
Abstract
Despite evidence that morphine-related pathologies reflect adaptations in NAc glutamate signaling, substantial gaps in basic information remain. The current study examines the impact of non-contingent acute, repeated, and withdrawal-inducing morphine dosing regimens on glutamate transmission in D1- or D2-MSNs in the nucleus accumbens shell (NAcSh) and core (NAcC) sub-regions in hopes of identifying excitatory plasticity that may contribute to unique facets of opioid addiction-related behavior. Following an acute morphine injection (10 mg/kg), average miniature excitatory postsynaptic current (mEPSC) amplitude mediated by AMPA-type glutamate receptors was increased at D1-MSNs in the both the NAcShl and NAcC, whereas only the frequency of events was elevated at D2-MSNs in the NAcSh. In contrast, spontaneous somatic withdrawal induced by escalating dose of repeated morphine twice per day (20, 40, 60, 80, 100 mg/kg) enhanced mEPSC frequency specifically at D2-MSNs in the NAcSh. Similar to previous findings, excitatory drive was elevated at NAcSh D1-MSNs after 10–14 days home cage abstinence. Following abstinence, an acute drug re-exposure produced a rapid and enduring endocytosis of GluA2-containing AMPARs at D1-MSNs in the shell, that when blocked by an intra-NAc shell infusion of the Tat-GluA23Y peptide, increased reinstatement of morphine place preference—a phenomenon distinctly different than effects previously found with cocaine. The present study is the first to directly identify unique circuit specific adaptations in NAc glutamate synaptic transmission associated with morphine-related acute reward and somatic withdrawal as well as post-abstinence short-term plasticity. Moreover, while differing classes of abused drugs (i.e., psychostimulants and opioids) produce seemingly similar bidirectional plasticity in the NAc following drug re-exposure, our findings indicate this plasticity has distinct behavioral consequences.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Opioids are the main class of drugs for pain management despite the risk for abuse (Wise 1989). In addition to their primary rewarding effects, repeated use of opioids can result in the development of physical dependence that manifests as debilitating somatic and psychological withdrawal symptoms that can perpetuate continued use (Koob et al. 1989; van Ree et al. 1999). Increasing evidence suggests that opioid-induced plasticity related to dependence and withdrawal verus plasticity responsible for establishing opioid-seeking behavior and drug-associations that can provoke craving and relapse occurs within divergent, as well as overlapping, neural circuits (Badiani et al. 2011; Graziane et al. 2016; Hearing 2019; Hearing et al. 2016, 2018; Russell et al. 2016; Zhu et al. 2016)—highlighting a major challenge towards identifying the neurophysiological bases of dependence and withdrawal versus adaptations responsible for enduring relapse risk.
Prior findings posit glutamate plasticity in the nucleus accumbens (NAc) as a significant factor in the acute rewarding effects of opioids (Baharlouei et al. 2015), conditioned opioid associations (Fujio et al 2005; Hearing et al. 2016; Siahposht-Khachaki et al. 2017), and relapse vulnerability (Bossert et al. 2005, 2006; Shen et al. 2011, 2014). Data also indicate that elevations in NAc glutamate transmission underlie somatic and affective withdrawal symptoms (Russell et al. 2016; Sepulveda et al. 2004; Zhu et al. 2016). However, the NAc is a heterogeneous area of the brain divided into NAc core (NAcC) and shell (NAcSh) sub-regions based in part, on anatomical connectivity. While the NAcC sub-region interacts with brain regions associated with motor circuitry, thus coordinating behavioral output, the NAcSh interacts with limbic and autonomic brain regions, indicating significant regulation of reward, emotional, and visceral responses to stimuli (Everitt et al. 1999; Heimer et al. 1991; Zahm and Brog 1992). Within each sub-region, the primary target of excitatory glutamate afferents is the principal medium spiny projection neurons (MSNs), which are categorically divided based on expression of type 1 (D1-MSNs) or type 2 dopamine receptors (D2-MSNs) (Le Moine and Bloch 1995; Lobo and Kennedy 2006; Smith et al. 2013).
Despite evidence that morphine-related pathologies reflect adaptations in NAc glutamate signaling, substantial gaps in basic information remain. For example, while acute morphine exposure transiently increases extracellular glutamate in the NAc (Desole et al. 1996; Enrico et al. 1998; Sepulveda et al. 2004), evidence supporting a role of AMPAR plasticity is lacking. Further, while a causal link between elevations in NAc shell GluA1-containing AMPA-type receptors pharmacologically-precipitated morphine withdrawal (Russell et al. 2016), it remains unclear whether similar changes occur during spontaneous withdrawal, and in what cell type these adaptations occur. Increasing evidence indicates the nature and locus of opioid-induced glutamate plasticity in the NAc dictates the relationship to behavior, with most findings to date highlighting adaptations to the NAcSh in opioid reward and aversion (Gracy et al. 2001; Graziane et al. 2016; Hearing et al. 2016; Russell et al. 2016; Svingos et al. 1997; Zhu et al. 2016). For example, abstinence from non-contingent morphine administration is associated with divergent plasticity in the NAcSh at D1- and D2-MSNs (Graziane et al. 2016; Hearing et al. 2016), but not in the NAcC (Hearing et al. 2016), with increased transmission at D1- and D2-MSNs contributing to opioid reward and aversion learning, respectively (Graziane et al. 2016; Hearing et al. 2016; Russell et al. 2016; Zhu et al. 2016). The current study examines the impact of non-contingent acute, repeated, and withdrawal-inducing morphine dosing regimens on glutamate transmission in D1- or D2-MSNs in the NAcSh and NAcC sub-regions in hopes of identifying excitatory plasticity that may contribute to differing facets of opioid addiction-related behavior.
Materials and methods
Animals
Adult (P49-72) male mice were a combination of heterozygous bacterial artificial chromosome (BAC) transgenic mice (Jackson Laboratories, Bar Harbor, ME, USA) expressing tdtomato or eGFP expression driven by either dopamine receptor DR1 (drd1a-tdtomato) or DR2 (drd2-eGFP), or double transgenics expressing tdtomato and eGFP. Mice were single or group housed on a 12-h light/dark cycle with food and water available ad libitum with experiments run during the light portion. All experiments were approved by the Marquette University and University of Minnesota Institutional Animal Care and Use Committees.
Stereotaxic intra-cranial cannula implantation
For surgical procedures, mice were anesthetized with ketamine and xylazine (100/10 mg/kg, respectively, i.p.). Depth of anesthesia was assessed prior to the subject being placed in the stereotaxic frame (Kopf Instruments, Tujunga, CA, USA). Measurements targeting implantation of the single barrel guide cannula (26ga, 5 mm pedestal, 3.5 mm projection; C315GS-5/SP, Plastics1, Roanoke, VA, USA) to the NAcSh region were taken with respect to bregma/midline (+ 1.50 a/p, ± 1.45 m/l, − 4.0) at a 14° angle. Cannula was cemented in place using Geristore (DenMat, Lompoc, CA, USA). Mice were allowed a minimum 5-day recovery period before beginning behavior testing.
Morphine-induced locomotion
Locomotor chamber apparatus was placed under AnyMaze video tracking system (Stoelting, Wood Dale, IL, USA) and measurements were made automatically by the software as previously described (Hearing et al. 2016).
Acute morphine
Mice were given an injection (i.p.) of saline or morphine (10 mg/kg) and euthanized 3–4 h following injection for electrophysiological recordings. This time point was chosen for the purpose of performing the electrophysiological recordings while morphine was still present in tissue and serum. Our recordings were performed prior to reaching the approximate 5-h half-life of subcutaneous morphine administration (Hipps et al. 1976) and during a post-injection time point previously shown to observe elevated locomotor activity (Hearing et al. 2016). These experiments were performed at the University of Minnesota.
Morphine challenge
Mice were administered five daily injections of saline or morphine (10 mg/kg)—a regimen previously shown to augment glutamate transmission and promote sensitization (Hearing et al. 2016)—followed by 10–14 days of abstinence. Mice were then administered a saline or morphine (10 mg/kg) challenge injection. 16–24 h following challenge injection, mice were euthanized for subsequent electrophysiological recordings. These experiments were performed at both the University of Minnesota and Marquette University.
