
Abstract
Hyperinsulinaemic hypoglycaemia (HH) is a major cause of hypoglycaemia in the neonatal period, infancy and childhood. It is caused by unsuppressed insulin secretion in the setting of hypoglycaemia and carries a high risk of significant neurological sequelae, such as cognitive impairment. Genetic mutations have been implicated in the pathogenesis of the condition. Other causes include intra-uterine growth retardation, perinatal asphyxia, maternal diabetes mellitus and syndromes, such as Beckwith-Wiedemann. Based on the aetiology, the clinical presentation can range from absence of symptoms to the typical adrenergic symptoms and coma and even death. The diagnosis is based on biochemical findings and the gold-standard imaging technique is 18F-DOPA PET/CT scanning. Treatment options involve medications, such as diazoxide, nifedipine, glucagon and octreotide, as well as surgery. Novel treatment, such as long-acting octreotide, lanreotide and sirolimus, may be used as an alternative to pancreatectomy. Potential future medical treatments include exendin, a GLP-1 receptor antagonist, and glucagon infusion via a pump.
Conclusion: Advances in the fields of genetic testing, imaging techniques and medical treatment are beginning to provide novel insights into earlier detection, less invasive treatment approaches and fewer complications associated with the complex entity of hyperinsulinaemic hypoglycaemia.
What is Known: • HH is caused by dysregulated insulin release from the β cell due to genetic mutations and carries a risk for complications, such as neurocognitive impairment. 18F-DOPA PET/CT scanning is presented as the gold-standard imaging technique currently in children with hyperinsulinaemic hypoglycaemia. • Clinical presentation is heterogeneous and treatment options include medical therapy and pancreatectomy. | |
What is New: • 18F-DOPA PET/CT is indicated in suspected focal CHI due to paternal transmitted mutations in ABCC8 or KCNJ11. • Novel treatment options have been introduced, such as long-acting octreotide, lanreotide, sirolimus and selective nonpeptide somatostatin receptor subtype 5 (SSTR5) agonists. Future medical treatments include exendin, a GLP-1 antagonist, and glucagon infusion via a pump. However, all these options are off-label at present. |
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hyperinsulinaemic hypoglycaemia (HH) refers to a genetically and phenotypically heterogeneous disease. It is characterised by inappropriate, excessive secretion of insulin from the pancreatic β cell in the presence of low plasma glucose concentrations, causing hypoketotic, hypofattyacidaemic and hyperinsulinaemic hypoglycaemia. It is the commonest cause of mild to severe persistent hypoglycaemia in infants and children, placing them at risk for neurological complications, such as epilepsy, cerebral palsy and cognitive impairment [18]. HH usually presents soon after birth, but can also present in infancy, childhood or even adulthood, depending on the aetiology [1, 28].
Normally, insulin secretion is strictly regulated so that plasma glucose concentrations are maintained within a narrow physiological range (3.5–5.5 mmol/L) [18]. The molecular basis of HH includes defects in pathways that regulate insulin release from the β cell. More specifically, the underlying pathogenetic mechanism is dysregulated insulin secretion secondary to mutations in different genes that are involved in the regulation of insulin secretion from the β cells. The unsuppressed insulin secretion in the presence of low glucose concentrations results in severe and persistent hypoglycaemia [18].
Persistent form of HH is caused by mutations in the genes that regulate insulin secretion; however, hyperinsulinism may be transient due to risk factors, such as intra-uterine growth retardation, perinatal asphyxia, maternal diabetes mellitus or in the setting of syndromes, such as Beckwith-Wiedemann [31]. Hypoglycaemia typically occurs after a period of fasting, exercise or occasionally during the course of an illness, when there are increased metabolic demands. However, it can also manifest postprandially, following a protein or carbohydrate-rich meal.
The incidence of the condition is estimated as 1 in 40,000–50,000 in the general population to 1 in 2500 in populations with high rates of consanguinity [12, 23, 25].
Biochemical profile
The diagnosis is based on biochemical findings that involve recurrent non-ketotic hypoglycaemia accompanied by unsuppressed insulin, c-peptide and pro-insulin, as well as suppressed or low serum ketone bodies, free fatty acids and branched chain amino acids [61]. Critical samples should be obtained during a hypoglycaemic episode so that the diagnosis can be confirmed.
Clinical presentation
The clinical phenotype can be variable, ranging from absence of symptoms to coma or even death. Symptoms of hypoglycaemia such as poor feeding, drowsiness and lethargy are typical and can also include adrenergic symptoms such as tachycardia, palpitations, pallor, sweating, hunger and tremor, but also neuroglycopenic symptoms such as impaired speech, blurred vision, seizures and coma. In addition to symptoms related directly to hypoglycaemia, other reported clinical features include facial features, such as the high forehead and thin upper lip, and macrosomia, which is a common feature which reflects foetal hyperinsulinaemia [16]. Hepatomegaly and hypertrophic cardiomyopathy can also be present due to increased storage of glucose as glycogen [18]. The clinical heterogeneity of the syndrome may be attributed to differences in the underlying pathogenetic mechanisms.
Long-term clinical manifestations of HH include neurodevelopmental disorders, such as cognitive impairment, abnormalities in speech, motor and vision [44]. The simultaneous absence of glucose and ketones, which serve as alternative fuels for brain protection, results in neurobehavioural deficits. Neurodevelopmental delay has been strongly associated with early presentation and severe HH [6].
Aetiology of HH
There is an evolving understanding of the underlying mechanisms involved in HH. Mutations in genes that regulate insulin secretion from β cells have been implicated in monogenic forms of hyperinsulinism. However, genetic aetiology still remains unidentified in up to 50% of patients [23].
