
Abstract
Preconditioning exercise prior to stroke exerts neuroprotection, which is an endogenous strategy that leads the brain cells to express several intrinsic factors and inhibits their apoptosis. However, it is unclear how long these benefits last after exercise cessation. The aim of this study was to investigate the effects of detraining on preconditioning exercise-induced neuroprotective potential after stroke. Rats were trained using a treadmill for aerobic exercise 5 days each week for 3 weeks, and their neuroprotective effects were examined until 3 weeks after exercise cessation. Stroke was induced by 60 min of left middle cerebral artery occlusion at 3 days, 1, 2, and 3 weeks after exercise cessation. Infarct volume, neurological deficits, sensorimotor function, expression levels of brain-derived neurotrophic factor (BDNF), hypoxia-induced factor-1α (HIF-1α), glial fibrillary acidic protein (GFAP), and P2X7 receptors, and apoptosis activity were examined using immunohistochemical and western blot analyses. Preconditioning exercise significantly reduced infarct volume and ameliorated sensorimotor function after stroke, and its beneficial effects were observed until 2 weeks after exercise cessation. The expression level of BDNF in the ischemic brain was significantly upregulated at 3 days after exercise cessation; however, the expression levels of HIF-1α, GFAP, and P2X7 receptor were significantly increased until 2 weeks after exercise cessation; thereby, significant anti-apoptotic effects were lost at 3 weeks of detraining. Our findings suggest that preconditioning exercise-induced neuroprotective potential may be lost shortly after exercise cessation. Neuroprotection through intrinsic protective factors, such as BDNF and HIF-1α, may provide different neuroprotective mechanisms in a time-dependent manner during detraining.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Physical exercise is not only beneficial in improving cardiovascular health but may also help enhance neuroplasticity and slow down cognitive decline in the brain (Tolppanen et al. 2015). Exercise, which can be considered a mild stressor, is a prototypical preconditioning stimulus that has beneficial effects on cognitive function and brain health (Ploughman et al. 2015; Islam et al. 2017). Therefore, preconditioning exercise is known as brain tolerance, which is an endogenous process that provides robust neuroprotection after stroke (Zhang et al. 2011). Previous clinical studies indicated that patients with stroke who reported regular exercise before stroke onset had milder strokes and better functional outcomes after stroke (Kraup et al. 2008; Stroud et al. 2009; Deplanque et al. 2012; Ricciardi et al. 2014). Furthermore, several animal studies have demonstrated the beneficial effects of preconditioning exercise as a prophylactic exercise on stroke-induced brain injury (Zhang et al. 2011, 2014; Otsuka et al. 2016). Therefore, preconditioning exercise is an endogenous strategy that leads to the expression of several intrinsic factors to acquire tolerance in brain cells and involves inhibition of the apoptosis pathway or promotion of the cell survival pathway, to defend themselves from subsequent damage. To the best of our knowledge, in contrast to the reports on endogenous neuroprotective potential due to regular exercise training in clinical settings and animal experiments, the effects of neuroprotection on exercise cessation or detraining are less understood.
Detraining is defined as the complete or partial loss of training-induced physiological, anatomical, and performance adaptation as a consequence of training cessation or reduction (Mujika and Padilla. 2000). Regarding the influence of detraining on myocardial function or brain function due to exercise training, Bocalini et al. (2010) demonstrated that enhancement of myocardial contractility after an 8-week swimming training program was completely lost after a short period of detraining in rats. Waring et al. (2015) demonstrated that cardiac adaptation, produced after 4 weeks of intensity-controlled exercise training, is lost after a similar period of detraining. In addition, Radak et al. (2006) showed that the levels of nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) increased in the rat brain as a result of 8 weeks of swimming training, but decreased after 6 weeks of detraining. Furthermore, they showed that training-induced memory improvement disappeared with detraining (Radak et al. 2006). The reversibility of the exercise-induced adaptive process is well-known for its cardiovascular and brain functions. However, the reversibility of preconditioning exercise-induced neuroprotective potential after stroke is unclear. It would be clinically meaningful in the field of brain health promotion and rehabilitation to clarify how long neuroprotective effects last after exercise cessation. Therefore, we focused on the effects of exercise cessation or detraining on the neuroprotective effects due to preconditioning exercise-induced brain tolerance after stroke.
There are many potential neuroprotective mechanisms associated with preconditioning exercise (Kramer et al. 2019). A previous review article reported that preconditioning exercise could exert neuroprotective effects against acute stroke by promoting angiogenesis, increasing neurotrophic factors, mediating inflammatory responses, and inhibiting neuronal apoptosis (Zhang et al. 2011; Sakakima et al., 2019). In addition, intrinsic factors such as cytokines and transcriptional factors are present intracellularly and can protect cells from injury, including cerebral ischemia (Dong et al. 2010). Preconditioning exercise can enhance BDNF after stroke, which is widely distributed throughout the brain and plays a critical role in the neural repair process after stroke (Ding et al. 2004; Ploughman et al. 2009; Sakakima et al. 2012; Otsuka et al. 2016). BDNF expression increases in the surrounding lesion after stroke, which may have neuroprotective functions (Otsuka et al. 2016). In addition, preconditioning exercise induces an increase in intrinsic protective factors such as hypoxia-induced factor-1α (HIF-1α), which is a transcriptional factor implicated in the hypoxic or ischemic brain (Yang et al. 2018; Otsuka et al. 2019). Hirayama et al. (2015) demonstrated that astrocyte-derived HIF-1α which is involved in the P2X7 receptor, an ATP-gated cation channel, mediates ischemic tolerance in the late phase after ischemic preconditioning. In addition, they demonstrated that a P2X7 receptor-dependent neuroprotective mechanism could allow astrocytes to cause long-lasting HIF-1α expression, thereby leading to efficient induction of ischemic tolerance (Hirayama and Koizumi. 2017). Therefore, this study focused on the neuroprotective mechanism of P2X7 receptor-mediated upregulation of HIF-1α in astrocytes because this mechanism is long-lasting owing to ischemic preconditioning. However, the influence of detraining on the expression patterns of these protective factors induced by preconditioning exercise remains unclear. Therefore, we examined the neuroprotective mechanisms of intrinsic protective factors, such as BDNF and HIF-1α, in a time-dependent manner.