Spontaneous withdrawal
Across a period of 5 days, two injections (saline or morphine) were given each day in their home cage approximately 12 h apart, with doses for each day at 20, 40, 60, 80, and 100 mg/kg (10 injections total). Twenty-four hours or 14 days following the final injection, mice were placed into a clear plastic cage (16.5″ × 8″ × 7.5″) and examined for signs of somatic withdrawal during a 30-min period. Somatic measures were chosen based on previous works examining morphine withdrawal (Cruz et al. 2008; Papaleo and Contarino 2006; Schulteis et al. 1994; van der Laan and de Groot 1988; van der Laan et al. 1991). Jumps, tremors, paw flutters, wet dog shakes, piloerection, and grooming were hand scored with each measurement recorded as a score of one, including the singular possible observation of piloerection, to generate a global withdrawal score with the following equation: \(\frac{\text{jumps}}{3}\, + \,{\text{tremors}}\, + \,{\text{grooming}}\, + \,{\text{paw flutters}}\, + \,{\text{shakes}}\, + \,{\text{pilerection}}\) (Papaleo and Contarino 2006). Additional measurements included locomotor activity in the form of distance traveled (m) using AnyMaze video tracking software (Stoelting Company, Wood Dale, IL, USA). These experiments were performed at Marquette University.
Conditioned place preference
All conditioned place preference experiments employed a two-chamber apparatus (St. Albans, VT, USA) and were performed as previously described (Wydeven et al. 2014). For conditioning, subjects were injected with morphine (5 mg/kg) or vehicle, and after a 20-min delay were confined for 30 min in the corresponding CS+/CS− chamber. Morphine dosing for place preference training was chosen based on previous findings that this dose produces robust place preference (Hearing et al. 2016). Further, this dose in addition to extinction training following conditioning was shown to produce identical cell-specific plasticity observed following five daily injections of morphine and home cage abstinence (Hearing et al. 2016). A total of four morphine (5 mg/kg) and four saline trials were performed in alternating fashion, with only one trial performed per day and preference evaluated 24 h following the final conditioning session. Following conditioning, mice underwent six daily extinction sessions as previously described (Hearing et al. 2016), with animals confined to the CS+ and CS− compartment for 20 min each on days 1, 3, and 5 (extinction training) and allowed to freely explore on days 2, 4, and 6 (extinction testing). Day 6 data were used for two-way ANOVA analysis.
Intra-cranial GluA2 peptide and reinstatement of place preference
Experimental treatments for the reinstatement tests were assigned after extinction training and assignments were made to ensure that each treatment group had similar preference scores prior to and following extinction. Reinstatement of place preference was performed in five different experiments. To block endocytosis of GluA2-containing AMPA receptors in the NAcSh, a synthetic interference peptide designed to disrupt activity-dependent endocytosis without altering basal receptor trafficking, was used as previously described (Ahmadian et al. 2004; Brebner et al. 2005). Mice received an intra-NAcSh infusion of the active (Tat-GluA23Y) or inactive version (Tat-GluA23A) of the peptide diluted in ACSF (75 pmol; 0.5 μL/hemisphere; 0.1 μL/min) using a 32ga internal cannula with 1.2-mm projection beyond the guide. Following infusions, mice were returned to their home cage for 60 min, at which point they received an i.p. injection of morphine (5 mg/kg) or saline, followed by examination of preference behavior during a 20-min test. Electrophysiology recordings to confirm effects of Tat-peptide expression were done within 2 h following testing. These experiments were performed at the University of Minnesota.
Histological analysis
Histological examination of cannula targeting was done visually on the electrophysiology rig or post-mortem in tissue fixed with transcardial perfusion of 4% paraformaldehyde buffered in saline using an overdose of pentobarbital (650 mg/kg). Brains were cryoprotected, sliced at 40 μm, mounted, and cover-slipped with ProLong gold antifade mounting medium (Life Technologies, Eugene, OR, USA). Two mice were excluded from data analysis due to considerable tissue damage.
Electrophysiology
Sagittal (250 μm) sections of the NAcSh and NAcC were used for morphine challenge studies and acute morphine, with coronal slices (300 μm) used in morphine challenge and withdrawal studies as previously described (Hearing et al. 2016). Slices were collected in a high sucrose solution as previously described (Hearing et al. 2013) and allowed to recover for at least 45–60 min in ACSF solution saturated with 95% O2/5% CO2 containing (in mM) 119 NaCl, 2.5 KCl, 1.0 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, 26.2 NaHCO3 and 11 glucose. Electrophysiological recordings assessing miniature excitatory postsynaptic currents (mEPSCs) were performed in the presence of picrotoxin (100 μM) and lidocaine (700 μM) to block GABAergic neurotransmission and sodium-dependent action potentials, respectively, as previously described (Hearing et al. 2016; Jedynak et al. 2016). The majority of NAcSh recordings were from the medial portion with an equal blend along the rostro-caudal axis. Cells were visualized using infrared-differential contrast (IR-DIC) microscopy, and medium spiny neurons (MSNs) were identified by cell subtype-specific fluorophore (tdTomato or EGFP) in combination with capacitance (> 50 pF). Using a Sutter Integrated Patch Amplifier (Sutter Instruments, Novato, CA, USA) and/or Axon Instruments Multiclamp 700B (Molecular Devices, Sunnyvale, CA, USA), MSNs were voltage clamped at − 72 mV using electrodes (2.5–4 MΩ) with a cesium-methyl sulfonate-based internal solution containing (in mM) 120 CsMeSO4, 15 CsCl, 10 TEA-Cl, 8 NaCl, 10 HEPES, 5 EGTA, 0.1 spermine, 5 QX-314, 4 ATP-Mg, and 0.3 GTP-Na. Data were filtered at 1–2 kHz and digitized at 20 kHz via custom Igor Pro software (Wavemetrics, Lake Oswego, OR, USA) or Clampex 10.7 software (Molecular Devices, Sunnyvale, CA, USA). Series (10–40 MΩ) and input resistance were monitored using a depolarizing step (5 mV, 100 ms). Neurons with a holding current below − 150 pA were excluded from analysis. Data collection and analysis were performed as previously described (Hearing et al. 2016; Kourrich et al. 2007).
Notably, independent samples t tests between mEPSC metrics from morphine challenge study Sal–Sal mice recorded using sagittal (University of Minnesota) and coronal sections (Marquette University) were performed. We observed no impact of slice orientation/recording location on mEPSC metrics in NAcSh D1-MSNs (Amp t(20) = 0.4359, p = 0.67; Freq t(20) = 0.5078, p = 0.62), NAcSh D2-MSNs (Amp t(11) = 0.8798, p = 0.40; Freq t(12) = 1.937, p = 0.08), NAcC D1-MSNs (Amp t(18) = 0.9552, p = 0.35, Freq t(20) = 0.5279, p = 0.60), or NAcC D2-MSNs (Amp t(16) = 1.86, p = 0.08, Freq t(16) = 1.88, p = 0.08).
Drugs
Picrotoxin and lidocaine were purchased from Sigma-Aldrich (St. Louis, MO, USA). Morphine was purchased from the Boynton Pharmacy (University of Minnesota, Minneapolis, MN, USA) or Froedtert Hospital Pharmacy (Medical College of Wisconsin, Milwaukee, WI, USA).
Statistical analysis and data presentation
mEPSCs were analyzed with independent samples t tests or two-way ANOVAs using SigmaPlot (Systat Software, San Jose, CA, USA) or Graph Pad Prism (GraphPad Software, Inc., La Jolla, CA, USA). Appropriate post hoc analyses were used for pairwise comparisons as indicated. The threshold for statistical significance in all cases was p < 0.05. Electrophysiology data are represented utilizing standard column graphs displaying mean ± SEM, with adjacent scatter plots of individual data points. Sample size in experiments is presented as n and N, where n is the number of cells and N is the number of mice.
Results
Bidirectional changes in AMPA receptor synaptic transmission in the NAcSh and NAcC
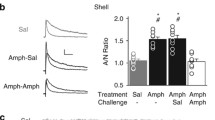
We recently demonstrated that prolonged withdrawal from repeated non-contingent morphine increases excitatory drive at NAcSh D1-MSNs, while reducing drive at D2-MSNs (Hearing et al. 2016). Our previous work shows that re-exposure to relapse-inducing stimuli (i.e., discrete cues, drug, stress) following abstinence or extinction promotes a transient reduction in synaptic AMPAR signaling in unidentified MSNs that drive cocaine-induced reinstatement of place preference (Benneyworth et al. 2019; Brebner et al. 2005; Ebner et al. 2018; Famous et al. 2008; Ingebretson et al. 2018; Jedynak et al. 2016; Kourrich et al. 2007; Rothwell et al. 2011; Schmidt et al. 2013, 2015; Thomas et al. 2001). While the cell-type selectivity of this plasticity remains unclear, previous reports indicate that this reduction occurs in D2-MSNs (Ortinski et al. 2015). Initial studies sought to determine whether re-exposure to opioids promotes a similar bimodal shift in synaptic strength using ex vivo recordings of miniature excitatory postsynaptic currents (mEPSCs)—a direct measure of synaptic AMPAR function—24 h following a post-abstinence (10–14 days) challenge injection. To identify effects of acute morphine exposure based on previous drug experience and ensure effects of challenge injection are specific for drug re-exposure, this experiment contained four experimental groups. Two groups received five daily injections of saline and later received a saline or morphine priming injection (Sal–Mor, Mor–Mor), and two groups receiving five injections of morphine followed by a saline or morphine challenge (Mor–Sal, Mor–Mor), with recordings performed 24 h following the challenge injection. This approach permitted us to distinguish effects of repeated morphine, morphine re-exposure, and acute morphine.