Physiological regulation of insulin secretion in the pancreatic β cell
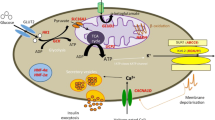
Insulin is a major regulator of plasma glucose concentration and, vice versa, the rate of insulin secretion from the β cells is dependent on glucose concentration [5]. This equilibrium is maintained by β cell ATP-sensitive potassium (KATP) channels, which are composed of four sulfonylurea receptor 1 (SUR1) subunits and four inward-rectifier potassium channel Kir6.2 subunits [34]. As part of normal physiology, when glucose concentrations increase, glucose enters the pancreatic β cell via facilitative glucose transporters, glucose transporter 2 (GLUT2) in particular. Subsequently, it is converted to glucose-6-phosphate through phosphorylation by glucokinase, so that it can enter the glycolytic pathway. The results increase in the ATP/ADP ratio leading to the closure of the KATP channels, depolarisation of the β cell membrane, activation of the intramembraneous calcium channels, influx of calcium into the β cell and insulin exocytosis (Fig. 1). This process of insulin secretion is dysregulated in HH [58].

Mechanism of insulin secretion in a β cell
Genetic mutations in hyperinsulinism
In the majority of the cases, the underlying genetic defect involves a mutation in one of the following genes (Table 1): ABCC8, KCNJ11, glucokinase (GCK), glutamate dehydrogenase (GLUD1), 3-hydroxyacyl-coenzyme A dehydrogenase (HADH), solute carrier family 16 member 1 (SLC16A1), hepatocyte nuclear factor 4-alpha (HNF4A), homeobox A (HNF1A), and can present with multiple phenotypes like in hexokinase 1 (HK1), phosphoglucomutase 1 (PGM1), forkhead box A2 (FOXA2), eukaryotic translation initiation factor 2 subunit gamma (EIF2S3) [19, 70]. The genetic basis is still obscure in the majority of the patients with diazoxide-responsive hyperinsulinaemic hypoglycaemia.
ABCC8 gene encodes for the SUR1 subunits of the KATP channels, whereas KCNJ11 encodes for the Kir6.2 subunits. Four sulphonylurea receptor 1 (SUR1) subunits and four Kir6.2 proteins form the KATP channel complex. The Kir6.2 forms the pore of the channel and the SUR1 acts as a regulatory subunit. Homozygous, compound heterozygous and heterozygous recessive inactivating mutations (missense, frameshift, nonsense, insertions/deletions {microdeletion}, splice site and regulatory mutations) have been reported in ABCC8 and KCNJ11 [26, 73, 74]. So far, more than 150 mutations have been reported in ABCC8 and 25 in KCNJ11. The ABCC8 and KCNJ11 genes are located on chromosome 11p15.1 and mutations in these two genes account for a significant percentage (40–45%) of all HH cases. Mutations in the remaining genes account for 5–10% of the patients, whereas there is no identified aetiology for the remaining 45–55% of cases [48].
Specifically, heterozygous activating GCK mutations cause HH that is usually responsive to medical treatment [15, 22, 63]. GLUD1 gene encodes the intra-mitochondrial enzyme glutamate dehydrogenase (GDH), which also regulates amino acid and ammonia metabolism in β cells, the liver and brain [53]. Gain-of-function mutations in the GLUD1 gene cause the hyperinsulinism and hyperammonia (HI/HA) syndrome [69]. Loss-of-function mutations in the HADH gene are associated with HH and protein sensitivity, with normal ammonia levels [33, 37, 42]. Mutations in the promoter region of SCL16A1 gene have been described in patients with exercise-induced HI [50, 52]. Mutations in the HNF4a gene, which encodes the hepatocyte nuclear factor 4 alpha (HNF4A), can cause transient or persistent hyperinsulinaemic hypoglycaemia, associated with macrosomia. Following a remission of the HI, patients develop maturity-onset diabetes of the young type 1 [36]. Mutations in HNF1A causing hyperinsulinism have also been reported [60].
Various genetic syndromes have been associated with HH, including Beckwith-Wiedemann syndrome, Kabuki syndrome, Turner syndrome, Sotos syndrome, Costello syndrome, Simpson-Golabi-Behmel syndrome, trisomy 13, Timothy syndrome and Usher syndrome [19].
Histology of HH
Histologically, two major morphological forms of HH have been recognized, the diffuse, which accounts for almost 50% of the cases, and focal form (40%). Atypical forms have also been reported (10%) [38]. The diffuse form is characterized by the increased nuclear size of the pancreatic β cells throughout the pancreas. It occurs secondary to biallelic recessive or dominant mutations in one of the involved genes. In the focal form, also known as focal adenomatous hyperplasia, a focal area of abnormal β cells is present, most commonly in the pancreatic body and tail [57]. The focal form results from two independent events: a monoallelic, recessive paternally inherited mutation in one of the genes, KCNJ11 or ABCC8, which code for the two subunits of the KATP channel, as well as a somatic loss of the maternal 11p15 allele including the ABCC8 or KCNJ11 region [78]. Maternal loss of heterozygosity leads to the unmasking of the paternally inherited KATP channel mutation. Therefore, suspicion of focal form is possible through molecular analysis. Additionally, new non-invasive imaging techniques that can identify focal lesions have been developed, such as 18-Fluoro-L-dihydroxyphenylalanine (18F-L-DOPA) positron emission tomography and computed tomography (PET/CT) scanning, which is the recommended imaging technique for the identification of even small focal lesions [11, 51]. The sensitivity of the technique is reported as 85% and the specificity as 96% [40]. F-DOPA is a radiotracer of cells of neuroendocrine origin, such as the β cells of the pancreas and was initially used for the detection of neuroendocrine tumours. 18F-L-DOPA PET/CT scanning can detect focal lesions based on the rationale that they contain a concentrated number of β cells [40]. A more recent study by Antwi et al. demonstrated that 68Ga-DOTA-exendin-4 PET/CT scan performs better than MRI in the detection of insulinomas, with a lower radiation burden and shorter investigation time [3]. Insulinoma acts differently from CHI. So far, there is no proof, if a functional test as DOPA PET, which is the gold standard for focal congenital hyperinsulinism) can be replaced by a surface marker used by 68-DOTA-exendin-4 [77]. However, this technique may represent a promising imaging alternative for the localisation of focal lesions in HH. The histological differentiation is of utmost importance for determining the appropriate management.
