mean serum phosphorus level remained stable throughout the study period (Table 2), and no patient had a level above the upper limit of the normal range. The mean serum calcium level did not change and remained stable at 9.6 mg/dl. Mean ALP and PTH levels decreased, while the mean 1,25vit D level increased, from 48 ± 21 pg/ml at the start of burosumab treatment to 63 ± 24 pg/ml at 12 months later, p = 0.041. The mean level of 25-hydroxy vitamin D did not change during the study: 25 ± 7, 27 ± 8, 22 ± 8, and 22 ± 8 ng/ml, at − 12, 0, 12, and 24–48 months, respectively, p = 0.1.
The increase in serum phosphorus level was in parallel with a decrease in urinary phosphate loss: mean TRP increased from 82.0 ± 6.8% at burosumab initiation to 89.8 ± 8.6% and 90.1 ± 5.3% at 6 and 12 months, respectively (p = 0.041). The mean TmP/GFR level increased from 2.2 ± 0.6 mg/dl burosumab initiation to 3.1 ± 0.6 mg/dl and 3.3 ± 0.6 mg/dl at 6 and 12 months, respectively (p < 0.001). The urine calcium/creatinine ratio did not change: 0.10 ± 0.12 mg/mg, 0.14 ± 0.09 mg/mg, 0.10 ± 0.09 mg/mg at burosumab initiation, 6 and 12 months (p = 0.829).
RSS during burosumab treatment
The mean RSS improved from the initiation of burosumab to 12 and 24 months, as evident from a decrease from 1.7 ± 1.0 to 0.5 ± 0.6 and 0.3 ± 0.6, respectively, p < 0.001 (Fig. 1). A negative correlation was found between the change in serum phosphorus level and RSS during the first year of burosumab treatment (r = − 0.53, p = 0.05).

Rickets Severity Score (RSS) for a patient who started burosumab treatment at the age of 4.3 years. The radiographs were taken A at initiation of treatment, RSS = 2.5; B after 6 months, RSS = 1.5; C after 1 year, RSS = 1.0; D after 2 years, RSS = 0.5; E after 3 years, RSS = 0.5; and F after 4 years, RSS = 0. G The means and standard errors of RSS at burosumab initiation, after 12 months and after 24 months. The results show a significant decrease in RSS (p < 0.001)
Growth during burosumab treatment
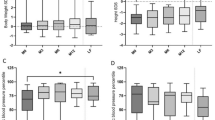
Anthropometric measurements were available for 24 children at the following time points: at burosumab initiation, after 12 months, after 24 months, and at the end of the study. Table 3 shows the mean height z-score, weight z-score, and BMI z-score for each time point. Both the mean height z-score and the mean weight z-score improved between the start of burosumab treatment and the end of the study (− 2.07 ± 1.05 vs. − 1.72 ± 1.04, p < 0.001, and − 0.51 ± 1.12 vs. − 0.11 ± 1.29, respectively, p = 0.001) (Fig. 2).

The means and standard errors of height z-score, weight z-score, and BMI z-score for 24 patients who received burosumab treatment. The measurements were taken at burosumab initiation, after 12 months, after 24 months, and at the end of the study. A displays the total height z-score, while B and C show the height z-score without and with combined recombinant human growth hormone (rhGH) treatment, respectively. D shows the weight z-score, and E displays the BMI z-score. The results indicate statistically significant increases in height z-score and weight z-score (p = 0.001). The significance level is indicated with an asterisk (*p < 0.05)
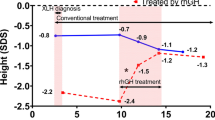
Of the 35 children, eight were treated with GH. Six of these children began rhGH treatment while on conventional treatment, and the other two started rhGH treatment while being treated with burosumab (Table 4). The mean age at rhGH initiation was 8.6 ± 2.9 years, and the mean height z-score at the start of rhGH was − 2.98 ± 0.84. Two patients stopped rhGH treatment during the study period as they reached their final adult height, while two other patients stopped rhGH treatment after 1 year, per their parents’ requests. We analyzed the height z-scores during burosumab treatment separately for those who received rhGH treatment and those who did not (as shown in Table 3 and Fig. 2). The mean height z-scores improved between burosumab initiation and the end of the study: from − 2.01 ± 1.01 to − 1.66 ± 1.01 among those not treated with rhGH, p = 0.001, and from − 2.33 ± 1.27 to − 1.94 ± 1.24 among those treated, p = 0.042. As mentioned above, two of our patients initiated rhGH treatment while receiving burosumab treatment. One patient showed poor growth, potentially due to poor adherence, and the parents stopped the rhGH treatment after 1 year. The second patient began rhGH treatment after having received burosumab for 2 years. Her height z-score improved by 0.7 during the 2 years of concomitant treatment (Fig. 3).