The purpose of the present study was to investigate the duration of the neuroprotective effect after exercise cessation. Therefore, we examined the neuroprotective potential of detraining on brain damage and improvement of sensorimotor function, as well as the expression levels of protective factors after stroke.
Materials and methods
Animals
A total of 52 male 7-weeks-old Sprague–Dawley rats (mean body weight 272.3 ± 19.0 g) were obtained from CLEA Japan, Inc. (Shizuoka, Japan) animal supply. Efforts have been made to reduce the number of animals used. The rats were pair-housed under temperature-controlled conditions (23.0 ± 1.0 ℃) on a 12-h light/dark cycle, with food and water available ad libitum. The experimental protocol was approved by the Ethics Board of the Institute of Experimental Animal Science of Kagoshima University.
Preconditioning exercise (PE)
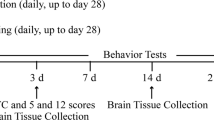
The experimental design is illustrated in Fig. 1. Rats were trained using a motor-driven treadmill (MK-680, MUROMACHI KIKAI CO., LTD, Japan) for 3 days (familiarization: running speed, 15–25 m/min; running duration, 10 min/day). After familiarization, the rats were randomly divided into eight groups: no exercise training and ischemia (No-Ex, n = 9), detrained 3 days after exercise and ischemia (n = 9), detrained 1 week after exercise and ischemia (n = 6), detrained 2 weeks after exercise and ischemia (n = 5), detrained 3 weeks after exercise and ischemia (n = 5), detrained 3 days after exercise and no ischemia (n = 5), detrained 2 weeks after exercise and no ischemia (n = 5), and detrained 3 weeks after exercise and no ischemia (n = 5). In addition, three normal rat brains were examined as intact control (normal control). Aerobic preconditioning exercise was performed 5 days a week for 3 weeks (running speed, 25 m/min; running duration, 30 min/day), as described previously (Otsuka et al. 2016, 2019). The rats in the No-Ex group ran freely in their cages for 3 weeks. Before the middle cerebral artery occlusion (MCAO) procedure, exercise was performed at room temperature during the daytime. Body weight was measured periodically to monitor the stress induced by treadmill running. It decreased until 3 days after MCAO; however, it significantly increased in all groups. The body weight of the Ex group (342.2 ± 38.9 g, mean ± standard deviation) was significantly increased compared to that of the No-Ex group (303.9 ± 22.3 g) at 3 weeks after MCAO. All rats in each group ran freely in their cages after cessation of exercise.

Experimental protocols. Schematic showing the timeline after exercise cessation. The rat in the No-Ex group only ran freely in their cages for 3 weeks, and stroke was induced by left MCAO (a No-Ex + MCAO group, n = 9). Preconditioning exercise (PE) groups were subjected to treadmill exercise as anerobic exercise for 3 weeks b. To determine the effects of detraining after ischemia, stroke was induced at 3 days (n = 9), 1 week (n = 6), 2 weeks (n = 5) and 3 weeks (n = 5) after PE cessation (Ex + MCAO group). The infarct volume, neurological deficits, and sensorimotor function were examined 2 days after MCAO a, b. In addition, to examine the effects of detraining and no ischemia, the rats were sacrificed at 3 days, 2 weeks, and 3 weeks after PE cessation (C Ex + No-MCAO group, n = 5 in each group)
Middle cerebral artery occlusion (MCAO) model
Rats were anesthetized using a combination of 0.3 mg/kg of medetomidine, 2.0 mg/kg of midazolam, and 2.5 mg/kg of butorphanol intraperitoneally. Rectal temperature was monitored throughout the surgical procedure and maintained at 37 ℃ using a heating blanket (KN-474, NATSUME, Tokyo, Japan). Stroke was induced by a 60-min left MCAO using an intraluminal filament, as described previously (Sakakima et al. 2012; Otsuka et al. 2016); thereafter, reperfusion was established by withdrawing the filament, and 2 days later, the rats were sacrificed. The brain, including the ischemic region, was analyzed histologically and immunohistochemically.
Evaluation of ischemic infarct
Rats were deeply anesthetized with sodium pentobarbital and transcardially perfused with physiological saline before decapitation. The brain was removed and cut into seven 2-mm-thick coronal sections from the frontal tip using a brain slicer. The slices were then immersed in a 1% solution of TTC in phosphate-buffered saline (PBS, pH 7.4) at 37 ℃ for 15 min. After staining, the sections of each group (n = 5–6) were scanned to determine the ischemic infarct volume. The infarctions were measured using Scion Image software BETA 4.0.3 (Scion Corp, Frederick, MD). The total infarct area (mm3) was multiplied by the thickness of the brain sections to obtain the infarct volume. The quantitative analysis of infarct volume was performed by two individuals and the mean infarct volume in each animal was analyzed.
Evaluation of neurological scores, motor behavior, locomotor function, and sensorimotor function in MCAO mice
Animals in each group (n = 5–6) were evaluated for neurological scores, sensorimotor dysfunction, and motor function using the beam-walking test, rotarod task, and sticky tape-removal test, at 2 days after MCAO. After acclimatization with the beam walking and rotarod tasks, all the animals were evaluated. A neurological grading system with a 5-point scale (0–4) was used as described previously (Sakakima et al. 2012; Otsuka et al. 2016). In the motor behavior test, the rats were examined using a beam-walking task, which was graded with a 6-point score (0–5), as previously described (Otsuka et al. 2016). For the motor function and balance test, rats were examined using a rotarod task (MK-670, MUROMACHI KIKAI CO, LTD, Japan), as described previously (Otsuka et al. 2016). The rotation speed was increased from 0 to 25 rpm in increments of 2.5 rpm every 6 s. The trial ended if the animal fell-off the cylinder. Each animal was given three trials. The best latency to fall (in seconds) for each animal was used in the analysis. Sensorimotor dysfunction was assessed using an adhesive sticky tape removal test, as described previously (Otsuka et al. 2016). Square sticky labels were used as ipsilateral tactile stimuli occupying the palmar surface of the right forepaw. The time to remove the label from the forelimbs was recorded in two trials for each forepaw. The better-recorded time in the two trials was used for the analysis. The maximum time required to remove labels was 120 s. Quantitative analysis of all evaluations was performed and checked by two individuals.