Because of the unsuppressed insulin secretion, glycogenolysis, gluconeogenesis, lipolysis and ketogenesis are suppressed in the presence of hypoglycaemia. The absence of alternative energy substrates (ketones, amino acids) results in the patients’ exposure to significant brain injury and disorders in brain development and function [77]. Considering that the brain lacks fuel stores and that the rate of glucose consumption is significantly higher in neonates and infants compared to adults, young patients with HH are vulnerable to severe neurological damage, developmental delay and mental retardation [18]. Therefore, aggressive and prompt treatment is required to prevent poor neurological outcome [12].
Treatment options for HH
The management of HH involves both medical and surgical therapies. The severity of the clinical presentation can vary from the absence of symptoms to severe hypoglycaemia, unresponsive to medical therapy and requiring a near-total pancreatectomy [30]. The neurological prognosis of patients undergoing surgery and patients treated medically shows no differences [55].
Emergency management of hypoglycaemia includes oral feeds or intravenous fluids as a bolus of 2 ml/kg of 10% glucose. In emergency situations, particularly when intravenous access is not obtainable, glucagon can be administered intramuscularly or subcutaneously. Plasma glucose is increased within few minutes through the induction of glycogenolysis, gluconeogenesis and lipolysis. The restoration of normoglycaemia is achieved by intravenous glucose administration at the rate of > 8 mg/kg/min, oral high-calorie carbohydrate diet or by enhancing endogenous glucose production by administering glucagon. Frequent blood glucose monitoring is extremely important and prolong periods of fasting should be avoided [46].
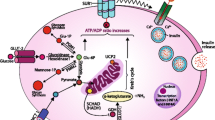
Medical treatment of HH (Fig. 2)
Response to pharmacological treatment can be variable. Conservative medical management aims at the suppression of insulin secretion and includes diazoxide as first-line treatment and somatostatin agonists (octreotide) as second-line treatment. Subcutaneous injection of octreotide is used in cases where diazoxide is ineffective [7].

Treatment of hyperinsulinaemic hypoglycaemia
Diazoxide activates KATP channels and inhibits depolarisation of the β cell membrane and insulin secretion. It binds to the SUR1 subunit of KATP channels, which means that intact KATP channels are required so that responsiveness is achievable. The vast majority of unresponsive to diazoxide patients are likely to harbour mutations in the ABCC8 or KCNJ11 genes. As mentioned above, the ABCC8 and KCNJ11 genes encode the SUR1 and Kir6.2 subunits of the ATP-sensitive K+ (KATP) channel, respectively [79]. Recessive inactivating mutations in ABCC8 and KCNJ11 genes affect the channel biogenesis, turnover, trafficking from the endoplasmic reticulum and Golgi apparatus to the plasma membrane and the sensitivity to magnesium ATP and ADP [35, 45, 56]. By contrast, in dominant ABCC8 or KCNJ11 mutations, it is only the channel activity that is impaired [56]. Dominant inactivating mutations in ABCC8 and KCNJ11 produce a milder phenotype compared to recessive inactivating ABCC8 and KCNJ11 mutations, with regard to responsiveness to diazoxide, need for pancreatectomy and time of presentation [79]. Treatment dose varies from 5 to 20 mg/kg/day 8 hourly [14].
Diazoxide is administered in conjunction with chlorothiazide, so that a synergistic effect is achieved. Chlorothiazide activates a different potassium channel and its diuretic action counteracts the salt and water retention caused by diazoxide. The suggested dose of chlorothiazide is 7–10 mg/kg/day in 2 divided doses. Diazoxide may cause hypotension in a normotensive patient with HH, as part of its antihypertensive action. Pulmonary hypertension has also been reported in patients treated with diazoxide. The mechanism remains unclear and may involve the ATP-sensitive channel agonistic action of diazoxide [17, 43, 76]. Other side effects include hypertrichosis, coarsening of the face, decreased serum immunoglobulin G (IgG) levels and hyperosmolar non-ketotic comas. Rarely, leukopenia and thrombocytopenia may be noted [29].
Somatostatin analogues are used in diazoxide-unresponsive hyperinsulinaemic hypoglycaemia. Octreotide binds to somatostatin receptors SSTR2 and SSTR5 and the mechanisms of action include activation of KATP channels, activation of a G-protein regulated K channel, inhibition of voltage-gated Ca2+ channel and direct inhibition of the process of exocytosis. The recommended dose varies from 5 to 40 micrograms/kg/day via the subcutaneous route. It can also be administered via a pump. The efficacy of the octreotide in the treatment of HH has long been recognized [4, 6, 8, 24, 32, 62, 66, 75]. GH, TSH and ACTH suppression, steatorrhoea, cholelithiasis, abdominal distension, diarrhoea and decreased linear growth may be important adverse effects of octreotide [29]. Long-acting release octreotide (LAR octreotide) may also be used as a monthly intramuscular injection. LAR octreotide maintains octreotide plasma concentrations stable over at least 4 weeks, simplifying medical care [41].