Height growth curves of two patients treated with concomitant burosumab (B) and growth hormone (GH). A—a female patient who was switched from conventional therapy to burosumab at 6 years of age, and 2 years later GH treatment was added. B—a male patient who initiated GH treatment at 4.5 years of age during the conventional treatment that was switched to burosumab a year later
Among 33 patients with available data, no correlations were found between the change in height z-score during the first year of burosumab treatment and the changes in serum phosphorus levels, ALP, PTH, 1,25vit D, and TmP/GFR.
Discussion
Our findings demonstrated that during a follow-up period of up to 4 years, the administration of burosumab to children with XLH resulted in improvement in laboratory metrics, rickets, and growth. The therapy was shown to be both safe and effective in a real-life clinical environment.
Conventional therapy for XLH improves serum phosphorus levels, but it may not completely heal rickets or correct skeletal deformities, and this can lead to persistent short stature and impaired mobility [24]. Additionally, the treatment regimen can be challenging to some patients and thus result in low adherence [2]. In the current study, despite treatment with conventional therapy for a mean 5.4 years, some patients still presented with persistent rickets (as indicated by a RSS of 1.7) and short stature (height z-score of − 2.07) at the initiation of burosumab treatment.
Burosumab represents a novel therapeutic strategy for managing XLH, by addressing the underlying pathophysiology of the disorder [25]. Our study demonstrated that transition from conventional therapy to burosumab injections significantly improved phosphorus homeostasis, thus corroborating results of previous trials [15, 26,27,28]. Phosphorus levels improved relatively quickly. Indeed, 71% of the cohort achieved levels within the normal range for age by the third month of treatment and 80% by the sixth month. Serum phosphorus levels improved due to improved phosphate absorption in the kidneys, as indicated by the increase in TRP and TmP/GFR. Additionally, ALP and PTH decreased significantly, while 1,25vit D increased. The decrease in ALP is an important marker of rickets healing and is consistent with previous research on burosumab [28]. Only minor dose adjustments were needed to maintain these targets throughout the study period; the mean burosumab initial dose was 0.8 ± 0.3 mg/kg, and the maximal dose was 1.1 ± 0.4 mg/kg. Of note, 1,25vit D increased but remained within the normal range, and urinary calcium excretion did not worsen. Elevated 1,25vit D above the normal range may be a sign of overtreatment.
The current study demonstrated significant decrease of RSS values over the study period. This finding is consistent with research that demonstrated decreased RSS after 64 weeks of burosumab treatment, which correlated to the decrease in ALP levels [23]. The sustained improvement in rickets healing during burosumab treatment suggests greater effectiveness than conventional therapy for treating XLH.
As impaired growth is a hallmark of XLH, the main objective of this study was to assess parameters of children’s growth. Prior research on untreated XLH showed normal birth length but stunted growth during infancy and childhood [29]. Conventional therapy has shown limited impact on growth. One study revealed a decline in annual growth during early childhood in children who were treated with conventional therapy, compared to a healthy population [30]. Growth curves for children with XLH treated with conventional therapy have been generated based on the height measurements of that cohort [30]. Starting conventional treatment during infancy may lead to better growth outcomes [30, 31].