Two-way ANOVAs were performed on mEPSC amplitude and frequency with daily drug and challenge exposures as between-subject factors; Tukey post hoc analyses were performed when appropriate. In NAcSh D1-MSNs, mEPSC amplitude and frequency were elevated in Mor–Sal mice [amplitude: 14.85 ± 0.83), frequency: (6.63 ± 0.66)] compared to Sal–Sal controls [amplitude: (11.24 ± 0.65), frequency: (5.99 ± 1.01)], and that a morphine challenge (Mor–Mor) returned amplitude and frequency to control levels [amplitude: (11.27 ± 0.35), frequency: (3.66 ± 0.55)] [amplitude: interaction, F(1,65) = 4.288, p < 0.05]; [frequency: interaction, F(1,65) = 14.08, p < 0.001] (Fig. 1b). While acute morphine exposure produced a trend toward increased mEPSC frequency compared to Sal–Sal mice, these effects were not significant. In contrast to D1-MSNs, no significant main effects or interactions were observed for D2-MSN mEPSCs amplitude [daily, F(1,44) = 0.3648; challenge, F(1,44) = 2.197, p = 0.15; interaction, F(1,44) = 0.2447] or frequency [daily, F(1,44) = 0.0204; challenge, F(1,44) = 1.468; interaction, F(1,44) = 2.646, p = 0.11] (Fig. 1c). These data show that acute morphine exposure does not promote lasting alterations in glutamate plasticity in the NAcSh and that morphine re-exposure promotes a bimodal effect on AMPAR signaling akin to that observed following re-exposure to psychostimulants and associated stimuli (Benneyworth et al. 2019; Brebner et al. 2005; Ebner et al. 2018; Famous et al. 2008; Ingebretson et al. 2018; Jedynak et al. 2016; Kourrich et al. 2007; Rothwell et al. 2011; Schmidt et al. 2013, 2015; Thomas et al. 2001); however unlike cocaine, these effects may be confined to D1-MSNs (Ortinski et al. 2015).

Cell- and region-specific effect of post-abstinence morphine exposure on nucleus accumbens AMPA receptor transmission. a Experimental timeline including 5 days of saline or morphine (10 mg/kg; i.p.), a 10–14-day withdrawal period, and challenge injection of saline or morphine. Electrophysiological recordings performed in NAcSh or NAcC D1- or D2-MSNs 24 h post-challenge injection in either coronal or sagittal slices (no significant difference was observed—see “Materials and methods”). b (1) Representative miniature excitatory postsynaptic current (mEPSC) traces and (2) mean mEPSC amplitude (left) and frequency (right) in NAcSh D1-MSNs from saline + saline challenge (Sal–Sal, white; n = 20/N = 11), saline + morphine challenge (Sal–Mor, black; n = 11/N = 6), morphine + saline challenge (Mor–Sal, gray; n = 22/N = 10) and morphine + morphine challenge (Mor–Mor, striped gray; n = 15/N = 5). c (1) Representative traces and (2) mean mEPSCs in NAcSh D2-MSNs [Sal–Sal (n = 15/N = 8), Sal–Mor (n = 8/N = 5), Mor–Sal (n = 12/N = 8), Mor–Mor (n = 13/N = 7)]. d (1) Representative traces and (2) mean mEPSCs in NAcC D1-MSNs [Sal–Sal (n = 19/N = 11), Sal–Mor (n = 12/N = 7), Mor–Sal (n = 16/N = 10), Mor–Mor (n = 9/N = 7). e (1) Representative traces and (2) mean mEPSCs in NAcC D2-MSNs [Sal–Sal (n = 17/N = 11), Sal–Mor (n = 7/N = 6), Mor–Sal (n = 13/N = 10), Mor–Mor (n = 6/N = 5) Scale bar = 20 pA/100 ms; Tukey post hoc: *p < 0.05, **p < 0.01 vs Sal–Sal, #p < 0.05, ##p < 0.01 vs Mor–Mor
In the NAcC, mEPSC amplitude in D1-MSNs from Sal–Sal (12.47 ± 0.59) and Sal–Mor (14.13 ± 0.97) was significantly higher compared to Mor–Sal (11.86 ± 0.54) and Mor–Mor (11.53 ± 0.66) (main effect of daily, F(1,57) = 6.447, p < 0.05), whereas no effects were observed for D1-MSN mEPSC frequency (daily, F(1,55) = 0.0696; challenge, F(1,55) = 2.483, p = 0.12; interaction, F(1,55) = 0.0693). Examination of D2-MSNs showed that mEPSC frequency was reduced in Mor–Sal (3.06 ± 0.19) compared to Sal–Sal (4.77 ± 0.48), and that a morphine challenge (Mor–Mor) returned mEPSC frequency to Sal–Sal control levels (5.44 ± 0.22) (interaction: F(1,40) = 13,96, p < 0.001) (Fig. 1c). No effects were observed for NAcC D2-MSN mEPSC amplitude (daily, F(1,43) = 0.0178; challenge, F(1,43) = 0.682; interaction, F(1,43) = 0.6136). These data align with our previous findings that repeated morphine reduces excitatory drive at NAcC D2-MSNs without altering transmission at D1-MSNs, and that similar to plasticity in NAcSh D1-MSNs, this plasticity is bimodal in nature.
Effects of acute morphine on nucleus accumbens cell-specific AMPAR signaling
Despite ample evidence indicating that glutamate transmission at NAc MSNs contributes to the rewarding effects of opioids (Dworkin et al. 1988; Graziane et al. 2016; Hearing et al. 2016; Olds 1982; Vekovischeva et al. 2001), our data show that a single morphine injection has no significant impact on NAcSh or NAcC AMPAR signaling 24 h after exposure, however this may reflect plasticity that is transient in nature during early exposure. As there are no known data on the immediate effects of acute in vivo opioid exposure on AMPAR transmission in the NAc (Chartoff and Connery 2014), we next investigated AMPAR signaling approximately 3 h following an injection of saline or morphine. This timepoint was chosen to isolate reward versus withdrawal-related effects of morphine as it aligns with elevations in drug-induced motor activity (not shown) and resides within the approximate 5-h half-life of subcutaneous morphine administration (Hipps et al. 1976).
In the NAcSh, mEPSC amplitude and frequency were significantly elevated in D1-MSNs of morphine-treated animals compared to saline controls (Fig. 2b) [amplitude: Sal (11.40 ± 0.64), Mor (16.25 ± 1.28), t(16) = 3.07, p < 0.01); frequency: Sal (5.02 ± 0.46), Mor (9.90 ± 1.5), t(16) = 2.79, p < 0.05]. Alternatively, acute morphine increased mEPSC frequency but not amplitude at D2-MSNs (Fig. 2c) ([amplitude: Sal (15.2 ± 0.19), Mor (14.2 ± 0.81), t(6) = 1.22, p = 0.27; frequency: Sal (5.95 ± 0.53), Mor (10.73 ± 1.05), t(6) = 4.06, p < 0.01].

Cell-specific effects of acute morphine on nucleus accumbens AMPA receptor transmission. a Experimental timeline including an acute injection of saline or morphine (10 mg/kg; i.p.) and electrophysiological recordings performed 3–4 h post-injection. Recordings were performed in sagittal slices containing the NAcSh and NAcC. b (1) Representative AMPAR mEPSC traces and (2) mean mEPSC amplitude (left) and frequency (right) in D1-MSNs from saline (Sal, white; n = 8, N = 5 and morphine (Mor, black; n = 10/N = 5) injected mice. c (1) Representative traces and (2) mean mEPSCs in NAcSh D2-MSNs (Sal: n = 4/N = 4; Mor: n = 4/N = 4). d (1) Representative traces and (2) mean mEPSCs in NAcC D1-MSNs (Sal n = 9/N = 6; Mor: n = 8/N = 7). e (1) Representative mEPSC traces and (2) mean mEPSCs in NAcC D2-MSNs (Sal: n = 5, N = 5; Mor: n = 5/N = 4). *p < 0.5, **p < 0.01 vs Sal. Scale bars = 20 pA/100 ms
Similar to the NAcSh, acute morphine increased mEPSC amplitude and frequency at D1-MSNs in the NAcC (Fig. 2d) [amplitude: Sal (11.54 ± 0.55), Mor (15.43 ± 0.92), t(15) = 3.72, p < 0.01; frequency: Sal (3.24 ± 0.59), Mor (6.23 ± 1.3), t(15) = 2.23, p < 0.05]. However, neither amplitude nor frequency of mEPSCs was altered by acute morphine at NAcC D2-MSNs (Fig. 2e) [amplitude: Sal (11.33 ± 0.62), Mor (13.32 ± 0.73), t(9) = 2.022, p = 0.074; frequency: Sal (4.36 ± 0.66), Mor (4.74 ± 0.77), t(9) = 0.382, p = 0.71]. Taken together, these data indicate that acute morphine promotes a global augmentation of excitatory drive at D1-MSNs, whereas alterations in glutamate transmission at D2-MSNs are specific to the NAcSh, and that these adaptations do not persist or require a period of abstinence to manifest.
