
Abstract
Castleman disease (CD) is a rare poorly understood lymphoproliferative disorder. Pediatric onset CD has been reported before. However, most of them have benign unicentric pattern. Multicentric CD (MCD) is quite rare in children. Herein, we report a 13-year-old adolescent boy with MCD of the hyaline vascular variant presenting with pleural and pericardial effusion, which is an uncommon presentation.
Conclusion: MCD should be considered in the differential diagnosis of pleural and/or pericardial effusion with unexplained lymph nodes in children.
What is Known •Pediatric Castleman disease (CD) most commonly occurs in the unicentric form, which typically is asymptomatic and cured by lymph node excision. •The diagnosis of MCD can be difficult owing to the heterogeneity of presentation and potential for nonspecific multisystem involvement. |
What is New •A 13-year-old adolescent boy was diagnosed with MCD of the hyaline vascular variant presenting with pleural and pericardial effusion, which is an uncommon presentation. •In a pediatric patient with fever, pleural-pericardial effusion and multiple lymph nodes, MCD should be considered in differantial diagnosis. |
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Castleman disease (CD) is a rare disease characterized by a massive growth of lymphoid tissue of unknown etiology. It was first defined by Dr. Benjamin Castleman in 1956 as hyperplasia of lymphoid follicles with or without germinal center formation and marked capillary proliferation with endothelial hyperplasia [1]. CD is classified as hyaline vascular, plasma cell, and mixed type according to histology or as localized or multicentric disease based on localization. In children, CD commonly presents in abdomen, chest or neck, whereas it usually (70 %) occurs in the mediastinum in adults. It may be associated with other malignancies, including Kaposi sarcoma, non-Hodgkin lymphoma, Hodgkin lymphoma, and POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome [2, 3, 7].
Herein, we report a 13-year-old adolescent boy with MCD of the hyaline vascular variant presenting with pleural and pericardial effusion, which is an uncommon presentation.
Case report
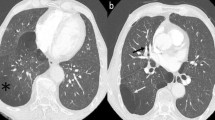
A 13-year-old boy was admitted to our pediatric clinic with chest pain, fever, and dyspnea. He had been investigated in another clinic for pericardial effusion and had received antibiotic and non-steroidal anti-inflammatory drugs for 10 days. On physical examination, he had 3/6 systolic murmur at the mesocardiac region. Laboratory investigation revealed the following findings; hemoglobin 9.7 gr/dl, white blood cell 22,800 mm/3, and platelets 319,000/mm3. Peripheral blood smear was normal with polymorph nuclear cell dominancy. Erythyrocyte sedimentation rate (ESR) was high with 69 mm/h; C-reactive protein (CRP) was high with 12.6 mg/dl (normal range, 0–0.8). Biochemical parameters were normal. Pleural and pericardial effusions were detected on chest x-ray and echocardiography. Abdominal ultrasonography revealed mild hepatosplenomegaly. Viral markers were negative (hepatic markers, human immunodeficiency virus, herpes simplex virus, Epstein-Barr virus, and cytomegalovirus). Anti-nuclear antibody, anti-double stranded DNA, and anti-nuclear cytoplasmic antibody were negative. Complement levels (C3 and C4) were within normal ranges. Abdominal and chest tomography showed multiple enlarged lymph nodes (the largest size 30 × 25 mm) (Fig. 1) in the posterior region of the stomach and para aortic region and on superior-anterior mediastinal, cardiophrenic recesses and axillary region with a bilateral pleural effusion. Bone marrow aspiration evaluation and flow cytometry were normal. A diagnostic thoracentesis was performed. Pleural fluid was compatible with exudate. Analyses of pleural fluid for bacterial, fungal agents and tuberculosis were negative. Naproxen sodium (15 mg/kg/day) and empiric ceftriaxone therapy were started. Because the tuberculin skin test was reactive (23 mm) and there was a family history of tuberculosis, anti-tuberculosis (anti-tbc) treatment was started (isoniazid, pyrazinamide, rifampin, and ethambutol). Since Familial Mediterranean fever (FMF) is very prevalent in our region, genetic testing for FMF was performed. Although FMF DNA revealed V726A heterozygous mutation, with clinical findings and persistent high acute phase reactant levels, colchicine treatment was added. Afterwards, his pleural and pericardial effusion started to dissolve. He was discharged with colchicine and anti-tbc therapy. Two weeks later, he was again presented with dyspnea, chest pain, and fever. He was hospitalized three times in the following 2-month period with the similar clinical and laboratory findings. A lymph node biopsy was performed to help for the diagnosis. Histopathological examination of the intraabdominal lymph node specimens revealed lymphoid follicles with marked vascular proliferation and hyalinization of abnormal germinal centers. There was concentric layering of lymphocytes in an onion-skin appearance. These findings corresponded to CD of hyaline vascular type (Fig. 2). After the diagnosis of CD was established, interleukin (IL) 6 level and human herpesvirus 8 (HHV-8) were studied. IL-6 level was slightly high (4.4 pg/ml, normal upper limit 4 pg/ml), and (HHV-8) was negative. Prednisolone 2 mg/kg/day was started. Anti-tbc regimen was continued with isoniazid and rifampin and stopped after 9 months. Three months after initiation of steroid therapy, he had no complaints, his chest x-ray was normal, and acute phase reactants returned to normal levels. Abdominal ultrasonography revealed multiple milimetric lymph nodes (largest one is 14 × 7 mm). Colchicine therapy was stopped. 5 mg/day prednisolone therapy was continued. At the time of the report, 1 year has passed since the diagnosis and he remains asymptomatic with normal levels of acute phase reactants.