The effect of burosumab treatment on growth is still being studied. Our research found notable improvement in growth during burosumab treatment. The mean height z-score improved from − 2.01 ± 1.01 to − 1.73 ± 1.01after 1 year and to − 1.66 ± 1.01 at the end of the study period. Other studies have shown similar trends of growth improvement, albeit to varying degrees [13,14,15, 26, 28]. In children aged 1–4 years, burosumab appeared to maintain growth rates [14], while in cohorts of children aged 5–12 years [13, 28] or 1–12 years [15, 26], modest improvements in height z-score were observed. A randomized trial reported improved mean height z-scores from baseline to week 64 and week 160 (− 1.72 ± 1.03, − 1.54 ± 1.13, and − 1.38 ± 1.06, respectively) [28]. A real-world retrospective study reported a mean increase of 0.94 in height z-scores after 24 months of burosumab treatment in 12 patients aged 2–18 years [27]. Overall, burosumab appears to be effective in improving height in children, yet the extent of improvement apparently varies with age. Burosumab versus conventional therapy was shown to be more effective in improving height z-scores among younger (aged < 5 years) than older (aged 5–12 years) children [26]. Other factors that may influence the extent of improvement are baseline height, pubertal stage, and skeletal deformities.
Our cohort included eight children treated with both burosumab and rhGH. Six of them began rhGH treatment while receiving conventional treatment, and the remaining two began rhGH treatment while receiving burosumab treatment. The mean height z-score improved from − 2.33 ± 1.27 at burosumab initiation to − 1.94 ± 1.24 at the end of the study. No side effects were reported by the patients during concomitant rhGH and burosumab treatments.
Varying benefit has been reported for the addition of rhGH therapy to conventional therapy [32,33,34,35,36,37]. A recent study included 34 individuals who received treatment with rhGH for persistent short stature, despite receiving adequate conventional treatment [37]. The mean height at rhGH initiation was − 2.4 ± 0.9 SDS, and the final adult height was − 1.3 ± 0.9 SDS. This suggests that the addition of rhGH therapy to conventional therapy allows for an improvement in the final height of children with XLH and growth failure.
To the best of our knowledge, there is only one study that assessed the effects of concurrent treatment with rhGH and burosumab on linear growth [38]. In that study, all the children initiated rhGH therapy during conventional treatment, at least 1 year before transitioning to burosumab [38]. During the first year of rhGH treatment, before initiating burosumab, height improved significantly (mean height gain of 1.0 ± 0.4 SDS). During the year of combined burosumab and rhGH therapies, the mean height continued to increase, albeit to a lesser extent (mean height gain of 0.2 ± 0.1 SDS).
Strength and limitations
The strengths of this multi-center study include a relatively large cohort of a rare disease, long duration of follow-up, and real-life methodology that enabled evaluating the feasibility and efficacy of treatment in a clinical practice setting. These factors provide valuable insights into the treatment of the disease in real-world situations. However, the study also had limitations. The laboratory tests, anthropometric measurements, and radiograph studies were performed at the clinicians’ discretion. This resulted in variations in the frequency of these assessments between patients and missing data at some time points for analysis. Only 24 patients had growth measurements at all the time points for analysis; thus, the cohort was too small to sub-analyze the change in height according to the pubertal status. Another limitation of the study was the lack of wrist radiographs at all the time points, for calculating the full RSS. The analysis was only based on lower extremity radiographs, which may not have fully reflected changes in disease progression or treatment efficacy. Lastly, only eight children received concurrent treatment with rhGH and burosumab. Further studies are needed to characterize the patients who may benefit from this combined treatment.
In conclusion
Our study provides evidence of the safety and effectiveness of burosumab treatment in a clinical practice setting. We observed improvements in laboratory parameters that were comparable to those reported in clinical trials. These improvements were sustained during the follow-up period of up to 4 years. We also observed improvement in height status. This indicates that burosumab treatment may have a positive impact on growth in individuals with XLH. To fully evaluate the effect of burosumab on final adult height, larger cohorts with longer treatment durations are needed. Of particular interest would be evaluation of the impact of initiating burosumab treatment during the first year of life, as early intervention may result in more significant improvements in growth. Furthermore, the concomitant use of rhGH and burosumab treatments should be evaluated, for individuals with severe short stature. Such investigation could elucidate the potential benefits and risks of combining these treatments and their impact on growth outcomes.