Spontaneous withdrawal enhances AMPAR signaling at D2-MSNs
In addition to our lack of knowledge regarding effects of acute in vivo opioids on NAc synaptic transmission, few studies to date have explored the impact of withdrawal-inducing morphine administration on glutamate transmission in a NAc sub-region and MSN subtype-specific manner. Notably, a single injection of morphine is sufficient to precipitate withdrawal symptoms 24 h after exposure (Rothwell et al. 2012) and prior studies indicate that NAcSh D2-MSNs potently regulate somatic withdrawal symptoms (Harris and Aston-Jones 1994; Russell et al. 2016; Zhu et al. 2016)—findings that are signfiicant given our observed plasticity 3 h, but not 24 h after a single injection of morphine. As no studies to date have examined cell- or region-specific NAc plasticity associated with spontaneous somatic withdrawal despite a purported role in relapse behavior, we next sought to determine whether the ostensible lack of plasticity 24 h following an acute exposure reflects the withdrawal period or the exposure type (i.e., acute versus repeated). To do so, we administered an escalating dose of morphine shown to produce dependence as measured by spontaneous withdrawal symptoms (Papaleo and Contarino 2006) and recorded metrics of spontaneous withdrawal 2 h prior to preparation for acute slice electrophysiology. Escalating doses of morphine significantly increased the number of jumps (t(17) = 2.217, p < 0.05), tremors (t(17) = 2.89, p < 0.05), wet dog shakes (t(17) = 2.503, p < 0.05), and piloerection (t(17) = 5.037, p < 0.001). No significant effect of morphine was observed on grooming behavior (t(17) = 0.729, p = 0.48) or paw flutters (t(17) = 0.527, p = 0.61). More specifically, morphine significantly increased the total withdrawal score (Fig. 3) [Sal (8.19 ± 1.57), Mor (20.06 ± 3.14, t(17) = 2.741, p < 0.05)], and decreased distance traveled in a novel context [Sal (85.73 ± 8.27), Mor (38.76 ± 2.13, t(17) = 6.906, p < 0.0001)]. Notably, in a separate cohort of mice, we examined whether somatic withdrawal symptoms persisted at 10–14 days post-drug exposure—a timepoint examined in drug challenge studies. Two-way day-by-drug ANOVA with day a repeated measure and drug exposure as a between-subject factor reveal significant main effects of day (F(1,13) = 10.52, p < 0.01), drug (F(1,13) = 4.481, p = 0.05), and a significant day-by-drug interaction (F(1,13) = 12.08, p < 0.01). Bonferroni post hoc analyses showed no significant impact of day on saline-exposed mice (p = 0.99) but a significant decrease in withdrawal score after 14-day abstinence in morphine-exposed mice (p < 0.001).

Cell-specific effects of morphine-induced spontaneous acute withdrawal on nucleus accumbens AMPA receptor transmission. a Experimental timeline including five twice-daily injections of escalating morphine administration (i.p.) and 24 h post-final injection behavior assessment. Recordings were performed in coronal slices approximately 2 h following behavior. b Mean (1) global withdrawal scores and (2) locomotor activity 24 h following repeated saline (Sal, white; N = 7) or morphine (Mor, black; N = 12) injections. c Mean mEPSC amplitude (left) and frequency (right) in NAcSh (1) D1-MSNs [Sal (n = 6/N = 5), Morn = 11/N = 7)] and (2) D2-MSNs [Sal (n = 6/N = 5), Mor (n = 12/N = 8)]. d Mean mEPSC amplitude and frequency in NAcC (1) D1-MSNs [Sal (n = 8/N = 7), Mor (n = 12/N = 9)] and D2-MSNs [Sal (n = 5/N = 4), Mor (n = 11/N = 8). *p < 0.05, ***p < 0.001 vs Sal
To identify plasticity that parallel withdrawal symptoms, we measured mEPSCs at D1- and D2-MSNs in the NAcC and NAcSh sub-regions 24 h following the final injection of morphine and 2 h post-behavior assessment. In the NAcSh, neither the amplitude nor frequency of mEPSCs was altered in D1-MSNs (Fig. 3c) [amplitude: Sal (12.75 ± 1.23), Mor (12.01 ± 0.61), t(15) = 0.612, p = 0.55; frequency: Sal (5.58 ± 0.72), Mor (4.84 ± 0.50), t(15) = 0.862, p = 0.40]. Conversely, a significant increase in frequency but not amplitude was observed in D2-MSNs (Fig. 3c): [amplitude: Sal (10.39 ± 1.41), Mor (11.12 ± 0.81), t(15) = 0.487, p = 0.63; frequency: Sal (2.94 ± 0.27), Mor (6.40 ± 0.86), t(15) = 2.876, p < 0.05]. In the NAcC, morphine had no effect on mEPSC amplitude or frequency in D1-MSNs (Fig. 3d) [amplitude: Sal (12.76 ± 0.94), Mor (13.49 ± 0.67), t(18) = 0.655, p = 0.52; frequency: Sal (3.01 ± 0.68), Mor (4.25 ± 0.47), t(18) = 1.558, p = 0.14] or D2-MSNs (Fig. 3d) [amplitude: Sal (9.01 ± 0.42), Mor (9.80 ± 0.60), t(14) = 0.832, p = 0.42; frequency: Sal (3.52 ± 1.34), Mor (1.9 ± 0.33), t(14) = 1.536, p = 0.15]. Taken together, these findings suggest that, similar to naloxone-precipitated withdrawal (Zhu et al. 2016), spontaneous somatic withdrawal selectively increases excitatory drive at D2-MSNs in the NAcSh and that these effects are more enduring than previously known. Further, these data support the notion that adaptations 10–14 days following a less robust morphine regimen (5 × 10 mg/kg) or lack thereof 24 h following acute exposure is not associated with enduring somatic withdrawal, and that the lack of plasticity observed 24 h following an acute injection is distinctly different from withdrawal associated plasticity at a similar timepoint.
Inhibiting endocytosis of AMPA receptors in the NAcSh enhances morphine-primed reinstatement of place preference
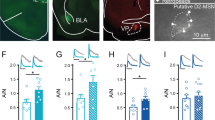
Inhibiting NAc GluA2-containing AMPAR trafficking disrupts amphetamine-induced sensitization and attenuates cocaine-induced reinstatement of cocaine seeking and place preference (Benneyworth et al. 2019; Brebner et al. 2005; Famous et al. 2008), suggesting that reductions in AMPAR signaling following drug re-exposure may reflect transient plasticity that triggers relapse-related behavior. Therefore, we examined whether morphine-induced depotentiation of AMPAR signaling involves receptor endocytosis and if this plasticity is causally involved in reinstatement of reward behavior. As plasticity associated with abstinence and drug re-exposure following repeated injections was largely confined to the NAcSh, we focused our efforts for this experiment in this sub-region. Using an approach previously shown to produce AMPAR plasticity and conditioned place preference (Hearing et al. 2016), all mice were initially conditioned with morphine (5 mg/kg). Mice were subsequently divided into three experimental groups, one group infused with the active peptide (Tat-GluA23Y) and receiving a saline priming injection (Mor/Tat(+)/Sal), a second group receiving the active peptide and a priming injection of morphine (Mor/Tat(+)/Mor), and a third infused with the inactive (Tat-GluA23S) isoform and receiving a morphine prime (Mor/Tat−)/Mor).