Histology and immunohistochemistry
After TTC staining, brains were immersed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) at 4 ℃ overnight. Rat brains that did not cause cerebral infarction were cut into seven 2-mm-thick coronal sections from the frontal tip using a brain slicer and immersed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) at 4 ℃ overnight without TTC staining. After fixation, the tissues (# 3–5 out of seven consecutive TTC sections from the cranial to the caudate region) were processed for histology and immunohistochemistry. The paraffin-embedded coronal brain sections (4-µm thick) were cut using a microtome (TU-213, YAMAMOTO KOHKI Industrial Co., LTD. Japan), affixed to APS adhesive slide glass (MATSUNAMI GLASS IND., LTD. Japan), and stained with hematoxylin and eosin for histological evaluation after MCAO.
The coronal brain sections were stained with the following antibodies: rabbit anti-BDNF antibody (Santa Cruz, CA, USA; sc-20981), rabbit anti-B-cell lymphoma 2-associated X protein (Bax; a marker of pro-apoptotic protein) antibody (Abcam plc, Cambridge, UK; ab32503), and rabbit anti-activated caspase-3 (a marker of apoptotic activity) antibody (Santa Cruz CA, USA; sc-7148). After deparaffinization and rehydration, endogenous peroxidase was blocked with methanol containing 0.9% hydrogen peroxide for 20 min. The sections were rinsed with phosphate-buffered saline (PBS, pH 7.6) 3 times for 10 min each and blocked with 10% skim milk in PBS for 20 min. These sections were individually incubated at 4 ℃ overnight with the following antibodies: rabbit anti-BDNF antibody (1: 50), rabbit anti-Bax antibody (1: 100), and rabbit anti-Caspase-3 antibody (1: 1000). Subsequently, the sections were washed in PBS three times for 10 min each and then incubated with goat anti-rabbit IgG conjugated to a peroxidase-labeled dextran polymer (EnVision; Dako, CA, USA) for 60 min. After the sections were rinsed with PBS, immunoreactivity was visualized using diaminobenzidine staining.
Semi-quantitative analysis of immunolabeled areas
The areas containing cells positive for BDNF, Bax, and activated Caspase-3 were semi-quantitatively measured in coronal sections (three out of seven consecutive TTC sections from the cranial to the caudate region, Fig. 2A) using Image J software 1.46r (NIH, USA). Two areas in the motor cortex (0.36 mm2 of each area) surrounding the lesion of each immunostained section were imaged at ×20 magnification using a microscope and a camera (n = 5–6, Fig. 2C). We calculated the mean ratio of each immunolabeled area (immunolabeled area/total area) from the two areas of the motor cortex. Semi-quantitative analysis of each immunolabeled area was performed by three individuals who were blinded to the treatment groups.

Representative TTC stained sections of the ischemic brain a in No-Ex + MCAO group (n = 6) and 3 days (3 D, n = 6), 1 week (1 W, n = 6), 2 weeks (2 W, n = 5), and 3 weeks (3 W, n = 5) of detraining + MCAO groups. HE staining shows the cerebral section of three out of seven consecutive TTC section in 3 days of detraining + MCAO b. Box–whisker plots represent that the infarct volume is significantly decreased until 2 W of detraining, but no significant reduction was observed in infarct volume at 3 W after exercise cessation c. The infarct volume at 3 W after exercise cessation was significantly increased compared to that of the 1 W of detraining. Two rectangles a, b in the motor cortex of the ischemic penumbra surrounding the lesion (*) were used for immunohistochemical analysis. *p < 0.05. Scale bar = 2 mm
Western blotting
Immunoblotting was performed to detect protein expression, as described previously (Otsuka et al. 2019). BDNF, hypoxia-induced factor-1α (HIF-1α), Bax, Caspase-3, glial fibrillary acidic protein (GFAP, a marker of activated astrocytes), and P2RX7 (a marker of P2X7 receptor, which is ATP, acts as a ligand-gated ion channel) were examined in the ipsilateral brain (cortex and striatum), including ischemic regions (n = 3 for each group). A previous report showed that TTC staining did not constrict quantitative gene and protein analyses (Kramer et al. 2010). Therefore, the ipsilateral brain after MCAO at 2 and 3 weeks of detraining was excised from the 2-mm-thick coronal sections of # 1, 2, and 6 out of seven consecutive TTC sections from the cranial to the caudate region (Fig. 2A). We analyzed the brains that included the motor cortex surrounding the ischemic regions, although the brain tissue after MCAO at 2 and 3 weeks of detraining did not correspond with the site of semi-quantitative analysis of immunohistochemistry. The protein level in the brain, which does not cause cerebral infarction, was analyzed in the left hemisphere (n = 3 for each group).