Data availability
All the data relevant to the study are included in this article and can be obtained by request. Further inquiries can be directed to the corresponding author.
Abbreviations
- ALP:
-
Alkaline phosphatase
- BMI:
-
Body mass index
- FGF23:
-
Fibroblast growth factor 23
- IGF-1:
-
Insulin-like growth factor 1
- PHEX:
-
Phosphate regulating endopeptidase homolog X-linked
- PTH:
-
Parathyroid hormone
- rhGH:
-
Recombinant human growth hormone
- RSS:
-
Rickets Severity Score
- TmP/GFR:
-
Tubular maximum transport of phosphate to glomerular filtration rate
- TRP:
-
Total reabsorption of phosphate
- 1,25vit D:
-
1,25-Dihydroxyvitamin D
- XLH:
-
X-linked hypophosphatemia
References
Baroncelli GI, Mora S (2021) X-linked hypophosphatemic rickets: multisystemic disorder in children requiring multidisciplinary management. Front Endocrinol (Lausanne) 12:688309. https://doi.org/10.3389/fendo.2021.688309
Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D et al (2019) Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol 15(7):435–455. https://doi.org/10.1038/s41581-019-0152-5
Kuro OM, Moe OW (2017) FGF23-alphaKlotho as a paradigm for a kidney-bone network. Bone 100:4–18. https://doi.org/10.1016/j.bone.2016.11.013
Imel EA, White KE (2019) Pharmacological management of X-linked hypophosphataemia. Br J Clin Pharmacol 85(6):1188–1198. https://doi.org/10.1111/bcp.13763
Zivicnjak M, Schnabel D, Billing H, Staude H, Filler G, Querfeld U et al (2011) Age-related stature and linear body segments in children with X-linked hypophosphatemic rickets. Pediatr Nephrol 26(2):223–231. https://doi.org/10.1007/s00467-010-1705-9
Santos Rodriguez F (2020) X-linked hypophosphataemic rickets and growth. Adv Ther 37(Suppl 2):55–61. https://doi.org/10.1007/s12325-019-01178-z
Quinlan C, Guegan K, Offiah A, Neill RO, Hiorns MP, Ellard S et al (2012) Growth in PHEX-associated X-linked hypophosphatemic rickets: the importance of early treatment. Pediatr Nephrol 27(4):581–588. https://doi.org/10.1007/s00467-011-2046-z
Glorieux FH, Marie PJ, Pettifor JM, Delvin EE (1980) Bone response to phosphate salts, ergocalciferol, and calcitriol in hypophosphatemic vitamin D-resistant rickets. N Engl J Med 303(18):1023–1031. https://doi.org/10.1056/NEJM198010303031802
Uday S, Shaw NJ, Mughal MZ, Randell T, Hogler W, Santos R et al (2021) Monitoring response to conventional treatment in children with XLH: value of ALP and Rickets Severity Score (RSS) in a real world setting. Bone 151:116025. https://doi.org/10.1016/j.bone.2021.116025
Collins M (2018) Burosumab: at long last, an effective treatment for FGF23-associated hypophosphatemia. J Bone Miner Res 33(8):1381–1382. https://doi.org/10.1002/jbmr.3544
Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM et al (2014) Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest 124(4):1587–1597. https://doi.org/10.1172/JCI72829
Imel EA, Zhang X, Ruppe MD, Weber TJ, Klausner MA, Ito T et al (2015) Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab 100(7):2565–2573. https://doi.org/10.1210/jc.2015-1551
Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A et al (2018) Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med 378(21):1987–1998. https://doi.org/10.1056/NEJMoa1714641
Whyte MP, Carpenter TO, Gottesman GS, Mao M, Skrinar A, San Martin J et al (2019) Efficacy and safety of burosumab in children aged 1–4 years with X-linked hypophosphataemia: a multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol 7(3):189–199. https://doi.org/10.1016/S2213-8587(18)30338-3
Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O et al (2019) Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet 393(10189):2416–2427. https://doi.org/10.1016/S0140-6736(19)30654-3