A Two-way ANOVA with test day as a repeated measure and treatment group as a between-subject factor revealed a significant day-by-treatment interaction (F(6,135) = 2.84, p < 0.05). Post hoc pairwise multiple comparisons showed that all three groups exhibited significant place preference compared to pre-test preference levels, and that preference did not significantly differ across all three groups during pre-test, preference, or extinction. For reinstatement testing, mice infused with the inactive Tat-GluA23S peptide receiving a morphine prime displayed a significant increase in preference compared to Tat-GluA23Y-infused mice injected with saline (Fig. 4b1) [Mor/Tat(+)/Sal (45.52 ± 83.7), Mor/Tat(−)/Mor (449 ± 79.05); p < 0.001], while morphine-primed mice infused with the active peptide (Mor/Tat(+)/Mor: 666 ± 87.8) showed a significant increase in preference reinstatement compared to both groups (p < 0.05 vs Mor/Tat(−)/Mor; p < 0.001 vs Mor/Tat(+)/Sal], indicating that blockade of AMPAR endocytosis enhanced reinstatement of place preference. Approximately 30–90 min following testing, ex vivo analysis of mEPSCs (Fig. 4b2) showed that mEPSC amplitude at D1-MSNs was significantly greater in mice infused with active Tat-GluA23Y compared to the inactive Tat-GluA23S [Mor/Tat(−)/Mor 16.76 ± 1.87, Mor/Tat(+)/Mor 11.24 ± 0.32, t(6) = 2.911, p < 0.05]. No significant effect of Tat peptide on mEPSC frequency was observed [Mor/Tat(−)/Mor (3.25 ± 0.48), Mor/Tat(+)/Mor (4.35 ± 0.95), t(6) = 1.032, p = 0.34]. Collectively, these data indicate that re-exposure to morphine drives endocytosis of AMPA receptors at D1-MSNs unlike cocaine (Benneyworth et al. 2019), and preventing such endocytosis exacerbates reinstatement of morphine-induced conditioned place preference.

Inhibiting AMPA receptor endocytosis in nucleus accumbens shell increases morphine-induced reinstatement of conditioned place preference. a (1) Representative Tat-peptide expression targeted to the NAc shell and (2) experimental timeline of conditioned place preference study including behavioral testing. Electrophysiological recordings were performed in Tat-expressing D1-MSNs fro sagittal slices approximately 30 min post-reinstatement test. All mice were conditioned with morphine. Following infusion with the active (Tat+) or inactive (Tat−) peptide, mice received a challenge injection of saline (Mor/Tat+/Sal, white), or morphine (Mor/Tat(−)/Mor, black; Mor/Tat(+)/Mor, gray), followed by a test of preference reinstatement 20 min later. b (1) Preference scores across test days in mice and (2) mEPSC amplitude (left) and frequency (right) from D1-MSNs expressing fluorescence following testing in a subset of mice receiving morphine injections with the active or inactive peptide. #p < 0.05 Mor/Tat(−)/Mor vs Mor/Tat(+)/Mor, ***p < 0.001 vs Mor/Tat+/Sal
Discussion
Here, we identify temporal- and region-specific changes in AMPAR signaling within discrete subpopulations of NAc MSNs associated with opioid reward, withdrawal, and relapse-like behavior. In agreement with our previous findings, we showed that protracted withdrawal from repeated morphine is associated with increases in synaptic drive at D1- and decreases at D2-MSNs predominantly in the NAcSh (Hearing et al. 2016). Similar to amphetamine and cocaine (Benneyworth et al. 2019; Jedynak et al. 2016; Kourrich et al. 2007), re-exposure to morphine produced bimodal plasticity; however, unlike cocaine, re-exposure to morphine triggered a reduction in drive at D1- and increased drive at D2-MSNs (Fig. 1; Ortinski et al. 2015). In contrast, acute morphine produced a transient increase in AMPAR-mediated neurotransmission at D1-MSNs in the NAcC and NAcSh (Fig. 2), whereas spontaneous withdrawal aligned with enhanced excitatory drive at D2-MSNs selectively in the NAcSh (Fig. 3). Unlike our previous findings with cocaine, blocking the underlying AMPAR endocytosis augmented rather than inhibited reinstatement of place preference following extinction training (Fig. 4), suggesting that similar forms of plasticity and post-drug experience may have distinct behavioral consequences across drug classes.
Acute morphine plasticity
Despite evidence that opioids acutely reduce glutamate release in the NAc (Martin et al. 1997; Sepulveda et al. 1998), relatively little is known regarding the role of NAc postsynaptic AMPAR and NMDAR signaling in the acute rewarding effects of opioids. Biochemical data have shown that expression of AMPARs is decreased in the NAcC 3 days following an acute morphine exposure (Jacobs et al. 2005) and that GluA1 surface expression is reduced in combined NAc tissue 24 h following acute exposure (Herrold et al. 2013). In the present study, we found that AMPAR-mediated mEPSC amplitude and frequency was elevated at D1-MSNs in the NAcC and NAcSh 3 h following an acute injection of morphine. Unlike previous studies, this time course aligns with residual motor activity following drug exposure as well as the acute rewarding effects of morphine rather than withdrawal (Rothwell et al. 2012). The NAcC plays a key role in initializing reward-related motor activity (Sesack and Grace 2010; Shiflett and Balleine 2011; Voorn et al. 2004) and the NAcSh in opioid-related reward and reinforcement learning (Graziane et al. 2016; Hearing et al. 2016; Heimer et al. 1997; Sesack and Grace 2010). Moreover, recent findings have shown that activation of NAc D1-MSN circuits promotes reward and addiction-related behavior (Dobi et al. 2011; Graziane et al. 2016; Hearing et al. 2016, 2018; Kim et al. 2011; Lobo et al. 2010; Ortinski et al. 2012, 2015; Pascoli et al. 2011; Smith et al. 2013; Suska et al. 2013). Thus, increased signaling at D1-MSNs in the NAcSh and NAcC likely contributes to the rewarding and psychomotor effects of acute morphine, respectively. This is reflected in enhanced drug-induced behavioral output observed after repeated and acute morphine (Hearing et al. 2016); behavior is blunted by depotentiation of postsynaptic AMPAR signaling.
It remains unclear whether elevations in AMPAR transmission at D1-MSNs in the NAcSh and NAcC reflect pre- or postsynaptic events. Accordingly, observed increases in mEPSC frequency may be attributed to increased receptor (or synapse) number rather than glutamate release probability (Graziane et al. 2016; Kerchner and Nicoll 2008) given the dampening effects of acute opioids on glutamate release in the nucleus accumbens (Sepulveda et al. 2004). Recent work has shown that a single cocaine exposure upregulates NAcSh GluA2-lacking AMPAR signaling at NAcSh D1R-MSN synapses—an adaptation observed 7, but not 1 day following drug exposure (Terrier et al. 2016). Although an in-depth comparison of cocaine-induced changes at the acute post-exposure period used here is lacking, acute morphine adaptations in the NAcSh also appear to require a period of withdrawal, as they were not observed 24 h following exposure. Alternatively, while mEPSC amplitude and frequency, and GluA1 surface expression are elevated in pooled MSNs and tissue punches during early withdrawal from acute amphetamine and repeated cocaine, no changes in mEPSCs were observed 24 h following acute morphine in the present study. Although the reason for this distinction is unclear, it may reflect a higher prevalence of mu-opioid receptors in the NAcSh compared to the NAcC (Svingos et al. 1997). Regardless, given increasing evidence that opioids and psychostimulants produce divergent neurophysiological and behavior effects, an important question moving forward will be to determine whether increased AMPAR signaling with acute morphine merely reflects a synaptic scaling event in response to reduced glutamate availability or if this plasticity persists.
Withdrawal-related AMPAR plasticity
In addition to reward, increased AMPAR signaling in the NAcSh has also been attributed to aversive effects of morphine withdrawal (Russell et al. 2016; Sepulveda et al. 2004). Indirect pharmacological evidence as well as direct measures of synaptic plasticity indicate that these adaptations may be confined to D2-MSNs in the NAc (Harris and Aston-Jones 1994; Russell et al. 2016; Zhu et al. 2016). In the present study, we show for the first time that, similar to naloxone-precipitated withdrawal, spontaneous somatic withdrawal aligns with increased glutamate transmission selectively at D2-MSNs in the NAcSh. Surprisingly, a similar phenomenon was also observed immediately following acute morphine exposure in both instances, though effects were confined to changes in mEPSC frequency. This may reflect increases in quantal release from pooled inputs, as withdrawal-related aversion memories are associated with increased signaling at thalamus but not prefrontal cortex or amygdala inputs at D2-MSNs (Zhu et al. 2016), but not definitively excluding a potential change in AMPAR expression (Graziane et al. 2016; Kerchner and Nicoll 2008). Importantly, we also observed that acute morphine increased signaling at NAcSh and NAcC D1-MSNs, possibly offsetting of the negative affect of a single post-morphine exposure.
Although a single exposure to morphine does not evoke spontaneous withdrawal, naloxone-precipitated withdrawal is possible 24 h after a single injection of morphine (Rothwell et al. 2012). Thus, our findings appear to agree with conclusions drawn by Russell et al. (2016) in that upregulation of glutamate transmission (at D2-MSNs in the present study) may reflect plasticity that primes NAc circuits for subsequent activation upon withdrawal (Russell et al. 2016). Although unclear, the apparent discrepancy between observed reductions in GluA2-lacking AMPAR surface expression immediately following precipitated withdrawal (Russell et al. 2016) and lack of changes to amplitude in the present study may reflect distinctions in the time of observation (30 min vs 24 h) or method of withdrawal (precipitated vs spontaneous). Alternatively, because mEPSCs likely reflect binding at receptors in the synapse, it is possible that reduced surface expression detected using biochemical measures (e.g., biotinylation) reflect sampling from synaptic and peri/extra-synaptic AMPARs that have been primed but not trafficked to or from the postsynaptic density.