Briefly, the brain was dissected on ice and homogenized in T-Per reagent (Pierce Protein Research Products, 78,510). Approximately 12 µg of protein in each sample was loaded in a 4–20% mini-protean precast gel (Bio-Rad, USA) and subsequently transferred to a nitrocellulose membrane. After blocking with Tris-buffered saline/Tween 20 buffer (0.01 M Tris–HCl, pH 7.5, 0.15 M NaCl, and 0.05% Tween 20) containing 3% skim milk for 1 h at 25 ℃, the membrane was incubated with a primary antibody overnight at 4 ℃, followed by a secondary horseradish peroxidase-labeled antibody. Detection was performed using the EzWestlumi plus detection system (ATTO, Tokyo, Japan). The following antibodies were used: rabbit anti-BDNF (1: 1000; Abcam plc, Cambridge, UK; ab108319), mouse anti-HIF-1α antibody (1: 400; Abcam plc, Cambridge, UK; H1alpha67 ab1), rabbit anti-Bax antibody (1: 5000; Abcam plc, Cambridge, UK; ab32503), rabbit anti-GFAP antibody (1: 1000; Cosmo Bio Co., Ltd. Tokyo; R01003), rabbit anti-P2RX7 antibody (1: 1000; Abcam plc, Cambridge, UK; ab109054), rabbit anti-caspase-3 antibody (1: 1000; Santa Cruz CA, USA; sc-7148), and mouse anti-α-tubulin antibody (1: 2000; Proteintech, USA; 66,031-1 g). Protein bands were visualized with chemical luminescence (WSE-6100 LuminoGraph I, ATTO, Tokyo, Japan) and measured using Image J 1.46r software (NIH, USA). α-tubulin was used as an internal loading control.
Statistical analysis
Statistical analyses were performed with parametric or non-parametric tests after the Shapiro–Wilk test. The Kruskal–Wallis test was used to analyze the neurological and beam-walking scores. An independent sample Student’s t test was used for the between-group analysis. The change in body weight before and after preconditioning exercise was analyzed using a paired t test. The infarct volume, rotarod test, sticky tape removal latency, and expressions of BDNF, HIF-1α, Bax, GFAP, P2RX7, and caspase-3 were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc honestly significant difference test for multiple comparisons. The infarct volume, and neurological and sensorimotor function were represented by box-whicker plots. Semi-quantitative analysis of immunohistochemistry and western blotting are represented by a bar graph because of the small sample size. Data are expressed as the mean ± standard error (SE). The threshold for statistical significance was set at p < 0.05. All data were analyzed using SPSS version 24 (IBM Corp., Armonk, NY, USA).
Results
Preconditioning exercise reduced infarct volume after stroke and its beneficial effect was observed until 2 weeks of detraining.
First, we examined the duration of preconditioning exercise-induced neuroprotective effects in ischemic stroke after exercise cessation. Extensive brain infarction was observed in the left hemisphere after MCAO (Fig. 2A, B). Quantitative analyses showed that the brain infarction volumes at 3 days (234.3 ± 47.1 mm3), 1 week (175.8 ± 42.1 mm3), and 2 weeks (188.8 ± 52.3 mm3) after exercise cessation were significantly decreased compared to those of the No-Ex groups (446.8 ± 52.5 mm3, p < 0.05, Fig. 2C). However, no significant reduction was observed in infarct volume at 3 weeks after exercise cessation (378.8 ± 24.9 mm3, Fig. 2C).
Preconditioning exercise ameliorated neurological and sensorimotor function after MCAO and its beneficial effects were lost after 3 weeks of detraining.
Second, we examined the duration of preconditioning exercise-induced beneficial effects such as improvement of neurological and sensorimotor function in ischemic stroke after exercise cessation. Animals in each group were evaluated for neurological and sensorimotor function after MCAO (Fig. 3). The neurological and sensorimotor dysfunction at 3 days, 1 week, and 2 weeks after exercise cessation significantly improved compared to those in the No-Ex and 3-week groups (p < 0.05). In particular, the sticky tape removal task was significantly improved at 3 days and 1 week after exercise cessation compared to those of No-Ex and 3 weeks after exercise cessation (Fig. 3B). The beam-waking score and waking time in the rotarod task were significantly improved 3 days after exercise cessation compared with those of No-Ex and 3 weeks (Fig. 3C, D). The beam-walking score at 3 weeks after exercise cessation was significantly lower than that at 3 days, 1 week, and 2 weeks. Interestingly, no significant difference was observed between No-Ex and 3 weeks after exercise cessation in term of neurological deficits and sensorimotor dysfunction. Furthermore, improvements in neurological and sensorimotor dysfunction are associated with decreased infarct volume.

Box–whisker plots representing significant difference in neurological scores a, the sticky tape removal test b, the beam-walking scores c, and rotarod walking times d in the No-Ex + MCAO (n = 6 for each test) and the Ex groups 3 D (n = 6), 1 W (n = 6), 2 W (n = 5), and 3 W (n = 5) after exercise cessation. The neurological scores and each sensorimotor function were improved until 2 W after exercise cessation. *p < 0.05, **p < 0.01
The expression of BDNF was upregulated in the early stage of detraining, but anti-apoptotic effects lasted until 2 weeks of detraining
Third, to investigate the neuroprotective mechanisms after exercise cessation, we examined the expression of BDNF and apoptotic activity in the motor cortex of the ischemic penumbra by immunohistochemistry (Figs. 2C and 4A). Semi-quantitative analyses showed that the BDNF immunoreactive area was significantly increased in the surrounding lesion after 3 days and 1 week compared to the No-Ex, 2 and 3 weeks after exercise cessation (p < 0.05, Fig. 4B). The increased BDNF immunoreactive area after MCAO was observed until 1 week, but not at 2 or 3 weeks after exercise cessation. In addition, we examined the expression of Bax and Caspase-3 in the motor cortex of the ischemic penumbra (Figs. 2C and 4A). The Bax and Caspase-3 immunoreactive areas were significantly decreased around the lesion 3 days, 1 week, and 2 weeks after exercise cessation compared to No-Ex and 3 weeks (p < 0.05, Fig. 4C, D). In contrast, the Bax and Caspase-3 immunoreactive areas were significantly increased 3 weeks after exercise cessation.