Lanreotide is another long-acting somatostatin analogue acting mainly on the somatostatin receptor 2, preventing the influx of calcium through the calcium channels in the β cells. In the pancreatic β cells, it activates the KATP channel leading to inhibition of suppression of insulin release. As opposed to octreotide, which requires multiple daily injections, lanreotide is administered as monthly injections, improving adherence to treatment and quality of life [32, 65].
Nifedipine is a calcium channel blocker that reduces the influx of calcium into β cells, which is a necessary step for insulin secretion. The recommended dose is 0.25–2.5 mg/kg/day every 8–12 h. Its effectiveness has been shown in several case reports [9, 20, 21, 39, 65, 67, 71], but this has not been repeated and its efficacy has been questioned by most centers and in a study of eleven patients with HH [27]. It has fewer adverse effects than the other medications.
Sirolimus is a mammalian target of rapamycin (mTOR) inhibitor reported to be used in children with the diffuse form of HH who are unresponsive to standard medical treatment. mTOR triggers protein synthesis and enhances mRNA translation [81]. The activation of the mTOR pathway has been implicated in the pathogenesis of HH [64], although this hypothesis has been questioned. An alternative mechanism of sirolimus action has been proposed; decreased insulin release through depletion of Ca2+ stores [82]. Sirolimus is an immunosuppressive medication with anti-proliferative properties and it is thought to be an alternative for pancreatectomy. In the diazoxide-unresponsive diffuse form of HH, subtotal pancreatectomy results in diabetes mellitus within 14 years in the vast majority of the patients, whereas hyperinsulinaemic hypoglycaemia will persist in a significant percentage of the patients [10]. Thus, the substitution of surgery with medical therapy is of paramount importance. Furthermore, sirolimus has been used additionally to other medications such as octreotide, in severe hyperinsulinism, in order to reduce the dependence on the medication and hypercaloric nocturnal feeds [2]. However, recent case series and long-term data on sirolimus use in HH have been reported suggesting that its use should be limited in view of long-term side effects. Short-term adverse events include stomatitis, infections, sepsis, renal dysfunction, gut dysmotility and anaemia [72], whereas potential long-term consequences include immunosuppressive effects and malignancy risk [54].
A new promising therapeutic option, exendin, has been reported for the treatment of HH due to mutations in KATP channel [13]. Exendin is a glucagon-like peptide-1 (GLP-1) receptor antagonist. GLP-1 is an incretin hormone produced in the small intestine and stimulating postprandial insulin secretion. Exendin inhibits GLP-1-induced insulin release and has a glucose-raising effect [13]. Continuous subcutaneous glucagon infusion via a pump represents a potential future treatment in children with HH. It has been used successfully in children with HH and has resulted in reduction or discontinuation of glucose infusion and in the avoidance of pancreatectomy in some cases [47, 49]. Newer drugs have been reported to reduce insulin secretion in in vitro studies, such as selective nonpeptide somatostatin receptor subtype 5 (SSTR5) agonists, which have been reported to suppress glucose- and sulfonylurea-induced insulin secretion in rats. These drugs could be promising agents for managing hyperinsulinism [59].
Non-medical treatment of HH
Frequent high-calorie carbohydrate feeds are important for avoiding or reducing the severity of hypoglycaemias. In addition, fish oil-derived polyunsaturated fatty acids (PUFA) have been used in a pilot study as a treatment adjunct for achieving greater glycaemic stability in children with diazoxide-responsive HI and the results are satisfactory. It has been suggested that PUFA possibly does not induce hyperglycaemia but reduces the risk of hypoglycaemia. PUFA have been reported to improve glycaemic control, thereby enabling the reduction of diazoxide dose or preventing second-line medical therapy. The mechanism of action remains obscure; however, stabilization of inappropriate electrical activity which is involved in HH pathophysiology has been hypothesised [68].
Surgical treatment of HH
Surgical treatment is indicated if a focal lesion is identified, if medical therapy fails to maintain normoglycaemia or if the patient’s adherence to treatment is not satisfactory. It is of major importance that the distinction between focal and diffuse lesions is made, so that unnecessary pancreatic resection is avoided and future development of diabetes mellitus is prevented.
Focal lesions may be cured by excision of the lesion (lesionectomy), unless the lesion is localised in the head of the pancreas, increasing the potential complexity of the surgery due to the risk of damaging the main pancreatic duct or the common bile duct. Therefore, the expertise of the surgeon is of critical importance and these operations should be held in centers with significant experience. Focal lesions in the body or tail of the pancreas are easily resected. Diffuse HI may be treated with subtotal/near-total pancreatectomy in patients with hypoglycaemia unresponsive to medical treatment. Residual hypoglycaemia, pancreatic exocrine insufficiency or iatrogenic diabetes mellitus are common complications [10]. In near-total pancreatectomy, the tail, body, uncinate process and part of the pancreatic head are resected. Up to 98% pancreatectomy is required so that recurrent hyperinsulinaemic hypoglycaemia, that may require repeat pancreatic resection, is less likely to occur [80]. Insulin-dependent diabetes mellitus develops in all HH patients treated by near-total pancreatectomy, and in a significant percentage, diabetes is present only few years after the surgery. It is of note that severe hypoglycaemia tends to be more common in patients with HI post-operatively as compared to patients with type 1 diabetes mellitus, particularly within the first year of insulin treatment [55]. Pancreatic enzyme replacement is often required after near-total pancreatectomy.