Region- and cell-specific bimodal AMPAR plasticity
Our previous findings show that 10–14 days after repeated morphine expression of GluA2-lacking AMPARs increases at pooled inputs to D1-MSNs while reducing excitatory drive at D2-MSNs in the NAcSh and NAcC (Hearing et al. 2016). In the present study, we also observed reductions in mEPSC frequency at NAcC D2-MSNs, but only a trend towards reduced signaling at NAcSh D2-MSNs. The prominence of plasticity in the NAcSh vs NAcC appears to contrast effects of repeated cocaine, but is consistent with findings following 10–14 days of withdrawal from repeated amphetamine (Jedynak et al. 2016); however, these effects have not been readily characterized in D1- vs D2-MSNs. In the current study, reductions in AMPAR signaling following morphine re-exposure were ostensibly isolated to D1-MSNs (Fig. 1b). In turn, morphine-treated mice infused with the active Tat peptide in the NAcSh exhibited increased mEPSC amplitudes compared to those infused with the inactive form (Fig. 4b). Thus, morphine re-exposure likely triggers endocytosis of synaptic GluA2-containing AMPARs in NAcSh D1-MSNs. As the Tat peptide inhibits activity-dependent rather than constitutive removal of synaptic GluA2-containing AMPARs, this endocytosis is more likely to reflect a rapid, LTD-like process than a slow and consistent removal of synaptic AMPARs over time (Ahmadian et al. 2004; Dong et al. 2015; Lee et al. 2002; Scholz et al. 2010; Wang et al. 2017; Yoon et al. 2009).
It should be noted that the precision of Tat injections in the current study was not specific with regard to the rostral–caudal and dorsal–ventral axes. This is significant as prior work has shown distinctions in how NAcSh cell subpopulations and AMPAR signaling along the dorsal–ventral (Al-Hasani et al. 2015) and rostro-caudal gradients (Reynolds and Berridge 2003) differentially regulate reward- and aversive-driven behavior. As our recordings were primarily, but not exclusively focused within the dorsal–medial region but even distribution along the rostral–caudal axis, it will be important for future studies to examine anatomical distinctions when identifying causality between plasticity and behavior.
Our current findings indicate that re-exposure to morphine promotes AMPAR endocytosis specifically at D1-MSN synapses previously potentiated during withdrawal from morphine; however, it is possible that reductions in synaptic strength may be occurring at adjacent, rather than previously potentiated synapses—a possibility difficult to demonstrate definitively. Although impossible to exclude, it is unlikely that inclusion of extinction produced alternate plasticity in CPP studies compared to those observed in challenge experiments, as our previous work showed identical cell-specific plasticity in mice following home cage abstinence and extinction. While the significance of this bimodal phenomenon is not yet clear, one intriguing possibility is that re-exposure to opioids may represent a temporary quelling of drug craving and in turn a trend towards returning to prior levels of D1-MSN excitation.
Role of bidirectional plasticity in reinstatement
Our previous work showed that in vivo reversal of morphine-induced pathophysiology at NAcSh D1-MSNs with optogenetic stimulation or treatment with the antibiotic, ceftriaxone, blocked reinstatement of morphine-evoked place preference (Hearing et al. 2016). One straightforward interpretation of these findings is that a progressive enhancement of AMPAR signaling during withdrawal serves as a common mechanism for driving addiction-related behavior (Kalivas 2009; Kalivas and Hu 2006), and that reducing synaptic strength prior to drug re-exposure impairs drug-induced behavior. This contention is also supported by numerous studies showing that re-exposure to drug or drug-associated cues induces a rapid potentiation (i.e., release) of NAc glutamatergic signaling in cocaine-, nicotine-, or heroin-withdrawn rats (Gipson et al. 2013a, b; Shen et al. 2011; Trantham-Davidson et al. 2012).
On the other hand, recent work by our group and others indicate that re-exposure to cocaine triggers a rapid reduction in synaptic strength in the NAc akin to LTD (Benneyworth et al. 2019; Ebner et al. 2018; Ingebretson et al. 2018; Jedynak et al. 2016), and that this short-term plasticity is necessary and sufficient to reinstate place preference (Benneyworth et al. 2019)—suggesting that decreases in excitatory drive onto NAc MSNs, particularly in the NAcSh, may promote reinstatement to drug seeking. Thus, our previously observed blockade of preference reinstatement may merely reflect an occlusion of short-term plasticity associated with morphine re-exposure and the ability to trigger behavior (Hearing et al. 2016; Pascoli et al. 2011, 2014). In the present study, blockade of AMPAR endocytosis augmented reinstatement of place preference; thus, unlike cocaine, increased expression of AMPAR during abstinence appears to be the primary driver of reinstatement, with internalization of AMPARs following morphine re-exposure perhaps reflecting a secondary synaptic scaling (Turrigiano 2008) in response to augmented glutamate release, but see (Siahposht-Khachaki et al. 2017). Regardless, these findings show that although two distinct drug classes can produce seemingly similar forms of plasticity, the behavioral consequences of this plasticity appear to be profoundly different.
Conclusion
Though psychostimulants and opioids share rewarding properties that can lead to uncontrollable drug use and relapse vulnerability, opioids are addictive substances with the ability to induce chemical dependence from which relapse is driven by attempts to alleviate somatic and psychological withdrawal symptoms. By modeling dosing regimens in a preclinical setting, we sought to parallel acute, repeated, and dependence-inducing opioid consumption. Analysis of AMPAR signaling from each dosing revealed unique and overlapping neuroplasticity to excitatory signaling at NAcSh and NAcC MSNs. Future therapeutic interventions should take into consideration that drug-induced neuroplasticity shared across drug classes does not inherently share functional consequences at the level of the neural circuit or in terms of behavior. Thus, more thorough characterization of opioid-induced plasticity is needed to provide more efficient and effective therapies for opioid use disorder.