The expression of BDNF, Bax, and Caspase-3 in the ischemic penumbra of rats in No-Ex + MCAO group (n = 6) and 3 D (n = 6), 1 W (n = 6), 2 W (n = 5), and 3 W (n = 5) of detraining + MCAO group was analyzed by immunohistochemistry a–d. Photomicrographs of each immunostaining were obtained from the motor cortex of the ischemic penumbra surrounding the lesion in the rectangle areas of Fig. 2C. BDNF immunoreactivity surrounding the lesion was significantly increased 3 D and 1 W after exercise cessation a, b. The Bax and caspase-3 immunoreactivities surrounding the lesion were significantly reduced 3 D, 1 W, and 2 W after exercise cessation a, c, d. *p < 0.05, **p < 0.01. Scale bar = 50 μm in all panels
In addition, we examined the protein levels of BDNF, Bax, and Caspase-3 in the ischemic brain of No-Ex group, 3 days, 2 weeks, and 3 weeks of detraining by western blotting (Fig. 5A). Because the preconditioning exercise-induced beneficial effects were lost after 3 weeks of detraining, we examined the protein level of the ischemic brain except in 1 week group after exercise cessation. The protein level of BDNF was increased 3 days after exercise cessation but did not increase at 2 or 3 weeks (p < 0.01, Fig. 5B). The protein levels of Bax and Caspase-3 significantly decreased until 2 weeks after exercise cessation (p < 0.05, Fig. 5C, D). These results suggest that anti-apoptotic effects after stroke were lost after 3 weeks of detraining.

Representative western blotting and semi-quantitative analyses of the expression of BDNF, Bax, and Caspase-3 in the ipsilateral brain of rats in the No-Ex + MCAO group and 3 days (3 D), 2 weeks (2 W) and 3 weeks (3 W) after detraining + MCAO group (n = 3 for each group) was analyzed by western blotting (A–D). Because the preconditioning exercise induced beneficial effects were recognized until 2 weeks of detraining, the western blotting was performed ischemic brain without 1 W after detraining + MCAO group. The protein level of BDNF was significantly increased 3 D after exercise cessation a, b. The protein levels of Bax and Caspase-3 were significantly decreased 3 D and 2 W after exercise cessation a, c, d. Data are presented as mean ± SE. *p < 0.05, **p < 0.01
The expressions of HIF-1α, GFAP, and P2X7 receptors in the ischemic brain were increased until 2 weeks of detraining
Fourth, to investigate the neuroprotective mechanism of P2X7 receptor-mediated upregulation of HIF-1α in astrocytes, we examined the protein levels of HIF-1α, GFAP, and P2X7 receptors in the ischemic brain after exercise cessation (Fig. 6A). The protein levels of HIF-1α were significantly increased in the ischemic brain until 2 weeks after exercise cessation compared with those in the No-Ex group (p < 0.05, Fig. 6B). The enhancement of HIF-1α protein levels was significantly decreased 3 weeks after exercise cessation (p < 0.05). In addition, the protein level of GFAP was significantly increased 2 weeks after exercise cessation compared with that of No-Ex and 3 days (p < 0.05, Fig. 6C). The protein levels of P2X7 receptor and HIF-1α were significantly increased in the ischemic brain until 2 weeks after exercise cessation compared with those in the No-Ex group (p < 0.05, Fig. 6D). The enhancement of P2X7 receptor proteins was significantly decreased 3 weeks after exercise cessation (p < 0.05).

Representative western blotting and semi-quantitative analysis of HIF-1α, GFAP, and P2X7 receptor in the ipsilateral brain in the No-Ex + MCAO group and 3 days (3 D), 2 weeks (2 W) and 3 weeks (3 W) after detraining + MCAO group (n = 3 in each group) a–d. The protein level of HIF-1α was significantly increased 3 D and 2 W after exercise cessation a, b. The protein levels of GFAP c and P2X7 d receptors were significantly increased until 2 W after exercise cessation. Data are presented as mean ± SE. *p < 0.05
Preconditioning exercise increased the expression of HIF-1α and P2X7 receptors in astrocytes until 2 weeks of detraining in the non-ischemic brain.
Finally, to confirm the change in intrinsic protective factors in the brain of normal control and exercise only groups after exercise cessation, we examined the protein levels of BDNF, HIF-1α, GFAP, and P2X7 receptor in the non-ischemic brain by western blotting (Fig. 7A). We examined the protein level of the brain except 1 week after exercise cessation, because the border of preconditioning exercise-induced beneficial effects was observed between 2 and 3 weeks of detraining. The protein level of BDNF after exercise cessation was not significantly different from that of the normal control group (Fig. 7B). In contrast, the protein level of HIF-1α significantly increased until 2 weeks after exercise cessation (p < 0.05, Fig. 7C). HIF-1α is involved in P2X7 receptor-mediated ischemic tolerance in astrocytes (Hirayama et al. 2015). Therefore, we examined the expression patterns of GFAP and P2X7 receptors after exercise cessation. The protein level of GFAP was significantly increased at 2 and 3 weeks after exercise cessation compared to that in the normal control group (p < 0.01, Fig. 7D). The protein level of P2X7 receptors was significantly increased until 2 weeks after exercise cessation as well as that of HIF-1α (p < 0.05, Fig. 7E), suggesting that the expression of HIF-1α may be dependent on the expression of P2X7 receptor in astrocytes.