Padidela R, Whyte MP, Glorieux FH, Munns CF, Ward LM, Nilsson O et al (2021) Patient-reported outcomes from a randomized, active-controlled, open-label, phase 3 trial of burosumab versus conventional therapy in children with X-linked hypophosphatemia. Calcif Tissue Int 108(5):622–633. https://doi.org/10.1007/s00223-020-00797-x
Brener A, Lebenthal Y, Cleper R, Kapusta L, Zeitlin L (2021) Body composition and cardiometabolic health of pediatric patients with X-linked hypophosphatemia (XLH) under burosumab therapy. Ther Adv Endocrinol Metab 12:20420188211001150. https://doi.org/10.1177/20420188211001150
Sandy JL, Simm PJ, Biggin A, Rodda CP, Wall CL, Siafarikas A et al (2022) Clinical practice guidelines for paediatric X-linked hypophosphataemia in the era of burosumab. J Paediatr Child Health 58(5):762–768. https://doi.org/10.1111/jpc.15976
Payne RB (1998) Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation. Ann Clin Biochem 35(Pt 2):201–206. https://doi.org/10.1177/000456329803500203
Derain Dubourg L, Aurelle M, Chardon L, Flammier S, Lemoine S, Bacchetta J (2022) Tubular phosphate handling: references from child to adulthood in the era of standardized serum creatinine. Nephrol Dial Transplant 37(11):2150–2156. https://doi.org/10.1093/ndt/gfab331
Brescia V, Fontana A, Lovero R, Capobianco C, Marsico SV, De Chirico T et al (2022) Determination of iFGF23 upper reference limits (URL) in healthy pediatric population, for its better correct use. Front Endocrinol (Lausanne) 13:1018523. https://doi.org/10.3389/fendo.2022.1018523
Thacher TD, Fischer PR, Pettifor JM, Lawson JO, Manaster BJ, Reading JC (2000) Radiographic scoring method for the assessment of the severity of nutritional rickets. J Trop Pediatr 46(3):132–139. https://doi.org/10.1093/tropej/46.3.132
Thacher TD, Pettifor JM, Tebben PJ, Creo AL, Skrinar A, Mao M et al (2019) Rickets severity predicts clinical outcomes in children with X-linked hypophosphatemia: utility of the radiographic Rickets Severity Score. Bone. 122:76–81. Epub 2019/02/18. https://doi.org/10.1016/j.bone.2019.02.010
Harada D, Ueyama K, Oriyama K, Ishiura Y, Kashiwagi H, Yamada H et al (2021) Switching from conventional therapy to burosumab injection has the potential to prevent nephrocalcinosis in patients with X-linked hypophosphatemic rickets. J Pediatr Endocrinol Metab 34(6):791–798. https://doi.org/10.1515/jpem-2020-0734
Mughal MZ, Baroncelli GI, de Lucas-Collantes C, Linglart A, Magnolato A, Raimann A et al (2022) Burosumab for X-linked hypophosphatemia in children and adolescents: opinion based on early experience in seven European countries. Front Endocrinol (Lausanne) 13:1034580. https://doi.org/10.3389/fendo.2022.1034580
Ward LM, Glorieux FH, Whyte MP, Munns CF, Portale AA, Hogler W et al (2022) Effect of burosumab compared with conventional therapy on younger vs older children with X-linked hypophosphatemia. J Clin Endocrinol Metab 107(8):e3241–e3253. https://doi.org/10.1210/clinem/dgac296
Paloian NJ, Nemeth B, Sharafinski M, Modaff P, Steiner RD (2022) Real-world effectiveness of burosumab in children with X-linked hypophosphatemic rickets. Pediatr Nephrol 37(11):2667–2677. https://doi.org/10.1007/s00467-022-05484-7
Linglart A, Imel EA, Whyte MP, Portale AA, Hogler W, Boot AM et al (2022) Sustained efficacy and safety of burosumab, a monoclonal antibody to FGF23, in children with X-linked hypophosphatemia. J Clin Endocrinol Metab 107(3):813–824. https://doi.org/10.1210/clinem/dgab729
Cagnoli M, Richter R, Bohm P, Knye K, Empting S, Mohnike K (2017) Spontaneous growth and effect of early therapy with calcitriol and phosphate in X-linked hypophosphatemic rickets. Pediatr Endocrinol Rev 15(Suppl 1):119–22. https://doi.org/10.17458/per.vol15.2017.crb.spontaneousgrowtheffect