References
Ahmadian G et al (2004) Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J 23:1040–1050. https://doi.org/10.1038/sj.emboj.7600126
Al-Hasani R et al (2015) Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron 87:1063–1077. https://doi.org/10.1016/j.neuron.2015.08.019
Badiani A, Belin D, Epstein D, Calu D, Shaham Y (2011) Opiate versus psychostimulant addiction: the differences do matter. Nat Rev Neurosci 12:685–700. https://doi.org/10.1038/nrn3104
Baharlouei N, Sarihi A, Komaki A, Shahidi S, Haghparast A (2015) Blockage of acquisition and expression of morphine-induced conditioned place preference in rats due to activation of glutamate receptors type II/III in nucleus accumbens. Pharmacol Biochem Behav 135:192–198. https://doi.org/10.1016/j.pbb.2015.06.004
Benneyworth MA et al (2019) Synaptic depotentiation and mGluR5 activity in the nucleus accumbens drives cocaine-primed reinstatement of place preference. J Neurosci. https://doi.org/10.1523/JNEUROSCI.3020-17.2019
Bossert JM, Busch RF, Gray SM (2005) The novel mGluR2/3 agonist LY379268 attenuates cue-induced reinstatement of heroin seeking. NeuroReport 16:1013–1016
Bossert JM, Gray SM, Lu L, Shaham Y (2006) Activation of group II metabotropic glutamate receptors in the nucleus accumbens shell attenuates context-induced relapse to heroin seeking. Neuropsychopharmacology 31:2197–2209. https://doi.org/10.1038/sj.npp.1300977
Brebner K et al (2005) Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 310:1340–1343. https://doi.org/10.1126/science.1116894
Chartoff EH, Connery HS (2014) It’s MORe exciting than mu: crosstalk between mu opioid receptors and glutamatergic transmission in the mesolimbic dopamine system. Front Pharmacol 5:116. https://doi.org/10.3389/fphar.2014.00116
Cruz HG, Berton F, Sollini M, Blanchet C, Pravetoni M, Wickman K, Luscher C (2008) Absence and rescue of morphine withdrawal in GIRK/Kir3 knock-out mice. J Neurosci 28:4069–4077. https://doi.org/10.1523/jneurosci.0267-08.2008
Desole MS et al (1996) Effects of morphine treatment and withdrawal on striatal and limbic monoaminergic activity and ascorbic acid oxidation in the rat. Brain Res 723:154–161
Dobi A, Seabold GK, Christensen CH, Bock R, Alvarez VA (2011) Cocaine-induced plasticity in the nucleus accumbens is cell specific and develops without prolonged withdrawal. J Neurosci 31:1895–1904. https://doi.org/10.1523/JNEUROSCI.5375-10.2011
Dong Z et al (2015) Long-term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Investig 125:234–247. https://doi.org/10.1172/JCI77888
Dworkin SI, Guerin GF, Goeders NE, Smith JE (1988) Kainic acid lesions of the nucleus accumbens selectively attenuate morphine self-administration. Pharmacol Biochem Behav 29:175–181
Ebner SR, Larson EB, Hearing MC, Ingebretson AE, Thomas MJ (2018) Extinction and reinstatement of cocaine-seeking in self-administering mice is associated with bidirectional AMPAR-mediated plasticity in the nucleus accumbens shell. Neuroscience 384:340–349. https://doi.org/10.1016/j.neuroscience.2018.05.043
Enrico P et al (1998) Effect of naloxone on morphine-induced changes in striatal dopamine metabolism and glutamate, ascorbic acid and uric acid release in freely moving rats. Brain Res 797:94–102
Everitt BJ, Parkinson JA, Olmstead MC, Arroyo M, Robledo P, Robbins TW (1999) Associative processes in addiction and reward the role of amygdala-ventral striatal subsystems. Ann N Y Acad Sci 877:412–438
Famous KR, Kumaresan V, Sadri-Vakili G, Schmidt HD, Mierke DF, Cha JH, Pierce RC (2008) Phosphorylation-dependent trafficking of GluR2-containing AMPA receptors in the nucleus accumbens plays a critical role in the reinstatement of cocaine seeking. J Neurosci 28:11061–11070. https://doi.org/10.1523/jneurosci.1221-08.2008
Fujio M et al (2005) Gene transfer of GLT-1, a glutamate transporter, into the nucleus accumbens shell attenuates methamphetamine- and morphine-induced conditioned place preference in rats. Eur J Neurosci 22:2744–2754. https://doi.org/10.1111/j.1460-9568.2005.04467.x
Gipson CD, Kupchik YM, Shen H, Reissner KJ, Thomas CA, Kalivas PW (2013a) Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron 77:867–872. https://doi.org/10.1016/j.neuron.2013.01.005
Gipson CD, Reissner KJ, Kupchik YM, Smith AC, Stankeviciute N, Hensley-Simon ME, Kalivas PW (2013b) Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci USA 110:9124–9129. https://doi.org/10.1073/pnas.1220591110
Gracy KN, Dankiewicz LA, Koob GF (2001) Opiate withdrawal-induced fos immunoreactivity in the rat extended amygdala parallels the development of conditioned place aversion. Neuropsychopharmacology 24:152–160. https://doi.org/10.1016/S0893-133X(00)00186-X
Graziane NM et al (2016) Opposing mechanisms mediate morphine- and cocaine-induced generation of silent synapses. Nat Neurosci 19:915–925. https://doi.org/10.1038/nn.4313
Harris GC, Aston-Jones G (1994) Involvement of D2 dopamine receptors in the nucleus accumbens in the opiate withdrawal syndrome. Nature 371:155–157. https://doi.org/10.1038/371155a0
Hearing M (2019) Prefrontal-accumbens opioid plasticity: implications for relapse and dependence. Pharmacol Res 139:158–165. https://doi.org/10.1016/j.phrs.2018.11.012
Hearing M, Kotecki L, de Velasco EMF, Fajardo-Serrano A, Chung HJ, Lujan R, Wickman K (2013) Repeated cocaine weakens GABA(B)-Girk signaling in layer 5/6 pyramidal neurons in the prelimbic cortex. Neuron 80:159–170. https://doi.org/10.1016/j.neuron.2013.07.019
Hearing MC et al (2016) Reversal of morphine-induced cell-type-specific synaptic plasticity in the nucleus accumbens shell blocks reinstatement. Proc Natl Acad Sci USA 113:757–762. https://doi.org/10.1073/pnas.1519248113
Hearing M, Graziane N, Dong Y, Thomas MJ (2018) Opioid and psychostimulant plasticity: targeting overlap in nucleus accumbens glutamate signaling. Trends Pharmacol Sci 39:276–294. https://doi.org/10.1016/j.tips.2017.12.004
Heimer L, Zahm DS, Churchill L, Kalivas PW, Wohltmann C (1991) Specificity in the projection patterns of accumbal core and shell in the rat. Neuroscience 41:89–125
Heimer L, Alheid GF, de Olmos JS, Groenewegen HJ, Haber SN, Harlan RE, Zahm DS (1997) The accumbens: beyond the core-shell dichotomy. J Neuropsychiatry Clin Neurosci 9:354–381. https://doi.org/10.1176/jnp.9.3.354
Herrold AA, Persons AL, Napier TC (2013) Cellular distribution of AMPA receptor subunits and mGlu5 following acute and repeated administration of morphine or methamphetamine. J Neurochem 126:503–517. https://doi.org/10.1111/jnc.12323
Hipps PP, Eveland MR, Meyer ER, Sherman WR, Cicero TJ (1976) Mass fragmentography of morphine: relationship between brain levels and analgesic activity. J Pharmacol Exp Ther 196:642–648
Ingebretson AE, Hearing MC, Huffington ED, Thomas MJ (2018) Endogenous dopamine and endocannabinoid signaling mediate cocaine-induced reversal of AMPAR synaptic potentiation in the nucleus accumbens shell. Neuropharmacology 131:154–165. https://doi.org/10.1016/j.neuropharm.2017.12.011
Jacobs EH, Wardeh G, Smit AB, Schoffelmeer AN (2005) Morphine causes a delayed increase in glutamate receptor functioning in the nucleus accumbens core. Eur J Pharmacol 511:27–30. https://doi.org/10.1016/j.ejphar.2005.02.009
Jedynak J et al (2016) Cocaine and amphetamine induce overlapping but distinct patterns of AMPAR plasticity in nucleus accumbens medium spiny neurons. Neuropsychopharmacology 41:464–476. https://doi.org/10.1038/npp.2015.168
Kalivas PW (2009) The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10:561–572. https://doi.org/10.1038/nrn2515
Kalivas PW, Hu XT (2006) Exciting inhibition in psychostimulant addiction. Trends Neurosci 29:610–616. https://doi.org/10.1016/j.tins.2006.08.008
Kerchner GA, Nicoll RA (2008) Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci 9:813–825. https://doi.org/10.1038/nrn2501
Kim J, Park BH, Lee JH, Park SK, Kim JH (2011) Cell type-specific alterations in the nucleus accumbens by repeated exposures to cocaine. Biol Psychiatry 69:1026–1034. https://doi.org/10.1016/j.biopsych.2011.01.013
Koob GF, Wall TL, Bloom FE (1989) Nucleus accumbens as a substrate for the aversive stimulus effects of opiate withdrawal. Psychopharmacology 98:530–534
Kourrich S, Rothwell PE, Klug JR, Thomas MJ (2007) Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci 27:7921–7928. https://doi.org/10.1523/JNEUROSCI.1859-07.2007
Le Moine C, Bloch B (1995) D1 and D2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. J Comp Neurol 355:418–426. https://doi.org/10.1002/cne.903550308
Lee SH, Liu L, Wang YT, Sheng M (2002) Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 36:661–674
Lobo DS, Kennedy JL (2006) The genetics of gambling and behavioral addictions. CNS Spectr 11:931–939
Lobo MK et al (2010) Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330:385–390. https://doi.org/10.1126/science.1188472
Martin G, Nie Z, Siggins GR (1997) μ-Opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. J Neurosci 17:11–22
Olds ME (1982) Reinforcing effects of morphine in the nucleus accumbens. Brain Res 237:429–440
Ortinski PI, Vassoler FM, Carlson GC, Pierce RC (2012) Temporally dependent changes in cocaine-induced synaptic plasticity in the nucleus accumbens shell are reversed by D1-like dopamine receptor stimulation. Neuropsychopharmacology 37:1671–1682. https://doi.org/10.1038/npp.2012.12