Representative western blotting and semi-quantitative analyses of the expression of BDNF, HIF-1α, GFAP, and P2X7 receptor (P2X7R) of the non-ischemic brain in the normal control, and in the 3 days (3 D), 2 weeks (2 W) and 3 weeks (3 W) after exercise cessation a–e (n = 3 in each group). No significant difference was observed in the expression of BDNF after exercise cessation b. The HIF-1α was significantly increased in the ischemic brain until 2 W after exercise cessation c. The GFAP was increased at 2 W and 3 W after exercise cessation d. The P2X7 receptor was significantly increased until 2 W after exercise cessation e. Data are presented as mean ± SE. *p < 0.05, **p < 0.01
Discussion
The present study demonstrates that neuroprotective effects due to 3-week preconditioning exercise-induced brain tolerance might be lost within a similar period after exercise cessation. In addition, the expression of intrinsic protective factors such as BDNF and HIF-1α after stroke varied during detraining. Our results suggest that long-lasting HIF-1α expression has a neuroprotective effect in the late stage of detraining; therefore, anti-apoptotic activity due to preconditioning exercise lasted 2 weeks after detraining. The loss of endogenous neuroprotective potential, such as reduced infarct volume and ameliorated sensorimotor dysfunction after stroke, was associated with the loss of expression of protective factors and anti-apoptotic pathways.
A number of intrinsic factors are present intracellularly and can be turned on to protect cells from stress and injury, including cerebral ischemia (Dong et al. 2010). The expression dynamics of instruct protective factors may be altered after exercise cessation. Our results showed that BDNF expression did not increase in the intact brain after exercise cessation. Huang et al. (2006) reported that rat hippocampal BDNF mRNA and protein levels were both elevated in response to 4 weeks of exercise training at 2 h after the last run, but not after 2 days. In addition, BDNF protein levels in the rat hippocampus increased depending on the distance of running achieved after 4 weeks of voluntary exercise (Adlard et al. 2004). Therefore, exercise-induced BDNF expression may increase in an exercise-dependent manner and decrease considerably for a short period after exercise cessation. Our findings showed that BDNF upregulation was observed after MCAO in the early stage of detraining, and that its upregulation in the ischemic brain did not persist during detraining. BDNF treatment inhibits caspase-3 activation following neuronal injury in vitro (Kim and Zhao 2005), and BDNF facilitates neuronal survival and development and modulates synaptic plasticity (Ho et al. 2011). Therefore, our results suggest that the upregulation of BDNF may act as a neuroprotective mechanism after stroke in the early stage of detraining.
In contrast, the expression of HIF-1α was increased until 2 weeks in the intact brain and ischemic brain after exercise cessation, which was associated with the expression of P2X7 receptor and GFAP. Our previous study demonstrated that preconditioning exercise induces ischemic tolerance through upregulation of HIF-α in cortical neurons and astrocytes, thereby leading to reduced infarct volume and sensorimotor deficits after stroke (Otsuka et al. 2019). Astrocyte-derived HIF-1α, which is involved in the P2X7 receptor, leads to long-lasting induction of ischemic tolerance (Hirayama et al. 2015; Hirayama and Koizumi. 2017). The upregulation of the P2X7 receptor in astrocytes was slow-onset and long-lasting at least until 2 weeks after ischemic preconditioning, which correlated with that of HIF-1α (Hirayama and Koizumi. 2017). Our previous immunohistochemical study demonstrated that P2X7 receptor-positive cells were co-localized with HIF-1α- and GFAP-positive cells in the brain after preconditioning exercise (Otsuka et al. 2019). Therefore, our results suggest that brain tolerance due to preconditioning exercise may be associated with astrocyte-derived HIF-1α, which is involved in the P2X7 receptor, thereby leading to long-lasting induction of brain tolerance after exercise cessation. Taken together, astrocyte-induced ischemic tolerance lasts for at least 2 weeks after exercise cessation and may exert neuroprotection after stroke. Our findings suggest that BDNF and HIF-1α induction may be modulated to exert different neuroprotective mechanisms in a time-dependent manner after exercise cessation. The mechanisms of preconditioning exercise-induced neuroprotection in the central nervous system are complex; therefore, further studies are needed to examine the neuroprotective mechanisms through several intrinsic protective factors after stroke.
Preconditioning exercise activates astrocytes and improves neurovascular integrity in the penumbra areas following brain ischemia, thereby regulating multiple factors such as insulin-like growth factor I (IGF-1) and vascular endothelial growth factor (VEGF) (Sakakima et al. 2019; Di Raimondo et al. 2020). Hirayama et al. (2015) demonstrated that the timing of ischemic preconditioning-induced activation of glial cells, such as microglia and astrocytes, differs between the cortex and stratum in the brain. These reports suggest that preconditioning exercise-induced neuroprotective mechanisms may differ from cortical and subcortical areas such as the stratum. MCAO models showed extensive ipsilateral brain infarctions, including the cortex and striatum. Therefore, we performed western blotting analysis of the ipsilateral brain. Our results did not characterize the difference in neuroprotective mechanisms between the cortex and subcortical areas. However, our western blotting analysis showed an obvious increase in GFAP protein levels in the cortex and stratum of preconditioning exercise animals. Further studies are needed to examine the differences in neuroprotective mechanisms through glial cell activation or neurotrophic factors in the cortex and subcortical areas.
Recently, Yang et al. (2020) demonstrated that ischemic preconditioning by transient MCAO provides long-lasting neuroprotection against subsequent ischemic stroke through the activation of the master gatekeeper of antioxidant defenses, which is nuclear factor erythroid 2-related factor 2 (Nrf2). Ischemic preconditioning is an interesting strategy for protecting neurons by inducing ischemic tolerance in the brain (Hirayama et al. 2015). Therefore, long-lasting neuroprotection, such as ischemic preconditioning, would be more clinically meaningful. However, such prophylactic treatments may be harmful and have poor feasibility for patients; therefore, safer and more feasible treatments have been sought. Generally, it is important that regular physical exercise continues to be done daily to allow it to exert its beneficial effects on brain health and cognitive function, especially in patients with neurological diseases such as Alzheimer’s disease (Nakanishi et al. 2021). Therefore, although preconditioning exercise may provide short-lasting neuroprotection that wanes after a decrease in the expression of intrinsic protective factors after exercise cessation, preconditioning exercise-induced neuroprotective effects would be clinically meaningful. In the clinical setting, our findings of short-lasting beneficial effects of prophylactic preconditioning exercise may be useful for patient education regarding brain health promotion or rehabilitation.