Enlarged lymph node (30 × 25 mm) on posterior region of stomach and para aortic region

Histology showed folliculer centers exhibiting prominent hyalinized vessels, surrounded by concentric layers of lymphocytes (onion-skin pattern)
Discussion
Childhood MCD is a rare and unexplained lymphoproliferative disorder. The pathogenesis of MCD is poorly understood. IL-6 has multiple effects such as proliferation of T and B lymphocytes and synthesis of acute phase reactants in liver. So, it supports hematopoiesis and dysregulation of immune responses. IL-6 has a key factor in the pathogenesis of CD. Most of the systemic symptoms seen in CD are due to the proinflammatory effects of IL-6 [10]. Our patient’s IL-6 level was only slightly elevated.
Unlike UCD, MCD is more commonly associated with HHV-8 and human immunodeficiency virus infection. Most patients present with nonspecific symptoms suggestive of an inflammatory illness. Fever is nearly universal, and most patients present with night sweats, weight loss, weakness or fatigue, and lymphadenopathy. A subset of patients have edema, body cavity effusions, skin, neurologic findings, and renal symptoms. Typical laboratory abnormalities in MCD include nearly universal anemia, thrombocytosis, hypoalbuminemia, polyclonal hypergammaglobulinemia, and an elevated erythrocyte sedimentation rate. Other findings include elevated serum levels of IL-6, vascular endothelial growth factor, lactate dehydrogenase, and CRP. The diagnosis is confirmed upon pathologic review of a biopsy of involved tissue, typically an excisional biopsy of a lymph node. The most abnormal node is selected for biopsy. The diagnosis of MCD requires the clinical features of active disease described above plus pathologic confirmation on biopsy [1, 2, 4].
Since CD is a rare condition in children, we had not considered this disease in our patient at first. He had fever, history of recurrent pleural and pericardial effusion, and high acute phase reactant levels. This raised our concerns about an infectious, oncologic, or rheumatologic disorder. His infectious work-ups were negative, and he did not respond to antibiotic and anti-tuberculosis therapy. On the other hand, colchicine therapy was added with the suspicion of FMF since it is frequent in our country. However, after 3 months of therapy, he had no improvement. Our patient’s persistent lymphadenopathies were the clue for our diagnosis. And after the biopsy, diagnosis of hyaline vascular variant of CD was established. We diagnosed MCD in our patient with the systemic symptoms plus multiple lymph node involvement.
Pleural and/or pericardial effusion is an unusual presentation for MCD. A child who had MCD presented with pleural effusion has been reported by Smith et al. But, this patient had a more severe disease onset with anasarca, abdominal ascites, and thrombocytopenia apart from our patient [8]. Recently, Takai et al. reported a new disease concept, TAFRO syndrome, named from thrombocytopenia, anasarca, fever, reticulin fibrosis of the bone marrow, and organomegaly [4]. Furthermore, Kojima et al. reported Japanese MCD cases with effusion and thrombocytopenia (Castleman-Kojima disease) [5]. A meeting report was published as the result of the discussions. The purpose of this meeting was to make a consensus document regarding the current understanding of this disease, and the findings suggest that this disease represents a novel clinical entity belonging to systemic inflammatory disorders with a background of immunological abnormality beyond the ordinal spectrum of MCD [4, 6]. Although our patient had pleural and pericardial effusion and fever, he did not have other features of TAFRO syndrome. Therefore, we did not consider him with this new disorder.