Ortinski PI, Briand LA, Pierce RC, Schmidt HD (2015) Cocaine-seeking is associated with PKC-dependent reduction of excitatory signaling in accumbens shell D2 dopamine receptor-expressing neurons. Neuropharmacology 92:80–89. https://doi.org/10.1016/j.neuropharm.2015.01.002
Papaleo F, Contarino A (2006) Gender- and morphine dose-linked expression of spontaneous somatic opiate withdrawal in mice. Behav Brain Res 170:110–118. https://doi.org/10.1016/j.bbr.2006.02.009
Pascoli V, Turiault M, Luscher C (2011) Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature 481:71–75. https://doi.org/10.1038/nature10709
Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, Luscher C (2014) Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509:459–464. https://doi.org/10.1038/nature13257
Reynolds SM, Berridge KC (2003) Glutamate motivational ensembles in nucleus accumbens: rostrocaudal shell gradients of fear and feeding. Eur J Neurosci 17:2187–2200
Rothwell PE, Kourrich S, Thomas MJ (2011) Synaptic adaptations in the nucleus accumbens caused by experiences linked to relapse. Biol Psychiatry 69:1124–1126. https://doi.org/10.1016/j.biopsych.2010.12.028
Rothwell PE, Thomas MJ, Gewirtz JC (2012) Protracted manifestations of acute dependence after a single morphine exposure. Psychopharmacology 219:991–998. https://doi.org/10.1007/s00213-011-2425-y
Russell SE, Puttick DJ, Sawyer AM, Potter DN, Mague S, Carlezon WA Jr, Chartoff EH (2016) Nucleus accumbens AMPA receptors are necessary for morphine-withdrawal-induced negative-affective states in rats. J Neurosci 36:5748–5762. https://doi.org/10.1523/jneurosci.2875-12.2016
Schmidt HD, Schassburger RL, Guercio LA, Pierce RC (2013) Stimulation of mGluR5 in the accumbens shell promotes cocaine seeking by activating PKC gamma. J Neurosci 33:14160–14169. https://doi.org/10.1523/jneurosci.2284-13.2013
Schmidt HD, Kimmey BA, Arreola AC, Pierce RC (2015) Group I metabotropic glutamate receptor-mediated activation of PKC gamma in the nucleus accumbens core promotes the reinstatement of cocaine seeking. Addict Biol 20:285–296. https://doi.org/10.1111/adb.12122
Scholz R, Berberich S, Rathgeber L, Kolleker A, Kohr G, Kornau HC (2010) AMPA receptor signaling through BRAG2 and Arf6 critical for long-term synaptic depression. Neuron 66:768–780. https://doi.org/10.1016/j.neuron.2010.05.003
Schulteis G, Markou A, Gold LH, Stinus L, Koob GF (1994) Relative sensitivity to naloxone of multiple indices of opiate withdrawal: a quantitative dose–response analysis. J Pharmacol Exp Ther 271:1391–1398
Sepulveda MJ, Hernandez L, Rada P, Tucci S, Contreras E (1998) Effect of precipitated withdrawal on extracellular glutamate and aspartate in the nucleus accumbens of chronically morphine-treated rats: an in vivo microdialysis study. Pharmacol Biochem Behav 60:255–262
Sepulveda J, Oliva P, Contreras E (2004) Neurochemical changes of the extracellular concentrations of glutamate and aspartate in the nucleus accumbens of rats after chronic administration of morphine. Eur J Pharmacol 483:249–258
Sesack SR, Grace AA (2010) Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology 35:27–47. https://doi.org/10.1038/npp.2009.93
Shen H, Moussawi K, Zhou W, Toda S, Kalivas PW (2011) Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors. Proc Natl Acad Sci USA 108:19407–19412. https://doi.org/10.1073/pnas.1112052108
Shen HW, Scofield MD, Boger H, Hensley M, Kalivas PW (2014) Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. J Neurosci 34:5649–5657. https://doi.org/10.1523/JNEUROSCI.4564-13.2014
Shiflett MW, Balleine BW (2011) Molecular substrates of action control in cortico-striatal circuits. Prog Neurobiol 95:1–13. https://doi.org/10.1016/j.pneurobio.2011.05.007
Siahposht-Khachaki A, Fatahi Z, Yans A, Khodagholi F, Haghparast A (2017) Involvement of AMPA/kainate glutamate receptor in the extinction and reinstatement of morphine-induced conditioned place preference: a behavioral and molecular study. Cell Mol Neurobiol 37:315–328. https://doi.org/10.1007/s10571-016-0371-2
Smith RJ, Lobo MK, Spencer S, Kalivas PW (2013) Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr Opin Neurobiol 23:546–552. https://doi.org/10.1016/j.conb.2013.01.026
Suska A, Lee BR, Huang YH, Dong Y, Schluter OM (2013) Selective presynaptic enhancement of the prefrontal cortex to nucleus accumbens pathway by cocaine. Proc Natl Acad Sci USA 110:713–718. https://doi.org/10.1073/pnas.1206287110
Svingos AL, Moriwaki A, Wang JB, Uhl GR, Pickel VM (1997) μ-Opioid receptors are localized to extrasynaptic plasma membranes of GABAergic neurons and their targets in the rat nucleus accumbens. J Neurosci 17:2585–2594
Terrier J, Luscher C, Pascoli V (2016) Cell-type specific insertion of GluA2-lacking AMPARs with cocaine exposure leading to sensitization cue-induced seeking, and incubation of craving. Neuropsychopharmacology 41:1779–1789. https://doi.org/10.1038/npp.2015.345
Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001) Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4:1217–1223. https://doi.org/10.1038/nn757
Trantham-Davidson H, LaLumiere RT, Reissner KJ, Kalivas PW, Knackstedt LA (2012) Ceftriaxone normalizes nucleus accumbens synaptic transmission, glutamate transport, and export following cocaine self-administration and extinction training. J Neurosci 32:12406–12410. https://doi.org/10.1523/jneurosci.1976-12.2012
Turrigiano GG (2008) The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135:422–435. https://doi.org/10.1016/j.cell.2008.10.008
van der Laan JW, de Groot G (1988) Changes in locomotor-activity patterns as a measure of spontaneous morphine withdrawal: no effect of clonidine. Drug Alcohol Depend 22:133–140
van der Laan JW, van’t Land CJ, Loeber JG, de Groot GV (1991) Validation of spontaneous morphine withdrawal symptoms in rats. Arch Int Pharmacodyn Ther 311:32–45
van Ree JM, Gerrits MA, Vanderschuren LJ (1999) Opioids, reward and addiction: an encounter of biology, psychology, and medicine. Pharmacol Rev 51:341–396
Vekovischeva OY et al (2001) Morphine-induced dependence and sensitization are altered in mice deficient in AMPA-type glutamate receptor-A subunits. J Neurosci 21:4451–4459
Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM (2004) Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci 27:468–474. https://doi.org/10.1016/j.tins.2004.06.006
Wang Q, Li D, Bubula N, Campioni MR, McGehee DS, Vezina P (2017) Sensitizing exposure to amphetamine increases AMPA receptor phosphorylation without increasing cell surface expression in the rat nucleus accumbens. Neuropharmacology 117:328–337. https://doi.org/10.1016/j.neuropharm.2017.02.018
Wise RA (1989) Opiate reward: sites and substrates. Neurosci Biobehav Rev 13:129–133
Wydeven N et al (2014) Mechanisms underlying the activation of G-protein-gated inwardly rectifying K+ (GIRK) channels by the novel anxiolytic drug, ML297. Proc Natl Acad Sci USA 111:10755–10760. https://doi.org/10.1073/pnas.1405190111
Yoon BJ, Smith GB, Heynen AJ, Neve RL, Bear MF (2009) Essential role for a long-term depression mechanism in ocular dominance plasticity. Proc Natl Acad Sci USA 106:9860–9865. https://doi.org/10.1073/pnas.0901305106
Zahm DS, Brog JS (1992) On the significance of subterritories in the “accumbens” part of the rat ventral striatum. Neuroscience 50:751–767
Zhu Y, Wienecke CF, Nachtrab G, Chen X (2016) A thalamic input to the nucleus accumbens mediates opiate dependence. Nature 530:219–222. https://doi.org/10.1038/nature16954
Acknowledgements
The behavioral work is done in part with the support by the Mouse Behavior Core at the University of Minnesota, which received funding from the National Institute for Neurological Disorders and Stroke (P30 NS062158). These studies were also supported by funding from the National Institute on Drug Abuse Grant K99 DA038706 (to M.H.), R00DA038706 (M.H.), R00DA038706-04S1 (A.C.M), R01DA019666 (M.J.T.), K02DA035459 (M.J.T.) and T32 DA007234 (A.E.I.).
Funding
The following funding sources made the study possible: National Institute for Neurological Disorders and Stroke (P30 NS062158); National Institute on Drug Abuse Grant K99 DA038706 (to M.H.), R00DA038706 (M.H.), R00DA038706-04S1 (A.C.M), R01DA019666 (M.J.T.), K02DA035459 (M.J.T.) and T32 DA007234 (A.E.I.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to disclose.
Ethical approval
The research in the current study used mice single or group housed on a 12-h light/dark cycle with food and water available ad libitum with experiments run during the light portion. All experiments were approved by the University of Minnesota and Marquette University Institutional Animal Care and Use Committee.
Informed consent
All authors have given their consent for manuscript submission.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Madayag, A.C., Gomez, D., Anderson, E.M. et al. Cell-type and region-specific nucleus accumbens AMPAR plasticity associated with morphine reward, reinstatement, and spontaneous withdrawal. Brain Struct Funct 224, 2311–2324 (2019). https://doi.org/10.1007/s00429-019-01903-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00429-019-01903-y