In conclusion, HH is the principal cause of transient or persistent hypoglycaemia in the neonatal and infancy period, resulting from inappropriate and unregulated insulin release from the β cells of the pancreas. Diazoxide is the mainstay of medical therapy. Methods of identifying focal disease include imaging techniques (18F-L-DOPA PET/CT scanning) and genetic studies. Surgery may be required in unresponsive cases of severe diffuse disease. It is imperative to identify children with the focal form of the disease, as management will be radically different and includes a limited pancreatectomy alone or with medical therapy. With the advances in medical therapy, more children are now managed medically and surgical treatment if preferred as a last resort if unresponsive to medical therapy. Surgical treatment should be undertaken after discussion with family and assessing its benefits and risks. HH carries a significant risk of complications, such as cognitive impairment, seizures and other neurological deficits, and can be life-threatening. This review aims to give an overview of the diagnosis, the pathogenesis, the associated genetic mechanisms, the clinical phenotype and the management options for this complex clinical entity, based on the latest data from the literature. Expectations have risen towards promising future genetic testing, imaging techniques and treatments that may enable earlier recognition of HH, more accurate differentiation between the two histological types, less invasive treatment approaches and fewer short-term and long-term complications.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormone
- ADP:
-
Adenosine diphosphate
- ATP:
-
Adenosine triphosphate
- CT:
-
Computed tomography
- EIF2S3:
-
Eukaryotic translation initiation factor 2 subunit gamma
- 18F-L-DOPA:
-
18-Fluoro-L-dihydroxyphenylalanine
- FOXA2:
-
Forkhead box A2
- GCK:
-
Glucokinase
- GH:
-
Growth hormone
- GLP-1:
-
Glucagon-like peptide-1
- GLUT2:
-
Glucose transporter 2
- GLUD1:
-
Glutamate dehydrogenase 1
- HA:
-
Hyperammonia
- HADH:
-
3-hyrdoxyacyl-coenzyme A dehydrogenase
- HH:
-
Hyperinsulinaemic hypoglycaemia
- HI:
-
Hyperinsulinism
- HK1:
-
Hexokinase 1
- HNF1A:
-
Homeobox A
- HNF4:
-
Hepatocyte nuclear factor 4-alpha
- IgG:
-
Immunoglobulin G
- KATP channels:
-
ATP-sensitive potassium channels
- Kir6.2:
-
Inward-rectifier potassium 6.2
- mTOR:
-
Mammalian target of rapamycin
- PET:
-
Positron emission tomography
- PGM1:
-
Phosphoglucomutase 1
- SLC16A1:
-
Solute carrier family 16 member 1
- SSTR:
-
Somatostatin receptor
- SUR1:
-
Sulfonylurea receptor 1
- TSH:
-
Thyroid-stimulating hormone
References
Ajala ON, Huffman DM, Ghobrial II (2016) Glucokinase mutation-a rare cause of recurrent hypoglycemia in adults: a case report and literature review. J Community Hosp Intern Med Perspect 6:32983
Amato LA, Quigley CA, Neville KA, Hameed S, Verge CF, Woodhead HJ, Walker JL (2015) Sirolimus treatment of severe congenital hyperinsulinism. Int J Pediatr Endocrinol 2015:123
Antwi K, Fani M, Heye T, Nicolas G, Rottenburger C, Kaul F, Merkle E, Zech CJ, Boll D, Vogt DR, Gloor B, Christ E, Wild D (2018) Comparison of glucagon-like peptide-1 receptor (GLP-1R) PET/CT, SPECT/CT and 3T MRI for the localization of occult insulinomas: evaluation of diagnostic accuracy in a prospective crossover imaging study. Eur J Nucl Med Mol Imaging 45:2318–2327
Arnoux JB, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V, Brunelle F, Fournet JC, Robert JJ, Aigrain Y, Bellanné-Chantelot C, de Lonlay P (2011) Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis 6:63
Ashcroft FM, Harrison DE, Ashcroft SJ (1984) Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature 312:446–448
Avatapalle HB, Banerjee I, Shah S, Pryce M, Nicholson J, Rigby L, Caine L, Didi M, Skae M, Ehtisham S et al (2013) Abnormal neurodevelopmental outcomes are common in children with transient congenital hyperinsulinism. Front Endocrinol (Lausanne) 4:60
Aynsley-Green A, Hussain K, Hall J, Saudubray JM, Nihoul-Fékété C, De Lonlay-Debeney P, Brunelle F, Otonkoski T, Thornton P, Lindley KJ (2000) Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed 82:F98–F107
Banerjee I, Avatapalle B, Padidela R, Stevens A, Cosgrove K, Clayton P, Dunne M (2013) Integrating genetic and imaging investigations into the clinical management of congenital hyperinsulinism. Clin Endocrinol 78:803–813
Baş F, Darendeliler F, Demirkol D, Bundak R, Saka N, Günöz H (1999) Successful therapy with calcium channel blocker (nifedipine) in persistent neonatal hyperinsulinemic hypoglycemia of infancy. J Pediatr Endocrinol Metab 12:873–878
Beltrand J, Caquard M, Arnoux J-B, Laborde K, Velho G, Verkarre V, Rahier J, Brunelle F, Nihoul-Fekete C, Saudubray JM, Robert JJ, de Lonlay P (2012) Glucose metabolism in 105 children and adolescents after pancreatectomy for congenital hyperinsulinism. Diabetes Care 35:198–203