Our study has some limitations. First, the adaptations that accompany exercise training are dependent on factors such as intensity, frequency, and duration of physical activity. In animal studies, exercise intensity and frequency prior to ischemia may be important contributing factors for stroke outcomes in rats (Terashi et al. 2019). A period of at least 2 or 3 weeks of moderate- or high-intensity exercise prior to ischemia is necessary to induce brain ischemic tolerance (Zhang et al. 2011; Sakakima. 2019), with a frequency of at least three times per week (Terashi et al. 2019). However, the duration of preconditioning exercise may be an important factor that exerts neuroprotective effects after detraining. Further studies are needed to examine the relationship between the factors of preconditioning exercise and detraining with neuroprotective potential. Second, this study focused on the upregulation of the protein levels of BDNF and HIF-1α, as well as the apoptotic activity in the ischemic brain during detraining. However, the mechanisms through which preconditioning exercise could protect the brain are vary (Di Raimondo et al. 2020). Therefore, further studies are needed to clarify preconditioning exercise-induced neuroprotective mechanisms such as multiple factors (i.e., IGF-1 and VEGF). Third, immunohistochemical analysis was performed on the motor cortex surrounding the lesions. However, we performed western blotting analysis in the ipsilateral brain, and included both ischemic and non-ischemic areas because we did not discriminate between ischemic and non-ischemic areas. Fourth, we used a small sample size in each group to minimize the number of animals used. Despite these limitations, this study demonstrated that 3 weeks of preconditioning exercise-induced neuroprotective effects might be completely lost after 3 weeks of detraining. In addition, our findings suggested that the enhancement of BDNF induction was lost in the early stage of detraining; however, enhancement of astrocyte-derived HIF-1α involved in the P2X7 receptor lasted for a long period during detraining.
Conclusion
The neuroprotective potentials due to 3-week preconditioning exercise-induced brain tolerance may be lost in a similar time period after exercise cessation. The present study demonstrated one of the underlying endogenous neuroprotective mechanisms of detraining after preconditioning exercise. Neuroprotection through intrinsic protective factors such as BDNF and HIF-1α may exert different neuroprotective mechanisms in a time-dependent manner. Our findings provide insight for further studies on the effects of preconditioning exercise training and detraining for human brain health promotion.
Data availability
Available from the corresponding author upon reasonable request.
Abbreviations
- BDNF:
-
Brain-derived neurotrophic factor
- NGF:
-
Nerve growth factor
- MCAO:
-
Middle cerebral artery occlusion
- HIF-1α:
-
Hypoxia-induced factor-1α
- TTC:
-
2,3,5–Triphenyltetrazorlium chloride
- GFAP:
-
Glial fibrillary acidic protein
- Bax:
-
Anti-B-cell lymphoma 2-associated X protein
References
Adlard PA, Perreau VM, Engesser-Cesar C, Cotman CW (2004) The timecourse of induction of brain-derived neurotrophic factor mRNA and protein in the rat hippocampus following voluntary exercise. Neurosci Lett 363(1):43–48
Bocalini DS, Carvalho EV, de Sousa AF, Levy RF, Tucci PJ (2010) Exercise training-induced enhancement in myocardial mechanics is lost after 2 weeks of detraining in rats. Eur J Appl Physiol 109(5):909–914
Deplanque D, Masse I, Libersa C, Leys D, Bordet R (2012) Previous leisure-time physical activity dose dependently decreases ischemic stroke severity. Stroke Res Treat 2012:614925
Di Raimondo D, Rizzo G, Musiari G, Tuttolomondo A, Pinto A (2020) Role of regular physical activity in neuroprotection against acute ischemia. Int J Mol Sci 21(23):9086
Ding Y, Li J, Luan X, Ding YH, Lai Q, Rafols JA, Phillis JW, Clark JC, Diaz FG (2004) Exercise pre-conditioning reduces brain damage in ischemic rats that may be associated with regional angiogenesis and cellular overexpression of neurotrophin. Neuroscience 124(3):583–591
Dong Y, Zhao R, Chen XQ, Yu AC (2010) 14-3-3gamma and neuroglobin are new intrinsic protective factors for cerebral ischemia. Mol Neurobiol 41(2–3):218–231
Hirayama Y, Koizumi S (2017) Hypoxia-independent mechanisms of HIF-1α expression in astrocytes after ischemic preconditioning. Glia 65(3):523–530
Hirayama Y, Ikeda-Matsuo Y, Notomi S, Enaida H, Kinouchi H, Koizumi S (2015) Astrocyte-mediated ischemic tolerance. J Neurosci 35(9):3794–3805
Ho VM, Lee JA, Martin KC (2011) The cell biology of synapticity. Science 334(6056):623–628
Huang AM, Jen CJ, Chen HF, Yu L, Kuo YM, Chen HI (2006) Compulsive exercise acutely upregulates rat hippocampal brain-derived neurotrophic factor. J Neural Transm (vienna) 113(7):803–811
Islam MR, Young MF, Wrann CD (2017) Neuroprotective potential of exercise preconditioning in stroke. Cond Med 1(1):27–34
Kim DH, Zhao X (2005) BDNF protects neurons following injury by modulation of caspase activity. Neurocrit Care 3(1):71–76
Kramer M, Dang J, Baertling F, Denecke B, Clarner T, Kirsch C, Beyer C, Kipp M (2010) TTC staining of damaged brain areas after MCA occlusion in the rat does not constrict quantitative gene and protein analyses. J Neurosci Methods 187(1):84–89
Kramer SF, Hung SH, Brodtmann A (2019) The impact of physical activity before and after stroke on stroke risk and recovery: a narrative review. Curr Neurol Neurosci Rep 19(6):28