There is no standard consensus for the treatment of MCD. Since it is a rare and heterogeneous disease, it is hard to make randomized clinical trials to compare the therapy options. Reported therapies for children with MCD have included steroids, chemotheratic agents, surgery, intravenous immunoglobulin, interferone-alfa, tocilizumab (IL-6 receptor antagonist), and rituximab (monoclonal antibody to CD20) [9]. Our patient responded very well to prednisolone therapy and had no need for further therapies.
In conclusion, MCD should be considered in the differential diagnosis of pleural and/or pericardial effusion in children with unexplained multiple lymph nodes.
Abbreviations
- Anti-tbc:
-
anti-tuberculosis
- CD:
-
Castleman disease
- CRP:
-
C-reactive protein
- ESR:
-
erythrocyte sedimentation rate
- FMF:
-
Familial Mediterranean fever
- HHV-8:
-
human herpesvirus 8
- IL-6:
-
interleukin 6
- MCD:
-
multicentric Castleman disease
- TAFRO:
-
Thrombocytopenia, anasarca, fever, reticulin fibrosis of the bone marrow and organomegaly
- UCD:
-
unicentric Castleman disease
References
Castleman B, Towne VW (1954) Case records of the Massachusetts General Hospital; weekly clinicopathological exercises; founded by Richard C. Cabot N Engl J Med 251(10):396–400
Dispenzieri A, Armitage JO, Loe MJ, Geyer SM, Allred J, Camoriano JK, Menke DM, Weisenburger DD, Ristow K, Dogan A, Habermann TM (2012) The clinical spectrum of Castleman’s disease. Am J Hematol 87(11):997–1002
Farruggia P, Trizzino A, Scibetta N, Cecchetto G, Guerrieri P, D’Amore ES, D’Angelo P (2011) Castleman's disease in childhood: report of three cases and review ofthe literature. Ital J Pediatr. 20;37:50
Kawabata H, Takai K, Kojima M, Nakamura N, Aoki S, Nakamura S, Kinoshita T, Masaki Y (2012) Castleman-Kojima disease (TAFRO syndrome): a novel systemic inflammatory disease characterized by a constellation of symptoms, namely, thrombocytopenia, Ascites (anasarca), microcytic anemia, myelofibrosis, renal dysfunction, and organomegaly : a status report and summary of Fukushima and Nagoya meetings. J Clin Exp Hematop 53(1):57–61
Kojima M, Nakamura N, Tsukamoto N, Yokohama A, Itoh H, Kobayashi S, Kashimura M, Masawa N, Nakamura S (2011) Multicentric Castleman’s disease representing effusion at initial clinical presentation: clinicopathological study of seven cases. Lupus 20(1):44–50
Masaki Y, Nakajima A, Iwao H, Kurose N, Sato T, Nakamura T, Miki M, Sakai T, Kawanami T, Sawaki T, Fujita Y, Tanaka M, Fukushima T, Okazaki T, Umehara H (2013) Japanese variant of multicentric castleman’s disease associated with serositis and thrombocytopenia—a report of two cases: is TAFRO syndrome (Castleman-Kojima disease) a distinct clinicopathological entity? J Clin Exp Hematop 53(1):79–85
Parez N, Bader-Meunier B, Roy CC, Dommergues JP (1999) Paediatric Castleman disease: report of seven cases and review of the literature. Eur J Pediatr 158(8):631–7
Smith C, Lee-Miller C, Dishop MK, Cost C, Wang M, Asturias EJ (2014) Multicentric Castleman disease presenting with fever. J Pediatr 165(6):1261–5
Soumerai JD, Sohani AR, Abramson JS (2014) Diagnosis and management of Castleman disease. Cancer Control 21(4):266–78
Van Rhee F, Fayad L, Voorhees P, Furman R, Lonial S, Borghaei H, Sokol L, Crawford J, Cornfeld M, Qi M, Qin X, Herring J, Casper C, Kurzrock R (2010) Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol 28(23):3701–8
Authors’ contribution
AOA, OB, DO, and NC clinically managed the patient. UH was responsible for the assesment of pathological analysis. AOA and OB wrote the manuscript. TS and NC critically reviewed the manuscript. All the authors approved the final copy of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
None declared.
Conflict of interest
The authors declare that they have no (financial) conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Communicated by David Nadal
Rights and permissions
About this article

Cite this article
Akman, A.O., Basaran, O., Ozyoruk, D. et al. Atypical presentation of multicentric Castleman disease in a pediatric patient: pleural and pericardial effusion. Eur J Pediatr 175, 873–876 (2016). https://doi.org/10.1007/s00431-015-2674-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-015-2674-6