Mao M, Carpenter TO, Whyte MP, Skrinar A, Chen CY, San Martin J et al (2020) Growth curves for children with X-linked hypophosphatemia. J Clin Endocrinol Metab 105(10):3243–3249. https://doi.org/10.1210/clinem/dgaa495
Makitie O, Doria A, Kooh SW, Cole WG, Daneman A, Sochett E (2003) Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab 88(8):3591–3597. https://doi.org/10.1210/jc.2003-030036
Baroncelli GI, Bertelloni S, Ceccarelli C, Saggese G (2001) Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets. J Pediatr 138(2):236–243. https://doi.org/10.1067/mpd.2001.108955
Haffner D, Nissel R, Wuhl E, Mehls O (2004) Effects of growth hormone treatment on body proportions and final height among small children with X-linked hypophosphatemic rickets. Pediatrics 113(6):e593–e596. https://doi.org/10.1542/peds.113.6.e593
Zivicnjak M, Schnabel D, Staude H, Even G, Marx M, Beetz R et al (2011) Three-year growth hormone treatment in short children with X-linked hypophosphatemic rickets: effects on linear growth and body disproportion. J Clin Endocrinol Metab 96(12):E2097–E2105. https://doi.org/10.1210/jc.2011-0399
Rothenbuhler A, Esterle L, Gueorguieva I, Salles JP, Mignot B, Colle M et al (2017) Two-year recombinant human growth hormone (rhGH) treatment is more effective in pre-pubertal compared to pubertal short children with X-linked hypophosphatemic rickets (XLHR). Growth Horm IGF Res 36:11–15. https://doi.org/10.1016/j.ghir.2017.08.001
Meyerhoff N, Haffner D, Staude H, Wuhl E, Marx M, Beetz R et al (2018) Effects of growth hormone treatment on adult height in severely short children with X-linked hypophosphatemic rickets. Pediatr Nephrol 33(3):447–456. https://doi.org/10.1007/s00467-017-3820-3
Andre J, Zhukouskaya VV, Lambert AS, Salles JP, Mignot B, Bardet C et al (2022) Growth hormone treatment improves final height in children with X-linked hypophosphatemia. Orphanet J Rare Dis 17(1):444. https://doi.org/10.1186/s13023-022-02590-5
Ertl DA, Le Lorier J, Gleiss A, Trabado S, Bensignor C, Audrain C et al (2022) Growth pattern in children with X-linked hypophosphatemia treated with burosumab and growth hormone. Orphanet J Rare Dis 17(1):412. https://doi.org/10.1186/s13023-022-02562-9
Funding
The authors received a grant from the Israel Society for Clinical Pediatrics to conduct this study.
Author information
Authors and Affiliations
Contributions
YLS, YL, LZ, and DT: conception and design of the study; YLS, SL, RR, SG, AB, DG, DS, AZ, ZZ, YB, RB, AT, ZZ, and MD: acquisition of data; and YLS and RR: analysis and interpretation of data and drafting the initial manuscript and editing it. All the authors reviewed the manuscript, revised it, and approved the final manuscript as submitted.
Corresponding author
Ethics declarations
Ethics approval
This study was performed in line with the principles of the Declaration of Helsinki. The study protocol was approved by the institutional review board of each individual medical center, with Sheba Medical Center as the primary site (approval number 5914-19-SMC). Written informed consent was not required by the ethics committee of Sheba Medical Center as the analysis did not use any identifying information of the individuals included in this retrospective study.
Competing interests
The authors declare no competing interests.
Additional information
Communicated by Peter de Winter
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Levy-Shraga, Y., Levi, S., Regev, R. et al. Linear growth of children with X-linked hypophosphatemia treated with burosumab: a real-life observational study. Eur J Pediatr 182, 5191–5202 (2023). https://doi.org/10.1007/s00431-023-05190-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-023-05190-y