Blomberg BA, Moghbel MC, Saboury B, Stanley CA, Alavi A (2013) The value of radiologic interventions and 18F-DOPA PET in diagnosing and localizing focal congenital hyperinsulinism: systematic review and meta-analysis. Mol Imaging Biol 15:97–105
Bruninig GJ (1990) Recent advances in hyperinsulinism and the pathogenesis of diabetes mellitus. Curr Opin Pediatr 2:758–765
Calabria AC, Li C, Gallagher PR, Stanley CA, De León DD (2012) GLP-1 receptor antagonist exendin-(9-39) elevates fasting blood glucose levels in congenital hyperinsulinism owing to inactivating mutations in the ATP-sensitive K+ channel. Diabetes 61:2585–2591
Cartier EA, Conti LR, Vandenberg CA, Shyng SL (2001) Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. PNAS 98:2882–2887
Christesen HB, Tribble ND, Molven A, Siddiqui J, Sandal T, Brusgaard K, Ellard S, Njolstad PR, Alm J, Brock Jacobsen B et al (2008) Activating glucokinase (GCK) mutations as a cause of medically responsive congenital hyperinsulinism: prevalence in children and characterisation of a novel GCK mutation. Eur J Endocrinol 159:27–34
de Lonlay P, Cornier-Daire V, Amiel J, Touati G, Goldenberg A, Fournet JC, Brunelle F, Nihoul-Fekete C, Rahier J, Junien C, Robert JJ, Saudubreay JM (2002) Facial appearance in persistent hyperinsulinemic hypoglycaemia. Am J Med Genet 111:130–133
Dehdashtian M (2014) Reversible pulmonary hypertension in an infant treated with diazoxide. APJMT 3:84–86
Demirbilek H, Hussain K (2017) Congenital hyperinsulinism: diagnosis and treatment update. J Clin Res Pediatr Endocrinol 9:69–87
Dunne MJ, Kane C, Shepherd RM, Sanchez JA, James RF, Johnson PR, Aynsley- Green A, Lu S, Clement JP IV, Lindley KJ et al (1997) Familial persistent hyperinsulinemic hypoglycemia of infancy and mutations in the sulfonylurea receptor. N Engl J Med 336:703–706
Durmaz E, Flanagan SE, Parlak M, Ellard S, Akcurin S, Bircan I (2014) A combination of nifedipine and octreotide treatment in an hyperinsulinemic hypoglycemic infant. J Clin Res Pediatr Endocrinol 6:119–121
Eichmann D, Hufnagel M, Quick P, Santer R (1999) Treatment of hyperinsulinaemic hypoglycaemia with nifedipine. Eur J Pediatr 158:204–206
Flanagan SE, Kapoor RR, Hussain K (2011) Genetics of congenital hyperinsulinemic hypoglycemia. Semin Pediatr Surg 20:13–17
Fournet JC, Junien C (2003) The genetics of neonatal hyperinsulinism. Horm Res 59:30–34
Glaser B, Hirsch HJ, Landau H (1993) Persistent hyper-insulinemic hypoglycemia of infancy: long-term octreotide treatment without pancreatectomy. J Pediatr 123:644–650
Glaser B, Thornton PS, Otonkoski T, Junien C (2000) The genetics of neonatal hyperinsulinism. Arch Dis Child 82:79–86
Gregory LC, Ferreira CB, Young-Baird SK, Williams HJ, Harakalova M, van Haaften G, Rahman SA, Gaston-Massuet C, Kelberman D, GOSgene, Qasim W, Camper SA, Dever TE, Shah P, Iain C.A.F., Robinson ICAF, Dattani MT (2019) Impaired EIF2S3 function associated with a novel phenotype of X-linked hypopituitarism with glucose dysregulation. EBioMedicine 42:470–480
Güemes M, Shah P, Silvera S, Morgan K, Gilbert C, Hinchey L, Hussain K (2017) Assessment of nifedipine therapy in hyperinsulinemic hypoglycemia due to mutations in the ABCC8 gene. J Clin Endocrinol Metab 102:822–830
Gutgold A, Gutgold DJ, Glaser B, Szalat A (2017) Diagnosis of ABCC8 congenital hyperinsulinism of infancy in a 20-year-old man evaluated for factitious hypoglycemia. J Clin Endocrinol Metab 102:345–349
Herrera A, Vajravelu ME, Givler S, Mitteer L, Avitabile CM, Lord K, De Leon DD (2018) Prevalence of adverse events in children with congenital hyperinsulinism treated with diazoxide. JCEM 103:4365–4372
Hussain K, Aynsley-Green A (2003) Hyperinsulinism in infancy: understanding the pathophysiology. Int J Biochem Cell Biol 35:1312–1317
Hussain K, Thornton PS, Otonkoski T, Aynsley-Green A (2004) Severe transient neonatal hyperinsulinism associated with hyperlactataemia in non-asphyxiated infants. J Pediatr Endocrinol Metab 17:203–209
Hussain K, Aynsley-Green A, Stanley CA (2004) Medications used in the treatment of hypoglycemia due to congenital hyperinsulinism of infancy (HI). Pediatr Endocrinol Rev 2:163–167
Hussain K, Clayton PT, Krywawych S, Chatziandreou I, Mills P, Ginbey DW, Geboers AJ, Berger R, van den Berg IE, Eaton S (2005) Hyperinsulinism of infancy associated with a novel splice site mutation in the SCHAD gene. J Pediatr 146:706–708
Inagaki N, Gonoi T, Clement JP 4th, Namba N, Inazawa J, Gonzalez G, Aguilar- Bryan L, Seino S, Bryan J (1995) Reconstitution of KATP: an inward rectifier subunit plus the sulphonylurea receptor. Science 270:1166–1170
Kane C, Shepherd RM, Squires PE, Johnson PR, James RF, Milla PJ, Aynsley-Green A, Lindley KJ, Dune MJ (1996) Loss of functional KATP channels in pancreatic β-cells causes persistent hyperinsulinemic hypoglycemia of infancy. Nat Med 2:1344–1347
Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, Ellard S, Hussain K (2008) Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes 57:1659–1663