Krarup LH, Truelsen T, Gluud C, Andersen G, Zeng X, Kõrv J, Oskedra A, Boysen G, ExStroke Pilot Trial Group (2008) Prestroke physical activity is associated with severity and long-term outcome from first-ever stroke. Neurology 71(17):1313–1318
Mujika I, Padilla S (2000) Detraining: loss of training-induced physiological and performance adaptations. Part I: short term insufficient training stimulus. Sports Med 30(2):79–87
Nakanishi K, Sakakima H, Norimatsu K, Otsuka S, Takada S, Tani A, Kikuchi K (2021) Effect of low-intensity motor balance and coordination exercise on cognitive functions, hippocampal Aβ deposition, neuronal loss, neuroinflammation, and oxidative stress in a mouse model of Alzheimer’s disease. Exp Neurol 337:113590
Otsuka S, Sakakima H, Sumizono M, Takada S, Terashi T, Yoshida Y (2016) The neuroprotective effects of preconditioning exercise on brain damage and neurotrophic factors after focal brain ischemia in rats. Behav Brain Res 303:9–18
Otsuka S, Sakakima H, Terashi T, Takada S, Nakanishi K, Kikuchi K (2019) Preconditioning exercise reduces brain damage and neuronal apoptosis through enhanced endogenous 14-3-3γ after focal brain ischemia in rats. Brain Struct Funct 224(2):727–738
Ploughman M, Windle V, MacLellan CL, White N, Doré JJ, Corbett D (2009) Brain-derived neurotrophic factor contributes to recovery of skilled reaching after focal ischemia in rats. Stroke 40:1490–1495
Ploughman M, Austin MW, Glynn L, Corbett D (2015) The effects of poststroke aerobic exercise on neuroplasticity: a systematic review of animal and clinical studies. Transl Stroke Res 6(1):13–28
Radak Z, Toldy A, Szabo Z, Siamilis S, Nyakas C, Silye G, Jakus J, Goto S (2006) The effects of training and detraining on memory, neurotrophins and oxidative stress markers in rat brain. Neurochem Int 49(4):387–392
Ricciardi AC, López-Cancio E, Pérez de la Ossa N, Sobrino T, Hernández-Pérez M, Gomis M, Munuera J, Muñoz L, Dorado L, Millán M, Dávalos A, Arenillas JF (2014) Prestroke physical activity is associated with good functional outcome and arterial recanalization after stroke due to a large vessel occlusion. Cerebrovasc Dis 37(4):304–311
Sakakima H (2019) Endogenous neuroprotective potential due to preconditioning exercise in stroke. Phys Ther Res 22(2):45–52
Sakakima H, Khan M, Dhammu TS, Shunmugavel A, Yoshida Y, Singh I, Singh AK (2012) Stimulation of functional recovery via the mechanisms of neurorepairby S-nitrosoglutathione and motor exercise in a rat model of transientcerebral ischemia and reperfusion. Restor Neurol Neurosci 30(5):383–396
Stroud N, Mazwi TM, Case LD, Brown RD Jr, Brott TG, Worrall BB, Meschia JF, Ischemic Stroke Genetics Study Investigators (2009) Prestroke physical activity and early functional status after stroke. J Neurol Neurosurg Psychiatry 80(9):1019–1022
Terashi T, Otsuka S, Takada S, Nakanishi K, Ueda K, Sumizono M, Kikuchi K, Sakakima H (2019) Neuroprotective effects of different frequency preconditioning exercise on neuronal apoptosis after focal brain ischemia in rats. Neurol Res 41(6):510–518
Tolppanen AM, Solomon A, Kulmala J, Kåreholt I, Ngandu T, Rusanen M, Laatikainen T, Soininen H, Kivipelto M (2015) Leisure-time physical activity from mid- to late life, body mass index, and risk of dementia. Alzheimers Dement 11(4):434-443.e6
Waring CD, Henning BJ, Smith AJ, Nadal-Ginard B, Torella D, Ellison GM (2015) Cardiac adaptations from 4 weeks of intensity-controlled vigorous exercise are lost after a similar period of detraining. Physiol Rep 3(2):e12302
Yang J, Liu C, Du X, Liu M, Ji X, Du H, Zhao H (2018) Hypoxia inducible factor 1α plays a key role in remote ischemic preconditioning against stroke by modulating inflammatory responses in rats. J Am Heart Assoc 7(5):e007589
Yang T, Sun Y, Li Q, Li S, Shi Y, Leak RK, Chen J, Zhang F (2020) Ischemic preconditioning provides long-lasting neuroprotection against ischemic stroke: The role of Nrf2. Exp Neurol 325:113142
Zhang F, Wu Y, Jia J (2011) Exercise preconditioning and brain ischemic tolerance. Neuroscience 177:170–176
Zhang Q, Zhang L, Yang X, Wan Y, Jia J (2014) The effects of exercise preconditioning on cerebral blood flow change and endothelin-1 expression after cerebral ischemia in rats. J Stroke Cerebrovasc Dis 23(6):1696–1702
Acknowledgements
The authors would like to thank Mr. Yuki Itashiki and Mr. Keita Fukumaru for their assistance in this animal study.
Funding
This work was supported by grants from JSPS KAKENHI (Grant No. JP20H0403 to Harutoshi Sakakima and Grant No. JP19K24315 to Shotaro Otsuka).
Author information
Authors and Affiliations
Contributions
All authors contributed to this study. This study is based on the original ideas of HS, SO, HM, and IM. SO, AT, ST, KN (Kazuki Nakanishi), and KN (Kosuke Norimatsu) performed the experiments and carried out the animal, histochemical and biochemical studies, as well as the quantitative analysis. HS and SO performed the literature review and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Ethical approval
The experimental protocol was approved by the Ethics Board of the Institute of Laboratory Animal Sciences of Kagoshima University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Otsuka, S., Sakakima, H., Tani, A. et al. Effects of detraining on preconditioning exercise-induced neuroprotective potential after ischemic stroke in rats. Brain Struct Funct 226, 2169–2180 (2021). https://doi.org/10.1007/s00429-021-02317-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-021-02317-5