Kapoor RR, James C, Flanagan SE, Ellard S, Eaton S, Hussain K (2009) 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency and hyper-insulinemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensitivity. JCEM 94:2221–2225
Kapoor RR, Flanagan SE, James C, Shield J, Ellard S, Hussain K (2009) Hyperinsulinaemic hypoglycaemia. Arch Dis Child 94:450–457
Khawash P, Hussain K, Flanagan SE, Chatterjee S, Basak D (2015) Nifedipine in congenital Hyperinsulinism - a case report. J Clin Res Pediatr Endocrinol 7:151–154
Laje P, States LJ, Zhuang H, Becker SA, Palladino AA, Stanley CA, Adzick NC (2013) Accuracy of PET/CT scan in the diagnosis of the focal form of congenital hyperinsulinism. J Pediatr Surg 48:388–393
Le Quan Sang KH, Arnoux JB, mamoune A, Saint-Martin C, Bellanne-Chantelot C, Valayannopoulos V, Brassier A, Kayirangwa H, Barbier V, Broissand C, Fabreguettes JR, Charron B, Thalabard JC, de Lonlay P (2012) Successful treatment of congenital hyperinsulinism with long-acting release octreotide. Eur J Endocrinol 166:333–339
Li C, Chen P, Palladino A, Narayan S, Russell LK, Sayed S, Xiong G, Chen J, Stokes D, Butt YM, Jones PM, Collins HW, Cohen NA, Cohen AS, Nissim I, Smith TJ, Strauss AW, Matschinsky FM, Bennett MJ, Stanley CA (2010) Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem 285:31806–31818
Lord K, De León DD (2013) Monogenic hyperinsulinemic hypoglycemia: current insights into the pathogenesis and management. Int J Pediatr Endocrinol 2013:3
Ludwig A, Enke S, Heindorf J, Empting S, Meissner T, Mohnike K (2018) Formal neurocognitive testing in 60 patients with congenital hyperinsulinism. Horm Res Paediatr 89:1–6
Matsuo M, Trapp S, Tanizawa Y, Kioka N, Amachi T, Oka Y, Ashcroft FM, Ueda K (2000) Functional analysis of a mutant sulfonylurea receptor, SUR1-R1420C, that is responsible for persistent hyperinsulinemic hypoglycemia of infancy. J Biol Chem 275:41184–41191
Mazor-Aronovitch K, Gillis D, Lobel D, Hirsch HJ, Pinhas-Hamiel O, Modan-Moses D, Glaser B, Landau H (2007) Long-term neurodevelopmental outcome in conservatively treated congenital hyperinsulinism. Eur J Endocrinol 157:491–497
Mohnike K, Blankenstein O, Pfuetzner A, Potzsch S, Schober E, Steiner S, Hardy OT, Grimberg A, van Waarde WM (2008) Long-term non-surgical therapy of severe persistent congenital hyperinsulinism with glucagon. Horm Res 70:59–64
Nestorowicz A, Inagaki N, Gonoi T, Schoor KP, Wilson BA, Glaser B, Landau H, Stanley CA, Thornton PS, Seino S, Permutt MA (1997) A nonsense mutation in the inward rectifier potassium channel gene, Kir6.2, is associated with familial hyperinsulinism. Diabetes 46:1743–1748
Neylon OM, Moran MM, Pellicano A, Nightingale M, O’Connell MA (2013) Successful subcutaneous glucagon use for persistent hypoglycaemia in congenital hyperinsulinism. J Pediatr Endocrinol Metab 26:1157–1161
Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, Kere J, Sipila I (2003) Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes 52:199–204
Otonkoski T, Näntö-Salonen K, Seppänen M, Veijola R, Huopio H, Hussain K, Tapanainen P, Eskola O, Parkkola R, Ekstrom K et al (2006) Noninvasive diagnosis of focal hyperinsulinism of infancy with [18F]-DOPA positron emission tomography. Diabetes 55:13–18
Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, Parton LE, Schuit F, Quintens R, Sipila I, Mayatepek E et al (2007) Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic β cells. Am J Hum Genet 81:467–474
Palladino AA, Bennett MJ, Stanley CA (2008) Hyperinsulinism in infancy and childhood: when an insulin level is not always enough. Clin Chem 54:256–263
Pallet N, Legendre C (2013) Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf 12:177–186
Pierro A, Nah SA (2011) Surgical management of congenital hyperinsulinism of infancy. Semin Pediatr Surg 20:50–53
Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, Ganguly A, Shyng SL, Stanley CA (2008) Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest 118:2877–2886
Rahier J, Guiot Y, Sempoux C (2000) Persistent hyperinsulinaemic hypoglycaemia of infancy: a heterogeneous syndrome unrelated to nesidioblastosis. Arch Dis Child Fetal Neonatal Ed 82:F108–F112
Rahman SA, Nessa A, Hussain K (2015) Molecular mechanisms of congenital hyperinsulinism. J Mol Endocrinol 54:119–129
Rico-Bautista E, Kusnetzow AK, Fowler ΜΑ, Athanacio J, Kredel TA, Zhao J, Wang S, Markinson S, Fei Zhu Y, Struthers RS, Betz SF, Crinetics Pharmaceuticals, San Diego, CA (2019) Selective nonpeptide somatostatin receptor subtype 5 (sst5) agonists suppress induced insulin secretion in pancreatic islets from both rats and healthy human donors. Presented in Endo 2019
Rozenkova K, Malikova J, Nessa A, Dusatkova L, Bjørkhaug L, Obermannova B, Dusatkova P, Kytnarova J, Aukrust I, Najmi LA, Rypackova B, Sumnik Z, Lebl J, Njølstad PR, Hussain K, Pruhova S (2015) High incidence of heterozygous ABCC8 and HNF1A mutations in Czech patients with congenital hyperinsulinism. J Clin Endocrinol Metab 100:E1540–E1549
Sakakibara A, Hashimoto Y, Kawakita R, Hosokawa Y, Nagahara K, Hasegawa Y, Hoshino S, Nagasaka H, Yorifuji T (2018) Diagnosis of congenital hyperinsulinism: biochemical profiles during hypoglycaemia. Pediatr Diabetes 19:259–264
Salomon-Estebanez M, Flanagan SE, Ellard S, Rigby L, Bowden L, Mohamed Z, Nicholson J, Skae M, Hall C, Craigie R, Padidela R, Murphy N, Randell T, Cosgrove KE, Dunne MJ, Banerjee I (2016) Conservatively treated Congenital Hyperinsulinism (CHI) due to K-ATP channel gene mutations: reducing severity over time. Orphanet J Rare Dis 11:163
Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, Suchi M, Taub R, Grimsby J, Matschinsky FM, Stanley CA (2009) Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes 58:1419–1427
Senniappan S, Alexandrescu S, Tatevian N, Shah P, Arya V, Flanagan S, Ellard S, Rampling D, Ashworth M, Brown R et al (2014) Sirolimus therapy in infants with severe hyperinsulinemic hypoglycemia. N Engl J Med 370:1131–1137
Shah P, Rahman SA, McElroy S, Gilbert C, Morgan K, Hinchey L, Senniappan S, Levy H, Amin R, Hussain K (2015) Use of long-acting Somatostatin analogue (Lanreotide) in an adolescent with Diazoxide-responsive congenital Hyperinsulinism and its psychological impact. Horm Res Paediatr 84:355–360
Shah P, Rahman SA, Demirbilek H, Güemes M, Hussain K (2016) Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol 5:729–742
Shanbag P, Pathak A, Vaidya M, Shahid SK (2002) Persistent hyperinsulinemic hypoglycemia of infancy-successful therapy with nifedipine. Indian J Pediatr 69:271–272
Skae M, Avatapalle HB, Banerjee I, Rigby L, Vail A, Foster P, Charalambous C, Bowden L, Padidela R, Patel L, Ehthsham S, Cosgrove KE, Dunne MJ, Clayton P (2014) Reduced glycemic variability in diazoxide-responsive children with congenital hyperinsulinism using supplemental omega-3-polyunsaturated fatty acids; a pilot trial with MaxEPAR. Front Endocrinol 5:31
Stanley CA (2004) Hyperinsulinism/hyperammonemia syndrome: insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Mol Genet Metab 81:S45–S51
Stanley CA (2016) Perspective on the genetics and diagnosis of congenital hyperinsulinism disorders. JCEM 101:815–826
Suprasongsin C, Suthutvoravut U, Mahachoklertwattana P, Preeyasombat C (1999) Combined raw cornstarch and nifedipine as an additional treatment in persistent hyperinsulinemic hypoglycemia of infancy. J Med Assoc Thail 82:S39–S42
Szymanowski M, Estebanez MS, Padidela R, Han B, Mosinska K, Stevens A, Damaj L, Pihan-Le Bars F, Lascouts E, Reynaud R et al (2016) mTOR inhibitors for the treatment of severe congenital hyperinsulinism: perspectives on limited therapeutic success. J Clin Endocrinol Metab 101:4719–4729
Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, Aguilar-Bryan L, Gagel RF, Bryan J (1995) Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 268:426–429
Thomas P, Ye Y, Lightner E (1996) Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum Mol Genet 5:1809–1812
Thornton PS, Alter CA, Katz LE, Baker L, Stanley CA (1993) Short- and long-term use of octreotide in the treatment of congenital hyperinsulinism. J Pediatr 123:637–643
Timlin MR, Black AB, Delaney HM, Matos RI, Percival CS (2017) Development of pulmonary hypertension during treatment with diazoxide: a case series and literature review. Pediatr Cardiol 38:1247–1250
Tornehave D, Kristensen P, Rorner J, Knudsen LB, Heller RS (2008) Expression of the GLP-1 receptor in mouse, rat, and human pancreas. J Histochem Cytochem 56:841–851
Verkarre V, Fournet JC, de Lonlay P, Gross-Morand MS, Devillers M, Rahier J, Brunelle F, Robert JJ, Nihoul-Fekete C, Saudubray JM et al (1998) Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinism in focal adenomatous hyperplasia. J Clin Invest 102:1286–1291
Welters A, Lerch C, Kummer S, Marquard J, Salgin B, Mayatepek E, Meissner T (2015) Long-term medical treatment in congenital hyperinsulinism: a descriptive analysis in a large cohort of patients from different clinical centers. Orphanet J Rare Dis 10:150
Welters A, Meissner T, Grulich-Henn J, Frohlich-Reiterer E, Warncke K, Mohnike K, Blankenstein O, Menzel U, Datz N, Bollow E, Holl RW (2017) Characterization of diabetes following pancreatic surgery in patients with congenital hyperinsulinism. Orphanet J Rare Dis 13:230
Wullschleger S, Loewith R, Oppliger W, Hall MN (2005) Molecular organization of target of rapamycin complex 2. J Biol Chem 280:30697–30704
Yang SB, Lee HY, Rapamycin DM, Tien AC, Rowson-Baldwin A, Shu YY, Jan YN, Jan LY (2012) Rapamycin induces glucose intolerance in mice by reducing islet mass, insulin content, and insulin sensitivity. J Mol Med (Berl) 90:575–585
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Communicated by Peter de Winter
Rights and permissions
About this article

Cite this article
Kostopoulou, E., Shah, P. Hyperinsulinaemic hypoglycaemia—an overview of a complex clinical condition. Eur J Pediatr 178, 1151–1160 (2019). https://doi.org/10.1007/s00431-019-03414-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-019-03414-8